

Abstract
Interfacial water in the vicinity of lipids plays an important role in many biological processes, such as drug delivery, ion transportation, and lipid fusion. Hence, molecular‐level elucidation of the properties of water at lipid interfaces is of the utmost importance. We report the two‐dimensional heterodyne‐detected vibrational sum frequency generation (2D HD‐VSFG) study of the OH stretch of HOD at charged lipid interfaces, which shows that the hydrogen bond dynamics of interfacial water differ drastically, depending on the lipids. The data indicate that the spectral diffusion of the OH stretch at a positively charged lipid interface is dominated by the ultrafast (<∼100 fs) component, followed by the minor sub‐picosecond slow dynamics, while the dynamics at a negatively charged lipid interface exhibit sub‐picosecond dynamics almost exclusively, implying that fast hydrogen bond fluctuation is prohibited. These results reveal that the ultrafast hydrogen bond dynamics at the positively charged lipid–water interface are attributable to the bulk‐like property of interfacial water, whereas the slow dynamics at the negatively charged lipid interface are due to bound water, which is hydrogen‐bonded to the hydrophilic head group.
Keywords: hydrogen bonds, water–lipid interfaces, lipids, ultrafast spectroscopy, water dynamics
Membranes are the boundary of biological cells and are responsible for the transportation of various chemical species and ions from one side to the other, an essential process for all living things.1 The main body of the biological membrane is a bilayer of phospholipids, which are amphiphilic in nature: the hydrophobic tail is a nonpolar alkyl chain and the hydrophilic head consists of polar or charged moieties. Water around the lipid head group unquestionably plays an important role in the function of biomolecular assemblies. Various interesting membrane processes, such as permeation of water across membranes,2 formation of water channels that mediate ion transport,3 and interaction between two membranes, are dependent on the behavior of water in the vicinity of lipid membranes.4 Therefore, understanding the structure and dynamics of interfacial water at lipid interfaces is of the utmost importance.
For elucidating the dynamics of water at amphiphile–water interfaces, time‐resolved IR experiments have been intensively performed by controlling the number of water molecules in the reverse micelles and lamellar structures,5 which provides rich insight into the nature of interfacial water. Vibrational sum frequency generation (VSFG) can provide information from a different viewpoint because it has intrinsic interface selectivity (which IR spectroscopy lacks): it can give the vibrational spectrum of interfacial molecules without controlling and limiting the number of water molecules. The VSFG technique has been applied to lipid interfaces to investigate the properties of interfacial water.6 In particular, heterodyne‐detected (HD‐) VSFG spectroscopy enables measurement of the imaginary part of χ(2) (Imχ(2)), which provides interface‐selective vibrational spectra that can be directly compared to IR and Raman spectra.6c,6d, 7 Furthermore, the sign of the Imχ(2) signal gives information about the orientation of interfacial molecules.6c,6d, 8
Recently, HD‐VSFG spectroscopy has been extended to femtosecond time‐resolved measurements and 2D spectroscopy (2D HD‐VSFG), which allows us to measure the ultrafast vibrational dynamics of interfacial molecules.9 2D HD‐VSFG measures the pump‐frequency dependence of ΔImχ(2) (pump‐induced change in Imχ(2) spectra), which makes it an interface analogue of the 2D IR technique.10 In this study, we applied 2D HD‐VSFG spectroscopy to positively and negatively charged lipid monolayers on isotopically diluted water for clarification of the hydrogen bond dynamics of interfacial water.
We chose DPTAP (1,2‐dipalmitoyl‐3‐trimethyl‐ ammonium propane chloride salt) and DPPG (1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphorylglycerol sodium salt) as positively and negatively charged lipids, respectively. Figure 1 shows the steady‐state Imχ(2) spectra of the OH stretch of isotopically diluted water (HOD in D2O) at the positively charged DPTAP and negatively charged DPPG interfaces. Since the ratio of each water species is H2O:HOD:D2O=1:8:16, HOD is the predominant species that gives rise to the signal in the OH stretch region. Because the Fermi resonance and other couplings are suppressed by the isotopic dilution, the Imχ(2) spectra only exhibit one broad band, which is attributable to the OH stretch of hydrogen‐bonded water below the lipid head group.6c The sign of Imχ(2) is negative for the positively charged DPTAP interface but positive for the negatively charged DPPG interface, indicating that water has hydrogen (H‐) down orientation at the DPTAP interface and H‐up orientation at the DPPG interface. The peak wavenumbers and spectral features of the OH stretch bands in the two Imχ(2) spectra are similar to each other, except for their signs, indicating similar hydrogen bond strength for water at the DPTAP and DPPG interfaces.
Figure 1.
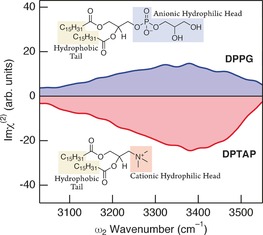
Steady‐state Imχ(2) spectra for the OH stretch of HOD of positively charged DPTAP (red line) and negatively charged DPPG (blue line) interfaces.
The steady‐state Imχ(2) spectra suggest that water at the DPTAP and DPPG interfaces have opposite orientation but similar hydrogen bond strength. At a glance, interfacial water at both lipid interfaces would look “bulk‐like” because the peak positions of the OH stretch bands are nearly equal to that of the IR spectrum of HOD in bulk D2O.6c However, 2D HD‐VSFG reveals that interfacial water at the DPTAP and DPPG interfaces is completely different from a dynamical viewpoint.
Figure 2 shows the 2D HD‐VSFG spectra of the OH stretch of HOD in D2O at the positively and negatively charged lipid interfaces at different time delays. The 2D HD‐VSFG spectrum shows the response (ΔImχ(2)) of the OH stretch at the lipid interfaces as a function of probe (ω 2, horizontal axis) and pump (ω pump, vertical axis) wavenumbers. The red color in the 2D HD‐VSFG spectrum in Figure 2 a represents the positive ΔImχ(2) associated with the bleaching of the negative OH stretch band (the 0–1 transition), and blue represents negative ΔImχ(2) associated with the appearance of the negative hot band (the 1–2 transition) for water at the DPTAP interface. The hot band is red‐shifted because of vibrational anharmonicity. The color of the positive and negative ΔImχ(2) spectral response of the 2D HD‐VSFG spectrum for DPPG (Figure 2 b) is opposite to DPTAP because of the different sign of the OH stretch band in the steady‐state spectra (Figure 1).
Figure 2.
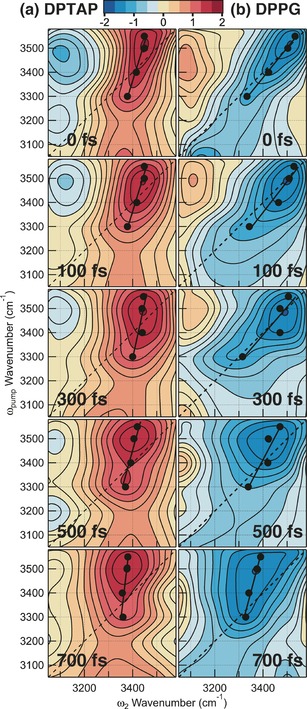
2D HD‐VSFG spectra of the OH stretch of HOD at the a) DPTAP and b) DPPG interfaces. The black, straight line in each 2D spectrum represents the slope of the bleach lobe. Spectra after 700 fs are not shown here because they are dominated by the spectral change arising from thermalization, which is not relevant to the present study.
The 2D HD‐VSFG spectrum of the DPTAP interface at 0 fs (Figure 2 a) shows a strong red lobe centered around (ω pump, ω 2)=(3500 cm−1, 3450 cm−1). The lobe is elongated along the diagonal line but the elongation is incomplete. The diagonal elongation indicates the memory of ω pump held by the OH stretch vibration in a distinct hydrogen bonding environment at the interface. As each OH vibration “forgets” the ω pump at which it has been photoexcited (that is, spectral diffusion), the diagonal elongation gradually vanishes with time. The degree of the diagonal elongation can be evaluated from the slope of the black straight line drawn in the 2D HD‐VSFG spectrum.11 The black line in Figure 2 is a fit for black solid markers that indicate the peak ω 2 wavenumbers of horizontal cuts at each ω pump wavenumber. Here the slope is defined in such a manner that the vertical line has zero slope, whereas the slope of the diagonal line is unity. As shown in Figure 3, the slope in the 2D HD‐VSFG spectrum of DPTAP is 0.29±0.04 at 0 fs, and it decays to 0.09±0.03 at 700 fs owing to spectral diffusion. In sharp contrast, the 2D HD‐VSFG spectrum of the DPPG interface at 0 fs (Figure 2 b) shows a blue lobe around (ω pump, ω 2)=(3500 cm−1, 3500 cm−1) that is almost completely elongated along the diagonal line. The slope for DPPG decays from 0.80±0.03 at 0 fs to 0.25±0.08 at 700 fs (Figure 3). The temporal change of the slope is reproduced by a single‐exponential decay with a time constant of 750±350 fs for both DPTAP and DPPG within S/N, as indicated by the solid lines in Figure 3.
Figure 3.
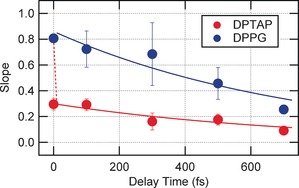
Slope of the bleach lobe plotted as a function of delay time for the DPTAP (red marker) and DPPG (blue marker) interfaces. Solid lines represent 750 fs single‐exponential decays. The red dotted line indicates the invisible ultrafast component of DPTAP.
Although the 2D HD‐VSFG spectra of DPTAP and DPPG exhibit spectral diffusion with a similar time constant of about 750 fs, the initial value of the slope at 0 fs for DPTAP is much smaller than that for DPPG. This suggests that nearly instantaneous dynamics, faster than approximately 100 fs, are hidden in the spectral diffusion data of DPTAP. Such an ultrafast process cannot be directly observed with the present time resolution (∼200 fs; see the Supporting Information), resulting in the reduced value of the initial slope in Figure 3. In other words, spectral diffusion for DPTAP consists of an invisible ultrafast process (<∼100 fs) and a visible sub‐picosecond process. (The invisible ultrafast component is suggested with a dotted line in Figure 3.) Such a bimodal temporal behavior is characteristic of the frequency correlation loss of bulk HOD in D2O reported by Tokmakoff and co‐workers with 2D IR, and the faster (60 and 130 fs) and slower (1.4 ps) dynamics of bulk HOD in D2O were attributed to hydrogen bond fluctuation and rearrangement, respectively.12 The present 2D HD‐VSFG experiment strongly suggests that the spectral diffusion of HOD at the DPTAP interface occurs in a similar manner, not only because of similar temporal behavior but also because the interfacial water at the DPTAP interface has almost the same hydrogen bond strength as bulk water.6c The difference in the time constant of the slow component, when comparing the interface and the bulk, may be due to the existence of the charge at the interface and/or the difference in the degree of the isotopic dilution. The 2D HD‐VSFG, as well as steady‐state HD‐VSFG spectra of HOD, suggests that χ(2)‐active water at the DPTAP interface is attributed to bulk‐like water in the electric double layer formed by the cationic lipid head group and the counterion. Notably, molecular dynamics (MD) simulations performed for the zwitterionic phosphatidylcholine suggested that water molecules form a clathrate structure around the choline group.13 If a clathrate structure also exists in the case of the positively charged DPTAP interface, such water molecules also contribute to the χ(2) signal, along with the water in the electric double layer that is not directly associated with the choline group. Nevertheless, it is considered that such water also exhibits the property of “bulk‐like water”, as shown by a recent vibrational spectroscopic study.14
In contrast to the DPTAP interface, the initial value of the slope for the DPPG interface is nearly unity. This implies that the inhomogeneity of the OH stretch band at the DPPG interface is well‐resolved by the time‐resolution of the present experiment because the frequency modulation is slow enough. In fact, the spectral diffusion dynamics of DPPG are dominated by the sub‐picosecond process, and this temporal behavior is completely different from the dynamics of bulk‐like water because the ultrafast process (<∼100 fs) is almost absent. This suggests that the χ(2)‐active water species is predominantly attributed to the bound water, which is hydrogen‐bonded to the head group of DPPG. Although the steady‐state spectrum of DPPG indicates that the hydrogen bond strength of interfacial water is similar to that of DPTAP (Figure 1), the 2D spectra clearly show that the hydrogen bond dynamics are drastically different. It implies that the time scale of the spectral‐diffusion dynamics does not have a direct correlation with the hydrogen bond strength estimated from the steady‐state measurement. In the case of DPPG, the interfacial water can be strongly hydrogen‐bonded to the phosphatidylglycerol head group: the phosphate group forms firm hydrogen bonds with water, and glycerol hydroxy groups at the terminal end can also provide additional hydrogen bonding sites.15 It is very likely that the hydrogen bond with the phosphatidylglycerol head group largely limits the fast fluctuation of interfacial water because of the slow motion of DPPG. Note that the phosphatidylglycerol group of DPPG can form hydrogen bonds with water, whereas the choline group of DPTAP cannot. The 2D spectra demonstrate that the dynamics of interfacial water are crucially dependent on whether the lipid head group can bind water and conformationally restrict the fluctuation of the hydrogen bond.
It is noteworthy that the peak height of the steady‐state Imχ(2) spectrum of HOD at the DPPG interface is about half of the peak height of DPTAP (Figure 1). This also suggests a much lower contribution from bulk‐like water in the electric double layer for DPPG. The multiple hydrogen bonding sites in the head group of DPPG may effectively lower the orientational order of water in the electric double layer, resulting in a dominant contribution from the bound water to the steady‐state and 2D spectra for DPPG. The orientation of water in the electric double layer of DPTAP seems not to be perturbed, as the head group of DPTAP does not have a hydrogen bonding site. Therefore, it is likely that SFG probes the DPTAP interface “more deeply” than DPPG.
Recently Skinner and co‐workers calculated 2D HD‐VSFG spectra using molecular dynamics simulations, and discussed the dynamics of isotopically diluted water at the DPTAP and DPPG interfaces.15 Their calculation showed that the spectral diffusion dynamics of the OH stretch at both interfaces are bimodal, but that fast dynamics (∼300 fs) are predominant at the positively charged DPTAP interface, whereas slow dynamics (∼10 ps) are dominant at the negatively charged DPPG interface. They concluded that the dynamics of water at the DPPG interface are slow compared to the DPTAP interface, mostly because of the conformational constraints on water imposed by the hydrogen bond with the head group of DPPG. Our experimental results are very consistent with their predictions about the bimodal feature and the change in the amplitudes of the fast and slow components, although the calculated time constants are considerably different. The 2D lobe shape, as well as the amplitude of the steady‐state spectrum16 calculated by the Skinner group, are also in qualitative agreement with the present experimental results for both the DPTAP and DPPG interfaces.
Bonn and co‐workers previously performed time‐resolved and 2D VSFG spectroscopy on the lipid–water interfaces using homodyne detection.17 They argued that two different water sub‐ensembles (that is, weakly and strongly hydrogen‐bonded water species), which show distinct vibrational dynamics, exist at the lipid interfaces17a,17b irrespective of the chemical nature of the head group.17c In the present heterodyne 2D VSFG study on HOD in D2O, we did not find any spectral responses that indicate such distinct types (two varieties) of interfacial water at either of the lipid interfaces. As shown in Figure 2, there is no discontinuity in the shape and dynamics of the bleach lobes observed for DPTAP and DPPG. We note that the previous homodyne experiments were performed in D2O, where the Fermi resonance significantly affects the spectral response.9e Moreover, the interpretation of time‐resolved VSFG spectra measured with homodyne detection is generally very difficult because it provides not Δχ(2) but Δ|χ(2)|2.
Lastly, we mention time‐resolved and 2D IR studies relevant to the present 2D HD‐VSFG work. Infrared vibrational echo and 2D‐IR experiments showed that the frequency correlation loss of the confined water in negatively charged reversed micelles exhibits the larger slow component, as the number of confined water molecules is reduced (that is, more interfacial water is monitored).18 This result looks consistent with our present observation about negatively charged DPPG, for which we observe the slow component almost exclusively. Actually, the interfacial water can be strongly hydrogen‐bonded to the head group of the surfactant and lipid. A similar trend has also been observed for 2D IR experiments of the micelles of neutral zwitterionic phosphatidylcholine (PC).19 However, the situation would be different between zwitterionic PC and negatively charged DPPG because the former is net neutral and a large dipole is induced between the phosphate and choline groups.
In summary, using interface specific, phase‐ and time‐resolved 2D HD‐VSFG spectroscopy, combined with the isotopic dilution technique, we have shown that water dynamics at lipid interfaces are essentially different, reflecting the chemical structure of the lipid. The observed distinct hydrogen bond fluctuation and dynamics of interfacial water may have a significant influence on cell membrane processes.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by JSPS KAKENHI Grant number JP25104005.
P. C. Singh, K.-i. Inoue, S. Nihonyanagi, S. Yamaguchi, T. Tahara, Angew. Chem. Int. Ed. 2016, 55, 10621.
References
- 1.
- 1a. Wood K., Plazanet M., Gabel F., Kessler B., Oesterhelt D., Tobias D. J., Zaccai G., Weik M., Proc. Natl. Acad. Sci. USA 2007, 104, 18049–18054; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Sehy J. V. B., Ackerman A. A., Neil J. J., Biophys. J. 2002, 83, 2856–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fitter J., Lechner R. E., Dencher N. A., J. Phys. Chem. B 1999, 103, 8036–8050. [Google Scholar]
- 3. Bordi F. C., Naglieri A., Biophys. J. 1998, 74, 1358–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skinner M. K., Tung P. S., Fritz I. B., J. Cell Biol. 1985, 100, 1941–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Fayer M. D., Acc. Chem. Res. 2012, 45, 3–14; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Moilanen D. E., Levinger N. E., Spry D. B., Fayer M. D., J. Am. Chem. Soc. 2007, 129, 14311–14318; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Moilanen D. E., Fenn E. E., Wong D., Fayer M. D., J. Am. Chem. Soc. 2009, 131, 8318–8328; [DOI] [PubMed] [Google Scholar]
- 5d. Costard R., Elsaesser T., J. Phys. Chem. B 2013, 117, 15338–15345; [DOI] [PubMed] [Google Scholar]
- 5e. Costard R., Heisler I. A., Elsaesser T., J. Phys. Chem. Lett. 2014, 5, 506–511; [DOI] [PubMed] [Google Scholar]
- 5f. Costard R., Levinger N. E., Nibbering E. T. J., Elsaesser T., J. Phys. Chem. B 2012, 116, 5752–5759. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Sovago M., Campen R. K., Wurpel G. W. H., Muller M., Bakker H. J., Bonn M., Phys. Rev. Lett. 2008, 100, 173901; [DOI] [PubMed] [Google Scholar]
- 6b. Watry M. R., Tarbuck T. L., Richmond G. L., J. Phys. Chem. B 2003, 107, 512–518; [Google Scholar]
- 6c. Mondal J. A., Nihonyanagi S., Yamaguchi S., Tahara T., J. Am. Chem. Soc. 2010, 132, 10656–10657; [DOI] [PubMed] [Google Scholar]
- 6d. Mondal J. A., Nihonyanagi S., Yamaguchi S., Tahara T., J. Am. Chem. Soc. 2012, 134, 7842–7850. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Ostroverkhov V., Waychunas G. A., Shen Y. R., Phys. Rev. Lett. 2005, 94, 046102; [DOI] [PubMed] [Google Scholar]
- 7b. Stiopkin I. V., Jayathilake H. D., Bordenyuk A. N., Benderskii A. V., J. Am. Chem. Soc. 2008, 130, 2271–2275; [DOI] [PubMed] [Google Scholar]
- 7c. Yamaguchi S., Tahara T., J. Chem. Phys. 2008, 129, 101102; [DOI] [PubMed] [Google Scholar]
- 7d. Nihonyanagi S., Yamaguchi S., Tahara T., J. Chem. Phys. 2009, 130, 204704; [DOI] [PubMed] [Google Scholar]
- 7e. Nihonyanagi S., Mondal J. A., Yamaguchi S., Tahara T., Annu. Rev. Phys. Chem. 2013, 64, 579–603; [DOI] [PubMed] [Google Scholar]
- 7f. Shen Y. R., Annu. Rev. Phys. Chem. 2013, 64, 129–150; [DOI] [PubMed] [Google Scholar]
- 7g. Nihonyanagi S., Kusaka R., Inoue K.-i., Adhikari A., Yamaguchi S., Tahara T., J. Chem. Phys. 2015, 143, 124707; [DOI] [PubMed] [Google Scholar]
- 7h. Yamaguchi S., J. Chem. Phys. 2015, 143, 034202. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Nihonyanagi S., Yamaguchi S., Tahara T., J. Am. Chem. Soc. 2010, 132, 6867–6869; [DOI] [PubMed] [Google Scholar]
- 8b. Chen X., Hua W., Huang Z., Allen H. C., J. Am. Chem. Soc. 2010, 132, 11336–11342; [DOI] [PubMed] [Google Scholar]
- 8c. Nihonyanagi S., Ishiyama T., Lee T.-k., Yamaguchi S., Bonn M., Morita A., Tahara T., J. Am. Chem. Soc. 2011, 133, 16875–16880; [DOI] [PubMed] [Google Scholar]
- 8d. Nihonyanagi S., Yamaguchi S., Tahara T., J. Am. Chem. Soc. 2014, 136, 6155–6158. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Nihonyanagi S., Singh P. C., Yamaguchi S., Tahara T., Bull. Chem. Soc. Jpn. 2012, 85, 758–760; [Google Scholar]
- 9b. Singh P. C., Nihonyanagi S., Yamaguchi S., Tahara T., J. Chem. Phys. 2012, 137, 094706; [DOI] [PubMed] [Google Scholar]
- 9c. Singh P. C., Nihonyanagi S., Yamaguchi S., Tahara T., J. Chem. Phys. 2013, 139, 161101; [DOI] [PubMed] [Google Scholar]
- 9d. Singh P. C., Nihonyanagi S., Yamaguchi S., Tahara T., J. Chem. Phys. 2014, 141, 18C527; [DOI] [PubMed] [Google Scholar]
- 9e. Inoue K.-i., Nihonyanagi S., Singh P. C., Yamaguchi S., Tahara T., J. Chem. Phys. 2015, 142, 212431. [DOI] [PubMed] [Google Scholar]
- 10. Hamm P., Zanni M., Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, Cambridge, 2011. [Google Scholar]
- 11. Kwak K., Park S., Finkelstein I. J., Fayer M. D., J. Chem. Phys. 2007, 127, 124503. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Fecko C. J., Eaves J. D., Loparo J. J., Tokmakoff A., Geissler P. L., Science 2003, 301, 1698–1702; [DOI] [PubMed] [Google Scholar]
- 12b. Loparo J. J., Roberts S. T., Tokmakoff A., J. Chem. Phys. 2006, 125, 194521; [DOI] [PubMed] [Google Scholar]
- 12c. Loparo J. J., Roberts S. T., Tokmakoff A., J. Chem. Phys. 2006, 125, 194522. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Pasenkiewicz-Gierula M., Takaoka Y., Miyagawa H., Kitamura K., Kusumi A., J. Phys. Chem. A 1997, 101, 3677–3691; [Google Scholar]
- 13b. Lopez C. F., Nielsen S. O., Klein M. L., Moore P. B., J. Phys. Chem. B 2004, 108, 6603–6610; [Google Scholar]
- 13c. Foglia F., Lawrence M. J., Lorenz C. D., McLain S. E., J. Chem. Phys. 2010, 133, 145103; [DOI] [PubMed] [Google Scholar]
- 13d. Wiener M. C., White S. H., Biophys. J. 1992, 61, 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Long J. A., Rankin B. M., Ben-Amotz D., J. Am. Chem. Soc. 2015, 137, 10809–10815. [DOI] [PubMed] [Google Scholar]
- 15. Roy S., Gruenbaum S. M., Skinner J. L., J. Chem. Phys. 2014, 141, 22D505. [DOI] [PubMed] [Google Scholar]
- 16. Roy S., Gruenbaum S. M., Skinner J. L., J. Chem. Phys. 2014, 141, 18C502. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Zhang Z., Piatkowski L., Bakker H. J., Bonn M., J. Chem. Phys. 2011, 135, 021101; [DOI] [PubMed] [Google Scholar]
- 17b. Livingstone R. A., Nagata Y., Bonn M., Backus E. H. G., J. Am. Chem. Soc. 2015, 137, 14912–14919; [DOI] [PubMed] [Google Scholar]
- 17c. Bonn M., Bakker H. J., Ghosh A., Yamamoto S., Sovago M., Campen R. K., J. Am. Chem. Soc. 2010, 132, 14971–14978. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Tan H.-S., Piletic I. R., Riter R. E., Levinger N. E., Fayer M. D., Phys. Rev. Lett. 2005, 94, 057405; [DOI] [PubMed] [Google Scholar]
- 18b. Fenn E. E., Wong D. B., Giammanco C. H., Fayer M. D., J. Phys. Chem. B 2011, 115, 11658–11670. [DOI] [PubMed] [Google Scholar]
- 19. Costard R., Greve C., Heisler I. A., Elsaesser T., J. Phys. Chem. Lett. 2012, 3, 3646–3651. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary