

Abstract
Research and therapeutic targeting of the phosphoserine/threonine phosphatases PP1 and PP2A is hindered by the lack of selective inhibitors. The microcystin (MC) natural toxins target both phosphatases with equal potency, and their complex synthesis has complicated structure–activity relationship studies in the past. We report herein the synthesis and biochemical evaluation of 11 MC analogues, which was accomplished through an efficient strategy combining solid‐ and solution‐phase approaches. Our approach led to the first MC analogue with submicromolar inhibitory potency that is strongly selective for PP2A over PP1 and does not require the complex lipophilic Adda group. Through mutational and structural analyses, we identified a new key element for binding, as well as reasons for the selectivity. This work gives unprecedented insight into how selectivity between these phosphatases can be achieved with MC analogues.
Keywords: inhibitors, medicinal chemistry, microcystin, protein phosphatases, structure–activity relationships
Protein phosphatases‐1 (PP1) and ‐2A (PP2A) are conserved protein Ser/Thr‐specific phosphatases (PSTPs) that share 50 % sequence identity1 and are major regulators of protein dephosphorylation.2, 3, 4 In order to elucidate the biological roles of PP1 and PP2A and to evaluate their therapeutic potential in diseases, it is necessary to develop specific inhibitors. However, this has been very challenging due to the high degree of conservation in the active sites of these PSTPs.5, 6 Natural toxins are strong inhibitors of these PSTPs but show limited selectivity.6 As an exception to this, Fostriecin shows about 104‐fold selectivity for PP2A over PP1.6 Since this compound suffers from low stability,6 new alternative approaches would be useful to address the problem of selectivity. Microcystins (MCs) are examples of non‐selective inhibitory natural toxins. In the past, structure–activity relationship (SAR) studies to achieve selectivity with MCs have been complicated by the complexity of the synthesis,6 which involves many steps, isomerization problems, and low yields.5, 6, 7, 8, 9, 10 In this work, we developed a faster synthesis of MC analogues, which enabled us to synthesize the unprecedented number of 11 cyclic MC analogues. When testing these analogues for their potency, we discovered the first highly selective MC‐based PP2A inhibitor. Our SAR study, combined with analysis of the crystal structures of PP1 and PP2A, as well as mutational analysis, provide a rationale for the selectivity.
MCs are cyclic heptapeptides with the typical structure cyclo[(d)Ala1‐X2‐β‐(d)MeAsp3‐Z4‐Adda5‐γ‐(d)Glu6‐Mdha7] (Figure 1, “MCs” with R′ and R′′=methyl), where Adda refers to (2S,3S,8S,9S)‐3‐amino‐9‐methoxy‐2,6,8‐trimethyl‐10‐phenyldeca‐4,6‐dienoic acid.11 The X and Z positions are occupied by natural l‐amino acids that are indicated in the name of the MC (e.g., MC‐LF (1) contains leucine and phenylalanine in positions 2 and 4, respectively12). The cyclic nature of the peptide,13, 14 the presence of the hydrophobic tail Adda,6 as well as the free carboxy groups of β‐(d)‐aspartic acid15 and γ‐(d)‐glutamic acid16, 17, 18 were found to be essential for the potency of MC. Furthermore, covalent linkage between Cys (Cys273 in PP11 and Cys269 in PP2A19) and Mdha1, 19 is not required for potency.20 Additionally, some MCs do not contain the N‐methyl group in Mdha (Dha, Figure 1: R′′=H), resulting in a slight decrease in the inhibition potency.18 In order to shed light on the potential effects of different residues in position 7 that cannot undergo a Michael addition with Cys thiols, MC analogues with alanine, glycine, and sarcosine were considered here. Since removing the methyl group of β‐(d)MeAsp3 did not have a strong effect on the potency,21 derivatives containing β‐(d)Asp in position 3 were chosen. With the exception of placing a cysteine in position 5,22 evaluation of the potency of MC analogues with shorter hydrophobic tails mimicking Adda has not yet been reported. To this end, analogues synthesized in this study include small lipophilic tails that are structurally similar to parts of Adda, and a small alkyl group in the α‐position with the same stereochemistry as Adda (8–12) or not (2–7; Figure 1).
Figure 1.

The general structure of MCs, where R′ and R′′ can be methyl groups or hydrogen and X and Z are natural l‐amino acids. Specific structures are shown for MC‐LF (1) and analogues with small lypophilic tails replacing Adda (shown in red) in the β‐ (2–7) or both α‐ and β‐position of residue 5 (8–12), and with glycine (5, 8, 10), alanine (2, 4, 6, 7, 9, 12), or sarcosine (11) in position 7 (shown in blue).
Amino acids 13, 14, and 15 (Scheme 1 A), which were required for the synthesis of 2, 5, 8 and 9, were obtained through Fmoc‐protection of the free amine group. The synthesis of Fmoc‐Amba [(2S,3S)‐3‐Fmoc‐amino‐2‐methyl‐butanoic acid, 20] was more challenging (Scheme 1 B). Starting from acetylated Evans’ oxazolidinone 16, 19 was synthesized in three steps by applying an Evans aldol reaction followed by an intramolecular Mitsunobu reaction in analogy to a published Adda synthesis.23 Exchange of the amino protecting group gave 20.
Scheme 1.
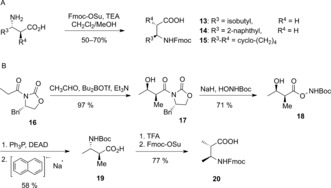
Synthesis of β‐amino acids. A) Fmoc protection. B) Novel synthesis of Fmoc‐Amba (20). Fmoc=9‐fluorenylmethyloxycarbonyl, Su=succinimidyl, TEA=triethylamine, Boc=tert‐butoxycarbonyl, DEAD=diethyl azodicarboxylate, TFA=trifluoroacetic acid.
Previous MC syntheses showed that the macrocyclization is the most challenging step of the synthesis owing to conformational, stereoelectronic, and steric issues.8, 9, 10 An additional challenge is competition between oligomerization and cyclization24 and isomerization.5, 8 We chose positions 4 and 5 for connection in the macrocyclization because it allowed us to attach the more synthetically demanding amino acids like Amba in the last step of the synthesis. Accordingly, using the standard Fmoc‐based strategy, we synthesized the linear peptides, cleaved them from the resin, and carried out macrocyclization in a highly diluted solution (see the Supporting Information). Final deprotection led to good overall yields, generally around 20–30 % after HPLC purification (Scheme 2) after 15–20 reactions (depending on the MC analogue). Compared to previous syntheses, our combined solution‐ and solid‐phase approach requires only a single purification step at the end of the synthesis and leads to improved yields, to which substituting the Adda group with simpler analogues also contributes.
Scheme 2.
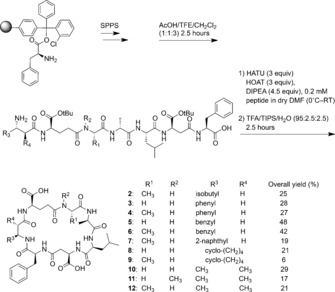
Novel synthesis of MC analogues 2–12. TFE=trifluoroethanol, HATU=O‐(7‐azabenzotriazol‐1‐yl)‐N,N,N′,N′‐tetramethyluronium hexafluorophosphate, HOAT=1‐hydroxy‐7‐azabenzotriazole, DMF=N,N‐dimethylformamide, TIPS=triisopropylsilane.
In all of the MC analogues, only one sharp peak with the correct mass was detected by UPLC/MS after the cyclization step (Figures S5–S15 in the Supporting Information). Nevertheless, in order to further analyze the possibility of epimerization as a previously encountered problem,5, 8 one MC analogue (6) was randomly selected and further analyzed. No evidence of epimerization was observed, since only one peak with the correct mass was detected by HPLC‐MS when using a column with a chiral stationary phase (Figure S17), and hydrolysis and analysis of 6 showed that the final compound was 99.5 % enantiomerically pure (Figure S18). The optimized conditions reported here thus avoid racemization.
The inhibitory effect of MC‐LF and the synthesized analogues was evaluated with an in vitro fluorescence assay using 6,8‐difluoro‐4‐methylumbelliferyl phosphate (DIFMUP) as a substrate.25, 26 First, 250 μm of the MC analogues (2–12) or 1 nm of MC‐LF (1) were incubated with the same amount of PP1 and PP2A under the same conditions to evaluate the percentage phosphatase activity (Table S2 and S3). In general, all of the new compounds were more potent toward PP2A, and the inhibitory activity decreased to a large extent in the absence of Adda, with compounds 2–7 showing some activity against PP2A while not being active against PP1. Analogues 8–12 were more potent, exhibiting activity against both phosphatases. This finding was rather surprising since they carry shorter lipophilic moieties in the β‐position of residue 5, which mimics Adda worse than the equivalent groups in compounds 2–7 (see Figure S1). Since the only other residue that differs between these compounds is the modified α‐position of residue 5, this modification could be the reason for their higher potency. Therefore, we measured the half maximal inhibitory concentration (IC50) of MC‐LF (1) and analogues 8–12 for both PP1 and PP2A (Table 1). The analogues containing Amba (10–12) were more potent than the ones with the cyclohexane ring (8,9) for both PP1 and PP2A. The presence and nature of a small alkyl group in the α‐position of residue 5 thus appears to modulate binding to both PP1 and PP2A. Similarly to nodularin binding,27 the differences in inhibitory potency could be due to solvent exclusion from the hydrophobic pocket. PP1 and PP2A crystal structures show the presence of a hydrophobic groove close to the active site, and in some crystal structures in this pocket, a water molecule is accommodated (Figure 2 A). The α‐methyl group of Adda replaces the water molecule when MC is bound (Figure 2 B). Okadaic acid and tautomycin also fill this pocket with a hydrophobic group.28, 29 Accordingly, the exclusion of solvent by the presence of a small alkyl group in this hydrophobic pocket could be important for binding and inhibiting PP1 and PP2A. This does not, however, exclude possible other effects of the α‐methyl group such as a potential effect on ring flexibility.27 To this end, more detailed NMR investigations of compounds 3 and 10 were carried out but the results gave no clear indication of whether the ring flexibility in the two compound classes differs (Figure S2 and the methods section of the Supporting Information). Compared to natural MCs,30, 31 the ring structures of both compounds are less compact. The strong exchange protection of four amide protons in MC‐LR31 is lost, thereby leading to an increase in the temperature dependence of the amide proton chemical shift to an average of 6.0 and 7.0 ppb K−1 compared to those observed in MC‐LR (4.3 ppb K−1),31 which could imply a higher flexibility in the analogue structures. Despite the fact that we obtained a converging structural ensemble under the assumption that the measured NOEs and dihedral angles derive from a single conformation (Figure S2A), the data are equally consistent with a larger pool of interconverting conformations (see the Supporting Information). Therefore, we generated a random pool of geometrically correct structures to examine whether the compounds can mimic MC. In this pool, we find conformers that closely resemble the interaction surface of MC with PP1 and PP2A (Figure S2B–D).
Table 1.
Inhibition potency of MC‐LF and the analogues toward PP2A and PP1.
| Compound | PP1 IC50 (μm) | PP2A IC50 (μm) | Selectivity PP2A/PP1 |
|---|---|---|---|
| 1 | (65.1±22.6)×10−6 | (7.5±1.3)×10−6 | 1:9 |
| 8 | 316.8±92.9 | 3.0±0.1 | 1:106 |
| 9 | 798.9±59.3 | 24.1±3.7 | 1:33 |
| 10 | 76.9±15.8 | 0.3±0.1 | 1:256 |
| 11 | 253.5±30.0 | 0.9±0.1 | 1:282 |
| 12 | 122.0±5.7 | 1.1±0.1 | 1:111 |
Figure 2.

Analysis of the conformational and binding properties of MCs by inspection of several phosphatase–ligand complexes. A) The presence of a molecule of water (ball and stick model) in a pocket close to the metal ions (pale purple spheres) in PP1 (PDB ID: 3HVQ). B) An overlay of two PP1 crystal structures, one co‐crystallized with MC (stick model; PDB ID: 2BDX) and the other with a water molecule in the hydrophobic groove (ball and stick model; PDB ID: 3HVQ). C) Alignment of crystal structures of PP1 (C atoms white) and PP2A (C atoms gray), showing a possible hydrogen bond between a carbonyl group of MC and Arg268 of PP2A (MC extracted from PDB ID 3FGA and merged into PP2A of PDB ID 4I5L). This H‐bonding is not possible in PP1 (PDB ID: 2BDX) as it has a Glu at the corresponding position. D) An alignment of Phe276 (PP1, PDB ID: 2BCD) and Cys269 (PP2A, PDB ID 2IE4), shown as ball and stick models, with the protein backbones shown as ribbon diagrams, and lines showing dihydro‐MC‐LA.
All of the MC analogues presented herein strongly surpass the selectivity of the natural toxin for PP2A over PP1. PP1 and PP2A differ in the region where they bind covalently to MC: Y272CGEF276 (PP1) structurally aligns with Y265CYRC269 (PP2A). Notably, these sequences align well in structures where PP1 is not covalently bound to toxins. When PP1 is covalently bound to MC, the bound Cys273 (PP1) aligns with Cys269 (PP2A) and this changes the conformation of this loop in PP1 (Figure S3).1, 19 Since our inhibitors do not bind covalently, it is more reasonable to compare the non‐covalently bound PP1–toxin structures with the ones for PP2A in this region.
As shown in Figure 2 C and Figure S4, a hydrogen bond could be formed between the carbonyl in position 7 and Arg268, which is only found in PP2A (PDB IDs: 3FGA, 4I5L) and has been reported to form van der Waals interactions with MC.19 In PP1, Glu275 aligns with Arg268 in PP2A, and this residue cannot participate in this additional interaction. Another reason for the selectivity is likely the steric bulk of Phe276 in PP1, which aligns with Cys269 in PP2A and thus adds steric bulk in close proximity to the amino acid in position 7 of MC (Figure 2 D). In order to validate this hypothesis, we performed site‐directed mutagenesis, replacing amino acids of PP1 with the corresponding amino acids of PP2A (E275R, F276C). The inhibitory potency of compounds 8–12 increased against this variant, but did not reach the level of potency against PP2A (Table S3), thus showing that these features indeed partly determine the selectivity. Additionally, using this PP1 double variant (E275R, F276C) and a PP1 single variant (E275R), we evaluated the IC50 of MC‐LF (17.1±2.9 pm and 35.0±3.8 pm, respectively) and compound 11 (12.0±2.6 μm and 46.1±0.8 μm, respectively; Figures S30 and S31). The results show that both residues play key roles in achieving selectivity between PP2A and PP1 in the MC analogues.
Since in the natural MCs, Mdha is found in position 7, it is reasonable to speculate that the strong affinity of the Adda tail for the PSTPs, in combination with the loop rearrangement in PP1 for covalent binding, mask features that fine‐tune the specificity between the phosphatases. Therefore, removing the Adda tail together with avoiding covalent binding led to a reduction in potency but enabled the achievement of strong selectivity.
In conclusion, with an efficient synthesis of MC analogues, we present an example of how synthetic simplicity can improve the overall efficiency of a challenging synthesis. The synthesis strategy enabled us to test 11 analogues, ultimately leading to an inhibitor (11) showing 282‐fold selectivity for PP2A over PP1, and revealing criteria for achieving selectivity between the two PSTPs in MC analogues. Notably, peptides 10 and 11 are the first MC analogues without the Adda tail that show potencies in the submicromolar range toward PP2A. The work described herein provides critical insight for future design and enables access to a large number of microcystin or potentially nodularin analogues to be tested for the selective inhibition of PSTPs.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
M.K. acknowledges a starting grant (336567) from the European Research Council (ERC).
M. Fontanillo, I. Zemskov, M. Häfner, U. Uhrig, F. Salvi, B. Simon, V. Wittmann, M. Köhn, Angew. Chem. Int. Ed. 2016, 55, 13985.
References
- 1. Goldberg J., Huang H. B., Kwon Y. G., Greengard P., Nairn A. C., Kuriyan J., Nature 1995, 376, 745–753. [DOI] [PubMed] [Google Scholar]
- 2. Shi Y., Cell 2009, 139, 468–484. [DOI] [PubMed] [Google Scholar]
- 3. Virshup D. M., Shenolikar S., Mol. Cell 2009, 33, 537–545. [DOI] [PubMed] [Google Scholar]
- 4. Brautigan D. L., FEBS J. 2013, 280, 324–345. [DOI] [PubMed] [Google Scholar]
- 5. Aggen J. B., Humphrey J. M., Gauss C. M., Huang H. B., Nairn A. C., Chamberlin A. R., Bioorg. Med. Chem. 1999, 7, 543–564. [DOI] [PubMed] [Google Scholar]
- 6. Sakoff J. A., McClusky A., Curr. Pharm. Des. 2004, 10, 1139–1159. [DOI] [PubMed] [Google Scholar]
- 7. Fagerholm A. E., Habrant D., Koskinen A. M. P., Mar. Drugs 2010, 8, 122–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Humphrey J. M., Aggen J. B., Chamberlin A. R., J. Am. Chem. Soc. 1996, 118, 11759–11770. [Google Scholar]
- 9. Taylor C., Quinn R. J., Bioorg. Med. Chem. Lett. 1996, 6, 2107–2112. [Google Scholar]
- 10. Maude B., Mehrotra A. P., Gani D. J., Chem. Soc. Perkin Trans. 1 1997, 2513–2526. [Google Scholar]
- 11. Carmichael W. W., Beasley V., Bunner D. L., Eloff J. N., Falconer I., Gorham P., Harada K., Krishnamurthy T., Yu M. J., Moore R. E., Toxicon 1988, 26, 971–973. [DOI] [PubMed] [Google Scholar]
- 12. Ward C. J., Codd G. A., J. Appl. Microbiol. 1999, 86, 874–882. [DOI] [PubMed] [Google Scholar]
- 13. Gulledge B. M., Aggen J. B., Chamberlin A. R., Bioorg. Med. Chem. Lett. 2003, 13, 2903–2906. [DOI] [PubMed] [Google Scholar]
- 14. Gulledge B. M., Aggen J. B., Eng H., Sweimeh K., Chamberlin A. R., Bioorg. Med. Chem. Lett. 2003, 13, 2907–2911. [DOI] [PubMed] [Google Scholar]
- 15. Pereira S. R., Vasconcelos V. M., Antunes A., FEBS J. 2013, 280, 674–680. [DOI] [PubMed] [Google Scholar]
- 16. Sainis I., Fokas D., Vareli K., Tzakos A. G., Kounnis V., Briaoulis E., Mar. Drugs 2010, 8, 629–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choi B. W., Namikoshi M., Sun F., Rinehart K. L., Carmichael W. W., Kaup M., Evans W. R., Beasley V. R., Tetrahedron Lett. 1993, 34, 7881–7884. [Google Scholar]
- 18. Rinehart K. L., Namikoshi M., Choi B. W., J. Appl. Phycol. 1994, 6, 159–176. [Google Scholar]
- 19. Xing Y., Xu Y., Chen Y., Jeffrey P. D., Chao Y., Lin Z., Li Z., Strack S., Stock J. B., Shi Y., Cell 2006, 127, 341–353. [DOI] [PubMed] [Google Scholar]
- 20. Kondo F., Ikai Y., Oka H., Okumura M., Ishikawa N., Harada K., Matsuura K., Murata H., Suzuki M., Chem. Res. Toxicol. 1992, 5, 591–596. [DOI] [PubMed] [Google Scholar]
- 21. Ufelmann H., Krüger T., Luckas B., Schrenk D., Toxicology 2012, 293, 59–67. [DOI] [PubMed] [Google Scholar]
- 22. Taylor C., Quinn R. J., Bioorg. Med. Chem. Lett. 1996, 6, 2113–2116. [Google Scholar]
- 23. Pearson C., Rinehart K. L., Sugano M., Costerison J. R., Org. Lett. 2000, 2, 2901–2903. [DOI] [PubMed] [Google Scholar]
- 24. El Haddadi M., Cavelier F., Vives E., Azmani A., Verducci J., Martinez J. J., Pept. Sci. 2000, 6, 560–570. [DOI] [PubMed] [Google Scholar]
- 25. Bouaïcha N., Maatouk I., Vincent G., Levi Y., Food Chem. Toxicol. 2002, 40, 1677–1683. [DOI] [PubMed] [Google Scholar]
- 26. Sotoud H., Gribbon P., Ellinger B., Reinshagen J., Boknik P., Kattner L., El-Armouche A., Eschenhagen T., J. Biomol. Screening 2013, 18, 899–909. [DOI] [PubMed] [Google Scholar]
- 27. O'Donnell M. E., Sanvoisin J., Gani D., J. Chem. Soc. Perkin Trans. 1 2001, 1696–1708. [Google Scholar]
- 28. Maynes J. T., Bateman K. S., Cherney M. M., Das A. K., Luu H. A., Holmes C. F., James M. N., J. Biol. Chem. 2001, 276, 44078–44082. [DOI] [PubMed] [Google Scholar]
- 29. Kelker M. S., Page R., Peti W., J. Mol. Biol. 2009, 385, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bagu J. R., Sonnichsen F. D., Williams D., Andersen R. J., Sykes B. D., Holmes C. F., Nat. Struct. Biol. 1995, 2, 114–116. [DOI] [PubMed] [Google Scholar]
- 31. Trogen G. B., Annila A., Eriksson J., Kontteli M., Meriluoto J., Sethson I., Zdunek J., Edlund U., Biochemistry 1996, 35, 3197–3205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary