

Abstract
The nonribosomal peptide synthetases (NRPSs) are one of the most promising resources for the production of new bioactive molecules. The mechanism of NRPS catalysis is based around sequential catalytic domains: these are organized into modules, where each module selects, modifies, and incorporates an amino acid into the growing peptide. The intermediates formed during NRPS catalysis are delivered between enzyme centers by peptidyl carrier protein (PCP) domains, which makes PCP interactions and movements crucial to NRPS mechanism. PCP movement has been linked to the domain alternation cycle of adenylation (A) domains, and recent complete NRPS module structures provide support for this hypothesis. However, it appears as though the A domain alternation alone is insufficient to account for the complete NRPS catalytic cycle and that the loaded state of the PCP must also play a role in choreographing catalysis in these complex and fascinating molecular machines.
Keywords: biosynthesis, enzymes, nonribosomal peptide synthetases, peptidyl carrier protein, protein structures
1. Structural and Biological Diversity of Nonribosomal Peptides
Secondary metabolites display a range of medicinally important activities including anticancer (bleomycin), antibiotic (vancomycin), antifungal (echinocandin), and immunosuppressive (cyclosporin) activity (Figure 1 A).1 Nonribosomal peptides (NRPs), which represent a particularly rich source of antimicrobial compounds,2 are synthesized independently of the ribosome by enzyme assemblies known as the nonribosomal peptide synthetases (NRPSs).3 By removing the constraints imposed by ribosome‐based synthesis, NRPs can be assembled from a range of monomers far greater than the standard proteinogenic amino acids: to date, more than 500 different monomers have been identified in NRPs, and these have dramatic effects on the structural and biological diversity of these compounds.4
Figure 1.
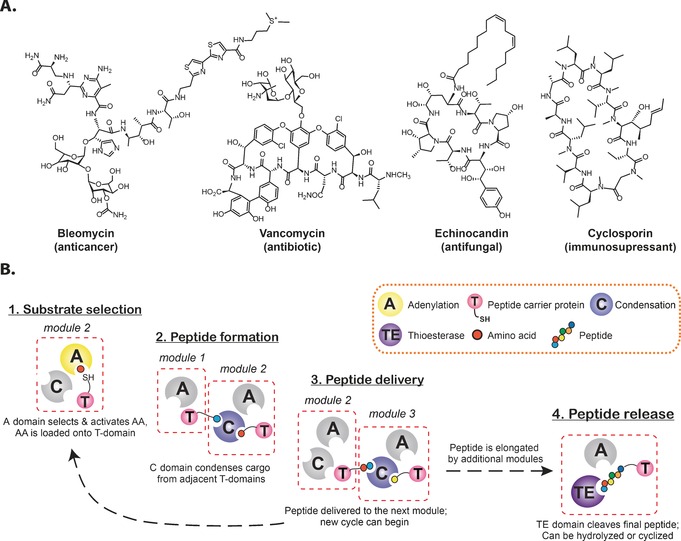
Nonribosomal peptide synthesis. A) Examples of bioactive NRPs. B) Linear NRPS biosynthesis is based on an assembly‐line‐like architecture, where substrate activation, modification, and peptide bond formation are catalyzed by a succession of active domains.
2. NRPS Assembly Lines
NRPSs utilize catalytic domains to perform different reactions during peptide synthesis. The majority of NRPS systems adopt a linear architecture, in which the catalytic domains are organized into modules that are each responsible for the incorporation of one amino acid into the growing peptide (non‐linear and iterative NRPS machineries are beyond the scope of this minireview).3 The minimal peptide extension module comprises 3 domains: adenylation (A), condensation (C), and peptidyl carrier protein (PCP, also known as thiolation, (T); Figure 1 B) domains.
The A domains select the amino acid monomer,5, 6 activate it by using ATP, and then load it onto the adjacent PCP domain. NRPS A domains comprise two subdomains, with the motions of the smaller (C‐terminal) subdomain allowing both selection/activation and PCP‐loading steps to occur within the same active site of the larger subdomain.7 Bacterial A domains have highly conserved structures, and their sequences can generally be used to predict the backbone peptide structure of the NRP. This is also a highly useful property for potential rational redesign of NRPSs.5, 6, 8
C domains catalyze peptide bond formation between PCP‐bound substrates: two PCPs bind to the C domain and the amine of a downstream aminoacyl‐PCP attacks the thioester of the upstream peptidyl‐PCP to form a new peptide bond. In this manner, the NRP is transferred downstream and elongated by one residue each cycle.9 The C domain plays an important role in the maintenance of the stereochemistry of the growing NRP,10 although our current understanding is limited by a lack of structures for substrate‐bound C domains.
Shuttling of intermediates between the A and C domains is performed by the PCP domain, a small (ca. 10 kDa) catalytically silent domain bearing an 18 Å phosphopantetheine (Ppant) arm that is added post‐translationally to a conserved serine residue at the N terminus of helix II (Figure 2 A).3 The Ppant moiety acts as a swinging arm to increase the “reach” of the PCP domain into the active sites of adjacent domains. Crucially, the Ppant arm terminates in a thiol moiety, which allows intermediates to be shuttled as thioesters whilst remaining reactive enough to support peptide bond formation in the C domain.
Figure 2.
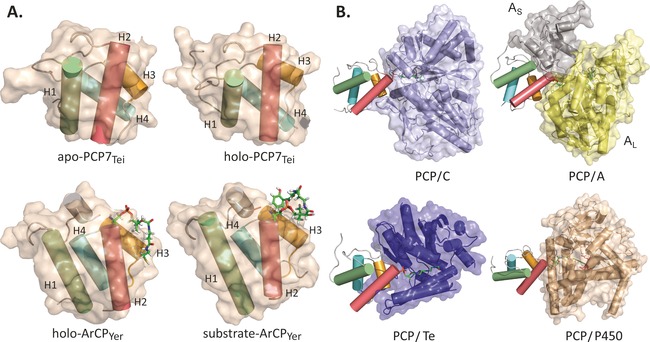
PCP structure and interactions. A) PCP7 from teicoplanin biosynthesis21 and ArCP from yersiniabactin biosynthesis23 show only subtle rearrangement of the typical four‐helix bundle upon Ppant linker and substrate loading, respectively. B) PCP interactions with adjacent domains are mediated mainly through helices II and III and the linkers preceding and following helix II (helices colored as in 2A). As=A domain, small subunit; AL=A domain, large subunit.12, 24, 26
The minimal C/A/PCP architecture is supplemented by an array of additional NRPS domains: the most common are epimerization (E) domains, which epimerize the l‐amino acids activated by A domains into their d‐form, and thioesterase (TE) domains, which release the completed NRP from the assembly line.3 Further diversity can be achieved through additional cis domains, such as formylation, oxidation, and methylation domains,3 or through interaction with enzymes that act in trans to the main NRPS machinery.11, 12, 13 In both cases, these interactions occur on PCP‐bound substrates, with PCP domains playing a major role in the recruitment of enzymes in trans.11, 12, 13 An exception to PCP‐driven recruitment is the X domain (an additional C/E‐type domain) from glycopeptide antibiotic biosynthesis, which is required to recruit cytochrome P450s to the PCP‐bound substrate to perform crosslinking of aromatic side chains.14, 15 Diversification of “standard” domain chemistry is also increasingly being observed in NRPS assembly lines: recent discoveries include β‐lactam formation,16 and TE‐catalyzed epimerization17 in norcardicin biosynthesis, and transesterification in salinamide biosynthesis.18
3. The Peptidyl Carrier Protein
The secondary and tertiary structures of PCP domains are highly conserved, with only minor deviations from the prototypical four‐helix bundle being documented to date.19, 20, 21, 22, 23, 24, 25, 26, 27 At the level of primary structure, PCPs are more variable,21 which gives rise to variations in local shape and charge distributions of the exposed and buried surfaces. This affects how individual PCPs interact with catalytic partners,21 especially in trans, where PCP recognition is crucial to selectivity.11, 20
An early hypothesis to explain the mechanism of NRPSs was based around large changes in PCP tertiary structure as a result of changes in PCP loading state, which would in turn affect and govern PCP interactions. This model was based on observations that a PCP domain from the tyrocidine synthetase exists in three different conformations (termed A, A/H, and H) depending on its loaded state.28 However, this has since been dismissed as an artifact of PCP domain excision from the larger synthetase since all other PCP structures elucidated to date adopt the A/H state.19, 20, 21, 22, 23, 24, 25, 26, 29, 30
The role of Ppant and substrate loading on PCP tertiary structure is therefore unclear. Analogous carrier proteins from polyketide synthetases can sequester their molecular cargo, although the biochemical relevance of this mechanism is not known.31, 32, 33 In the case of PCPs, the Ppant arm does not appear to interact appreciably with the protein core nor to alter the tertiary structure of the PCP in a significant way.21, 34 This observation supports the “swinging arm hypothesis”, in which the flexible Ppant arm delivers substrates to adjacent domains and the PCP domain serves as a largely rigid and chemically inert platform. Two recent NMR studies suggest that PCPs can interact weakly with the Ppant arm (Figure 2 A),23, 25 although these interactions were observed in atypical PCPs: the aryl‐acid‐loaded PCP from yersiniabactin synthetase23 and the pyrrole‐loaded PCP from pyoluteorin synthetase.25 In both cases, the Ppant arm was found to interact with helices II and III. While these findings suggest that the loaded state of the PCP could modulate NRPS interactions, our current understanding of PCP chain sequestration is limited by a lack of data on both prototypical and non‐excised PCP domains.
4. PCP Domain interactions With NRPS Catalytic Domains
Structural studies have shown that PCP domains use a similar protein surface in interactions with A, C, and TE domains,20 and that these interactions are mainly mediated by helices II/III and the loop connecting them (Figure 2 B). Hydrophobic interactions are commonly described,24, 26, 35, 36 but since cognate domains are selective to their partner PCP(s), further interactions, such as variable charge distribution,20 must also play important roles in recognition. Structures of holo‐PCPs in multidomain environments revealed that Ppant arm loading strengthens domain interactions.24, 26, 36 This appears to be especially crucial for PCP interactions in trans, for example with P450 enzymes.11, 12
Given the restricted motion available to PCP domains in a multidomain context, it is perhaps unsurprising that further interaction interfaces between PCPs and other partner domains have been demonstrated. For example, C and TE domains can interact with a PCP domain simultaneously at distinct interaction sites.35 Furthermore, in the recent multidomain AB3403 structure, the PCP domain is “packed” between the C and TE domains, revealing alternative helix I interactions with the TE domain when the Ppant arm is located in the binding site of the C domain.26 Given the subtle nature of substrate‐induced PCP conformational changes, it appears unlikely that this is the sole driving force for partner‐domain recruitment: clearly, another process must be driving NRPS catalysis.
5. Adenylation Domain Conformational Changes Guide PCP Interactions
The first structure of a complete NRPS module (SrfA‐C, C/A/PCP/TE) showed that while the Ppant linker site of the PCP is 16 Å from the C domain active site (and therefore within reach of its 18 Å Ppant arm), the same site is 57 and 43 Å away from the A and TE domain active sites, respectively.37 Substantial movements and domain rearrangements must thus take place within each module to allow the PCP domain to interact with all of its partners. It has been suggested that these PCP movements are coupled directly to the catalytic cycle of the A domains (Figure 3).7 During substrate activation, the small subdomain of the A domain is located close to the large subdomain (closed state),38 from where it rotates 140° to reveal the active site for subsequent PCP thiolation.39 The cycle is completed with a third, open state to allow substrate binding.37 Gulick and co‐workers identified a conserved LPxP motif in the linker region between the PCP and A domains that forms a stable interaction with the small subdomain of the A domain, thereby shortening the effective length of the linker.40 This interaction could serve to couple conformational changes in the A domain to movements of the PCP domain.40 In support of this model, the efficiency of A domains is enhanced by the presence of the PCP domain.41, 42 Further, A‐domain activity is faster in intact A/PCP di‐domains compared to excised domains,36 thus suggesting that the functions of the A and PCP domains are closely intertwined. The coupling of A‐domain activity to PCP motion has important implications for NRPS redesign, since alterations to the rate of A‐domain activity made during efforts to alter substrate selection would impact the overall efficiency of NRPS catalysis.
Figure 3.
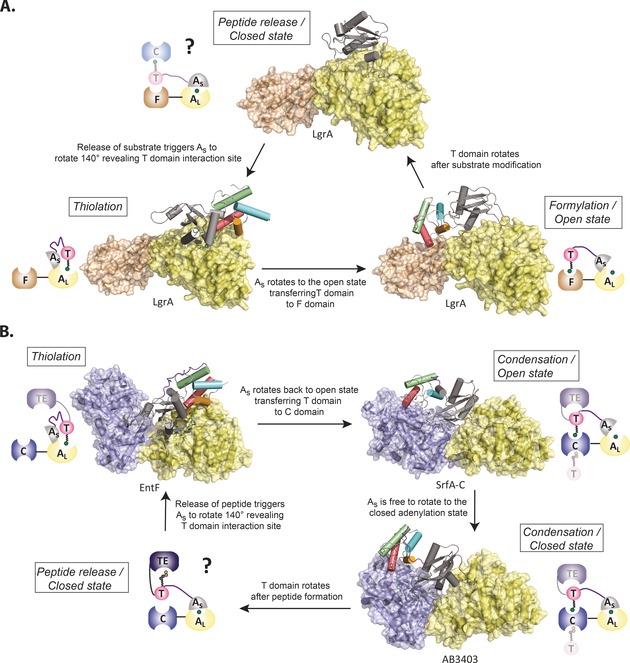
The A domain guides PCP domain movement. Large conformational rearrangements are needed for the PCP domain to move between the A‐ and C‐domain active sites. This movement is mediated by the domain alternation cycle of the A domain. Substrate loading state could mediate further movements to downstream domains. A) The catalytic cycle of an initiation module. B) The catalytic cycle of a termination module.26, 27, 37 Yellow=A, large subunit (AL); gray=A, small subunit (As); multicolored cylinders/pink circles=T (PCP) domain, brown= F domain, blue=C domain.
Recently published structures of two NRPS termination modules crystallized in different states (thiolation and condensation) further support the alternation theory (Figure 3).26 Nonetheless, the structures do raise questions as to how A and PCP domain movements are coupled to each other. For example, in the holo‐AB3404 structure (C/A/PCP/TE, condensation state), the A domain is in a closed state,26 which is inconsistent with the open state seen in the preceding SrfA‐C structure.37 Also, in the case of the recent gramicidin synthetase structures (LgrA, formylation (F)/A/PCP)27, the PCP interaction with the F domain would clash with the open state conformation of the A domain seen in the SrfA‐C structure. Unfortunately, the structure of the PCP is distorted in the LgrA structure, which displays a closed state of the A domain, thus leaving the question of PCP localization open to further analysis.
6. PCP Movements Guided by Substrate State
The apparent contradiction of PCP localization in these whole‐module structures appears to require an expansion of the theory that domain alternation is the sole driving force underlying NRPS catalysis. Conformational changes in C domains have been reported, but the function of these changes is unclear.43 The structure of AB340326 showed the C domain in a closed state, with strong interactions between the Ppant arm and the C domain, whilst in the apo‐SrfC‐A structure,37 the C domain is in an open state. Recent evidence shows that an A domain can catalyze two activation cycles (one to load the PCP and one to activate the next amino acid) on an intact module, which then halts in the absence of an upstream donor substrate.41 These observations, combined with the A‐domain states observed in complete‐module structures, suggest that interaction of the PCP with the upstream C domain “freezes” the A domain at the adenylation step if peptide is not available at the C‐domain donor site. This makes mechanistic sense, since further movement of the PCP to the thiolation state is not desirable before the bound substrate is released from the PCP domain.
A recent structure of a C domain bound to a tethered amino acid Ppant mimic revealed strong interactions between the binding pocket and the α‐amino group of the substrate.44 Since peptide bond formation alters this interaction, this could provide a sensor signal for C‐domain closing and PCP release, although this hypothesis needs further study. The mechanistic details that underpin C‐domain activity and the PCP interactions that support peptide bond formation are an area of great importance for future NRPS research, with biochemical studies to date lagging behind in vivo experiments.8, 45, 46
Although significant insight into the NRPS mechanism has been gained from recent complete‐module structures, knowledge of PCP domain interactions with downstream C and TE domains remains limited. Insight into these interactions can be gained however from a chemically trapped PCP/TE di‐domain structure.24 Overlaying the TE domain from this structure with the TE domain from the AB3403 structure shows how the lid region of the TE domain changes upon substrate binding (Figure 4). Strikingly, a simple rotation of the PCP domain appears to be sufficient to deliver the substrate from the C domain to the TE domain, and this could be accomplished without major structural rearrangement of the NRPS. Furthermore, a PCP/C di‐domain structure revealed the upstream PCP domain located close to the donor binding site of the C domain, but with the Ppant attachment site rotated away by 180° when the PCP domain is not loaded.47 This closely resembles the interaction between the PCP and TE domains in the AB3403 structure.26 Therefore, PCP loading state and acceptor‐domain flexibility, rather than A‐domain rearrangement, might well be the driving force for interaction between the PCP and C/TE domains. This model would be supported by the free movement of the TE domain seen in the EntF structure,26 since free rotation of the TE domain would facilitate binding of the substrate once it is released from the upstream C domain. A similar strategy could potentially be used to guide PCP domains to tailoring enzymes, but to date no structural information exists to examine this hypothesis.
Figure 4.
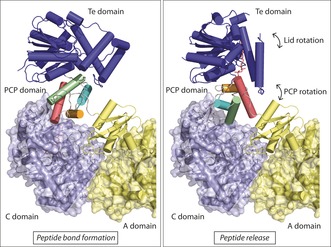
PCP interaction with the TE domain. Left: PCP interaction with the C domain from a whole‐module structure26 shows the TE domain in an open conformation but localized close to the PCP domain. Right: Overlaying the TE domain from a PCP/TE structure24 with the whole‐module TE domain shows lid closure upon PCP binding. Here, the PCP domain rotates but remains situated close to the C‐domain binding site.
7. Future Directions
Recent whole‐module structures provide insight into the mechanisms underpinning NRPS function and indicate that PCP domain movement is achieved through a combination of conformational changes and substrate loading states. The next challenge for the field is to understand the interactions between NRPS modules, since no structure of a multimodular NRPS has been resolved thus far. Marahiel recently proposed a model for a complete NRPS system based on multidomain structures, whereby a helical organization is achieved by rotating each module 120° along the helical axis, thereby leaving PCP domains close to the axis in such a way that intermediates would be protected from hydrolysis.48 A helical structure would orientate the C domains from adjacent modules in close proximity, thus allowing a simple rotation of the PCP domain to deliver substrate from one C domain to the next, which fits well with the current proposed catalytic model of an NRPS assembly line. While structural studies provide important insight into NRPS systems, they now need to be complemented with further biochemical characterization of complete modules, since it has become obvious that domain interactions both between and within modules play vital roles in catalysis.36, 41, 45, 46 Such studies are especially crucial to enable the effective redesign of NRPS machineries, since understanding the sources of substrate specificity for complete NRPS modules is currently lagging behind in vivo NRPS redesign.8 Given the importance of NRPs, these efforts are clearly needed to improve our access to derivatives of these medically relevant natural products.
Dedicated to Mohamed Marahiel
Biographical Information
Tiia Kittilä, born in 1984, studied biochemistry at the University of Helsinki, Finland, and completed her M.Sc. thesis at the Wihuri Research Institute. She joined the group of Max Cryle in 2013 as a DAAD scholar and is currently in the latter phase of her PhD studies. Her work focuses on the biosynthesis of the glycopeptide antibiotic teicoplanin.

Biographical Information
Aurelio Mollo, born in 1995, is studying undergraduate chemistry with a focus on biochemistry at Haverford College. He is currently working in the group of Dr. Louise Charkoudian, where he is involved in devising a chemoenzymatic route to the synthesis of the heptapeptide antibiotic complestatin.

Biographical Information
Louise K. Charkoudian, born in 1980, received her Ph.D. in bioinorganic chemistry at Duke University. After postdoctoral studies in bioorganic chemistry in the laboratory of Prof. Chaitan Khosla at Stanford University, she joined the faculty at Haverford College. Her research focuses on understanding and engineering natural product synthases.

Biographical Information
Max J. Cryle, born in 1979, obtained his Ph.D. in chemistry from the University of Queensland in 2006. He moved to the Max Planck Institute for Medical Research in Heidelberg as an HFSP Cross‐Disciplinary Fellow and later as an Emmy Noether group leader. He is now an EMBL Australia group leader in the Biomedicine Discovery Institute at Monash University, where he works on understanding antibiotic biosynthesis and developing new antibiotics.

Acknowledgements
The authors are grateful for financial support from Haverford College (to A.M. and L.K.C.), the Research Corporation for Science Advancement Cottrell Scholars Award (to L.K.C.), the Deutsche Akademischer Austausch Dienst (Graduate School Scholarship Program to T.K.) and Monash University, EMBL Australia and the Deutsche Forschungsgemeinschaft Emmy‐Noether Program, CR 392/1‐1 (to M.J.C.).
T. Kittilä, A. Mollo, L. K. Charkoudian, M. J. Cryle, Angew. Chem. Int. Ed. 2016, 55, 9834.
Contributor Information
Dr. Louise K. Charkoudian, Email: lcharkou@haverford.edu
Dr. Max J. Cryle, Email: max.cryle@monash.edu.
References
- 1. Newman D. J., Cragg G. M., J. Nat. Prod. 2007, 70, 461. [DOI] [PubMed] [Google Scholar]
- 2. Felnagle E. A., Jackson E. E., Chan Y. A., Podevels A. M., Berti A. D., McMahon M. D., Thomas M. G., Mol. Pharmaceutics 2008, 5, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hur G. H., Vickery C. R., Burkart M. D., Nat. Prod. Rep. 2012, 29, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walsh C. T., O'Brien R. V., Khosla C., Angew. Chem. Int. Ed. 2013, 52, 7098; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7238. [Google Scholar]
- 5. Challis G. L., Ravel J., Townsend C. A., Chem. Biol. 2000, 7, 211. [DOI] [PubMed] [Google Scholar]
- 6. Stachelhaus T., Mootz H. D., Marahiel M. A., Chem. Biol. 1999, 6, 493. [DOI] [PubMed] [Google Scholar]
- 7. Gulick A. M., ACS Chem. Biol. 2009, 4, 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Winn M., Fyans J. K., Zhuo Y., Micklefield J., Nat. Prod. Rep. 2016, 33, 317. [DOI] [PubMed] [Google Scholar]
- 9. Belshaw P. J., Walsh C. T., Stachelhaus T., Science 1999, 284, 486. [DOI] [PubMed] [Google Scholar]
- 10. Linne U., Marahiel M. A., Biochemistry 2000, 39, 10439. [DOI] [PubMed] [Google Scholar]
- 11. Kokona B., Winesett E. S., von Krusenstiern A. N., Cryle M. J., Fairman R., Charkoudian L. K., Anal. Biochem. 2016, 495, 42. [DOI] [PubMed] [Google Scholar]
- 12. Haslinger K., Brieke C., Uhlmann S., Sieverling L., Süssmuth R. D., Cryle M. J., Angew. Chem. Int. Ed. 2014, 53, 8518; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8658. [Google Scholar]
- 13. Uhlmann S., Süssmuth R. D., Cryle M. J., ACS Chem. Biol. 2013, 8, 2586. [DOI] [PubMed] [Google Scholar]
- 14. Haslinger K., Peschke M., Brieke C., Maximowitsch E., Cryle M. J., Nature 2015, 521, 105. [DOI] [PubMed] [Google Scholar]
- 15. Peschke M., Haslinger K., Brieke C., Reinstein J., Cryle M. J., J. Am. Chem. Soc., 2016, 138, 6746–6753. [DOI] [PubMed] [Google Scholar]
- 16. Gaudelli N. M., Long D. H., Townsend C. A., Nature 2015, 520, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaudelli N. M., Townsend C. A., Nat. Chem. Biol. 2014, 10, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ray L., Yamanaka K., Moore B. S., Angew. Chem. Int. Ed. 2016, 55, 364; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 372. [Google Scholar]
- 19. Tufar P., Rahighi S., Kraas F. I., Kirchner D. K., Lohr F. N., Henrich E., Köpke J., Dikic I., Güntert P., Marahiel M. A., Dötsch V., Chem. Biol. 2014, 21, 552. [DOI] [PubMed] [Google Scholar]
- 20. Lohman J. R., Ma M., Cuff M. E., Bigelow L., Bearden J., Babnigg G., Joachimiak A., G. N. Phillips, Jr. , Shen B., Proteins 2014, 82, 1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haslinger K., Redfield C., Cryle M. J., Proteins 2015, 83, 711. [DOI] [PubMed] [Google Scholar]
- 22. Zimmermann S., Pfennig S., Neumann P., Yonus H., Weininger U., Kovermann M., Balbach J., Stubbs M. T., FEBS Lett. 2015, 589, 2283. [DOI] [PubMed] [Google Scholar]
- 23. Goodrich A. C., Harden B. J., Frueh D. P., J. Am. Chem. Soc. 2015, 137, 12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y., Zheng T., Bruner S. D., Chem. Biol. 2011, 18, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jaremko M. J., Lee D. J., Opella S. J., Burkart M. D., J. Am. Chem. Soc. 2015, 137, 11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drake E. J., Miller B. R., Shi C., Tarrasch J. T., Sundlov J. A., Allen C. L., Skiniotis G., Aldrich C. C., Gulick A. M., Nature 2016, 529, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reimer J. M., Aloise M. N., Harrison P. M., Schmeing T. M., Nature 2016, 529, 239. [DOI] [PubMed] [Google Scholar]
- 28. Koglin A., Mofid M. R., Lohr F. N., Schafer B., Rogov V. V., Blum M., Mittag T., Marahiel M. A., Bernhard F. N., Dotsch V., Science 2006, 312, 273. [DOI] [PubMed] [Google Scholar]
- 29. Lai J. R., Koglin A., Walsh C. T., Biochemistry 2006, 45, 14869. [DOI] [PubMed] [Google Scholar]
- 30. Sundlov J. A., Shi C., Wilson D. J., Aldrich C. C., Gulick A. M., Chem. Biol. 2012, 19, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Crosby J., Crump M. P., Nat. Prod. Rep. 2012, 29, 1111. [DOI] [PubMed] [Google Scholar]
- 32. Beld J., Cang H., Burkart M. D., Angew. Chem. Int. Ed. 2014, 53, 14456; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14684. [Google Scholar]
- 33. Evans S. E., Williams C., Arthur C. J., Ploskon E., Wattana-amorn P., Cox R. J., Crosby J., Willis C. L., Simpson T. J., Crump M. P., J. Mol. Biol. 2009, 389, 511. [DOI] [PubMed] [Google Scholar]
- 34. Goodrich A. C., Frueh D. P., Biochemistry 2015, 54, 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frueh D. P., Arthanari H., Koglin A., Vosburg D. A., Bennett A. E., Walsh C. T., Wagner G., Nature 2008, 454, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mitchell C. A., Shi C., Aldrich C. C., Gulick A. M., Biochemistry 2012, 51, 3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanovic A., Samel S. A., Essen L., Marahiel M. A., Science 2008, 321, 659. [DOI] [PubMed] [Google Scholar]
- 38. Conti E., Stachelhaus T., Marahiel M. A., Brick P., EMBO J. 1997, 16, 4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yonus H., Neumann P., Zimmermann S., May J. J., Marahiel M. A., Stubbs M. T., J. Biol. Chem. 2008, 283, 32484. [DOI] [PubMed] [Google Scholar]
- 40. Miller B. R., Sundlov J. A., Drake E. J., Makin T. A., Gulick A. M., Proteins 2014, 82, 2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kittilä T., Schoppet M., Cryle M. J., ChemBioChem 2016, 17, 576. [DOI] [PubMed] [Google Scholar]
- 42. Henderson J. C., Fage C. D., Cannon J. R., Brodbelt J. S., Keatinge-Clay A. T., Trent M. S., ACS Chem. Biol. 2014, 9, 2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bloudoff K. I., Rodionov D., Schmeing T. M., J. Mol. Biol. 2013, 425, 3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bloudoff K. I., Alonzo D. A., Schmeing T. M., Cell Chem. Biol. 2016, 23, 331. [DOI] [PubMed] [Google Scholar]
- 45. Uguru G. C., Milne C., Borg M., Flett F., Smith C. P., Micklefield J., J. Am. Chem. Soc. 2004, 126, 5032. [DOI] [PubMed] [Google Scholar]
- 46. Thirlway J., Lewis R., Nunns L., Al Nakeeb M., Styles M., Struck A.-W., Smith C. P., Micklefield J., Angew. Chem. Int. Ed. 2012, 51, 7181; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7293. [Google Scholar]
- 47. Samel S. A., Schoenafinger G., Knappe T. A., Marahiel M. A., Essen L.-O., Structure 2007, 15, 781. [DOI] [PubMed] [Google Scholar]
- 48. Marahiel M. A., Nat. Prod. Rep. 2016, 33, 136. [DOI] [PubMed] [Google Scholar]