

Abstract
Can the structures of small to medium‐sized proteins be conserved after transfer from the solution phase to the gas phase? A large number of studies have been devoted to this topic, however the answer has not been unambiguously determined to date. A clarification of this problem is important since it would allow very sensitive native mass spectrometry techniques to be used to address problems relevant to structural biology. A combination of ion‐mobility mass spectrometry with infrared spectroscopy was used to investigate the secondary and tertiary structure of proteins carefully transferred from solution to the gas phase. The two proteins investigated are myoglobin and β‐lactoglobulin, which are prototypical examples of helical and β‐sheet proteins, respectively. The results show that for low charge states under gentle conditions, aspects of the native secondary and tertiary structure can be conserved.
Keywords: gas-phase reactions, IR spectroscopy, mass spectrometry, protein folding, protein structures
Owing to its accuracy, sensitivity, and speed, mass spectrometry (MS) coupled to fragmentation techniques is the method of choice for determining the primary structure of peptides and proteins.1 To obtain information on higher‐order structures, condensed‐phase spectroscopic or scattering techniques are usually employed. Often they have limited sensitivity and therefore require high sample densities but they can provide a wealth of information under close to physiological conditions. An important question is whether native solution structures can be maintained when the molecule is transported into the solvent‐free environment, and if so over what protein size range. If this transfer can be done with the native structure remaining intact, highly sensitive mass spectrometry techniques can be employed to interrogate these structures. For very large species, such as protein complexes2 or even an entire virus,3 there is unambiguous evidence that native structures can be retained in the absence of solvent. For smaller species, however, the situation is not so clear, and this is the region we wish to examine.
To investigate higher‐order structures of peptides and proteins in isolation, mass spectrometry can be coupled to hydrogen/deuterium exchange (HDX) measurements,4 non‐thermal dissociation techniques that rely on the absorption of UV photons or the capture of electrons,5 ion mobility spectrometry (IMS),6 or optical/IR spectroscopy.7 Of these, IMS and IR spectroscopy provide the most direct information on higher‐order structure. In IMS, the angle‐averaged collision cross‐section is determined with high accuracy. Together with modeling, this measurement can give information about the size and shape of the molecule, and IMS has been used extensively to investigate various proteins and protein aggregates. One common observation is that when the charge increases, the molecules undergo Coulomb‐induced unfolding to adopt extended helical structures8 and ultimately string‐like extended structures at very high charge states.9 At low charge states, however, the proteins usually remain compact, with sizes that are expected for condensed‐phase native structures.8 However, IMS is not directly sensitive to protein secondary structure, and observing a compact structure does not guarantee that the secondary structure accurately reflects that of the native form. IR spectroscopy, on the other hand, is sensitive to secondary structure. For proteins, the local environment of characteristic oscillators such as the C=O stretch (amide‐I) or N−H bend (amide‐II) modes influence band positions, and consequently IR spectroscopy has become a routine technique for probing secondary structure in the condensed phase.10
IR multiple‐photon dissociation (IRMPD) spectroscopy11 has been applied to a large number of small peptides or peptide complexes7b,7c in the gas phase. When combined with quantum chemical calculations, detailed structural information can be obtained. For very small systems containing only a few amino acids, gas‐phase structures that are distinctively different from those in the condensed phase are usually observed. However, when large barriers such as in a proline cis–trans isomerization are involved, condensed‐phase structural motifs can be retained.12 In any case, these gas‐phase “cleanroom” structures are still very useful because they can be used to calibrate theory and to test our general understanding of intrinsic intramolecular interactions in biological molecules.
There are only a few IR studies on isolated proteins in the absence of solvent.9a, 13 In mid‐IR spectra of larger species, individual oscillators cannot be spectrally resolved and, as for proteins in the condensed phase, amide‐I and amide‐II envelopes are observed.9a, 13b–13d The band positions in these studies suggest predominantly helical structures. However, in these early studies, the conditions employed and the relatively high charge states investigated9a, 13b–13d were not favorable for retaining native solution structures.
In this work, we used an approach in which IMS is used to select the global shape of proteins, followed by IR spectroscopy to probe their secondary structure (see the Supporting Information for details). Experiments were performed on the proteins myoglobin and β‐lactoglobulin. Myoglobin is a 153 amino acid protein with a native secondary structure that is mostly (ca. 85 %) α‐helical, while β‐lactoglobulin is a 162 residue protein that has a significant (ca. 60 %) proportion of β‐sheet secondary structure. The condensed‐phase structures of both proteins are shown in Figure 1.
Figure 1.
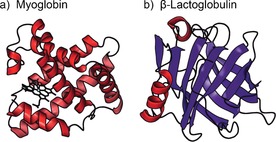
Condensed‐phase structures of a) myoglobin, a 153 amino acid protein with a native secondary structure that is mostly α‐helical and which contains a non‐covalently attached heme group (PDB ID: 1MBN) and b) β‐lactoglobulin, a 162 residue protein that has abundant β‐sheet secondary structure (PDB ID: 3BLG).
Figure 2 a, b shows electrospray ionization (ESI) mass spectra of myoglobin and β‐lactoglobulin sprayed from solvents of different compositions. The lower spectra in green are obtained with spraying from aqueous solutions with ammonium acetate buffer at a concentration of 10 mmol L−1. Under these conditions, narrow charge distributions with maxima at 8+ are observed. For β‐lactoglobulin, additional mass peaks resulting from adducts of palmitic acid are found and are marked in Figure 2 b as 9+′, 8+′ and 7+′. Changing the solvent to water/methanol (1:1) resulted in the mass spectra shown in blue. The charge‐state distributions are broader and shifted to higher charge states. For myoglobin, all labeled mass peaks in the two spectra correspond to the heme‐containing holo form. The mass spectra shown here are similar to those reported by others.14
Figure 2.
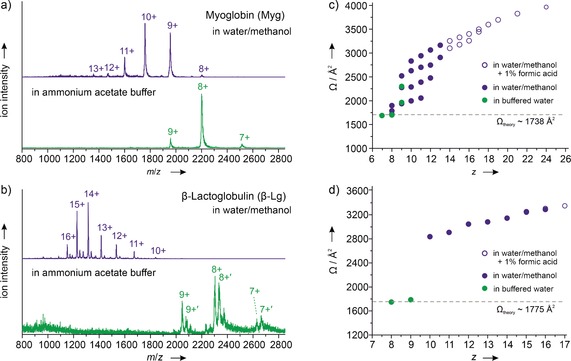
Mass spectra and collision cross‐sections for myoglobin and β‐lactoglobulin. a, b) Mass spectra obtained when spraying from buffered (ammonium acetate, pH≈7) aqueous or water/methanol solutions (green and blue, respectively). Prominent peaks are labeled, and in the case of myoglobin, they correspond to the holo form. For low charge states of β‐lactoglobulin, adducts with palmitic acid can be observed (labeled 7+′, 8+′ and 9+′). c, d) Cross‐sections as a function of charge. Solid green circles stem from ions sprayed from buffered aqueous solution, solid blue circles from ions from water/methanol solution, and open blue circles from ions from water/methanol solution with 1 % formic acid.
The resulting collision cross‐sections as a function of mass/charge as measured by IMS are shown in Figure 2 c, d. The uncertainties in the cross sections are in all cases smaller than the size of the corresponding symbol. Green circles correspond to ions sprayed from aqueous solution while blue circles correspond to ions sprayed from water/methanol solutions. In the case of myoglobin, multiple conformers are observed for most charge states and, correspondingly, more than one cross‐section value per charge state is shown. Cross‐sections expected for condensed‐phase structures (Figure 1) have been calculated using the EHSS algorithm15 and the values are shown as dotted lines in Figure 2 c, d.
As expected, the collision cross‐sections increase with increasing charge. At low charge states, compact ions are observed that have cross‐sections consistent with those expected for native structures. When the charge increases, the cross‐sections increase, thus indicating unfolding of the protein. For myoglobin several conformers with different degrees of unfolding coexist for all but the lowest and highest charge states.16 β‐Lactoglobulin shows different behavior. For each charge state, only a single conformer is observed, and furthermore, a large jump in cross‐section occurs when the solvent is changed to water/methanol, and the minimum observable charge state increases to 10+.
In order to probe the secondary structure, we performed IR spectroscopy experiments using the Fritz‐Haber‐Institut free‐electron laser (FHI‐FEL).17 Figure 3 shows IR spectra for several mass/charge states for both proteins. In all of the spectra, amide‐I and ‐II bands can be observed. In Figure 3 c (green lines) IR‐spectra for ions with a low cross‐section and low charge state are shown.
Figure 3.
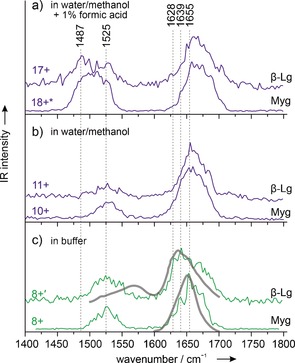
Infrared spectra for gas‐phase and condensed‐phase myoglobin and β‐lactoglobulin. a, b)Spectra for ions sprayed from water/methanol solutions. The positions and shapes of the amide‐I bands around 1655 cm−1 indicate helical secondary structures. c) Spectra for samples sprayed from aqueous solutions (green lines) and solution phase FTIR spectra (gray lines; data reproduced from Ref. 18 for β‐lactoglobulin and Ref. 19 for myoglobin). The amide‐I band for myoglobin 8+ indicates a helical secondary structure, while that of β‐lactoglobulin shows a clear β‐sheet signature.
For both 8+ ions, the amide‐II bands are found around 1525 cm−1, a value that is typical for proteins in the condensed phase.10 The amide‐I band for myoglobin 8+ is roughly symmetrical and has a maximum at 1655 cm−1, which is typical for a helical secondary structure.10 The IR spectrum of β‐lactoglobulin in the 8+ charge state, on the other hand, exhibits a rather different signature. There, the amide‐I band is asymmetric, with a maximum around 1639 cm−1, which is typical for β‐sheet‐rich proteins.10
Figure 3 a, b shows IR spectra for higher charge states. For myoglobin in the 10+ charge state, the spectrum shown in Figure 3 b results from a low‐cross‐section conformer (see Figure 2 for the cross‐section distribution), however the spectra for all of the myoglobin conformers in this charge state are essentially identical (see Figure S3). The positions of the amide‐I and amide‐II bands are almost unchanged for the 8+ and 10+ charge states, and only the width of the amide‐I band increases slightly. For β‐lactoglobulin, on the other hand, the shape and position of the amide‐I band changes dramatically between the 8+ and 11+ charge states. This transition is accompanied by a very large change in cross‐section between these two charge states (Figure 2). For the unfolded and higher charged 11+ conformer, the amide‐I band is similar in shape and position to that of 10+ myoglobin, which indicates a helical structure for the β‐lactoglobulin 11+ ions as well.
When the charge increases further to 17+ for β‐lactoglobulin and 18+ for myoglobin, the shapes of the amide‐I bands do not change and only show a slight shift to higher wavenumbers. The amide‐II bands, on the other hand, gain in relative intensity and shift significantly to lower wavenumbers. This observation can be attributed to the coulomb‐driven unraveling of helices to form extended string‐like structures.9
From a visual inspection of the spectra in Figure 3, it appears that gas‐phase β‐lactoglobulin in the 8+ charge state has a predominantly β‐sheet secondary structure, while myoglobin has an essentially helical secondary structure, which is consistent with the native solution‐phase structures of both proteins. As the charge increases, however, both proteins exhibit helical structures, and finally at the highest charge states, even these helices unravel. It is interesting to compare these spectra with results from the condensed phase. The circular dichroism (CD) spectrum was measured for both proteins using the same solvents as used for the spectroscopy experiments on the isolated species. All of the CD spectra for myoglobin and the CD spectrum of β‐lactoglobulin in water/methanol have signatures that are typical for helical structures (see Figure S2).20 On the other hand, the CD spectrum of β‐lactoglobulin in buffered aqueous solution is quite different and clearly indicates a β‐sheet structure.
In Figure 3 c), the gas‐phase spectra (green lines) are compared to the condensed‐phase FTIR spectra (gray lines) for β‐lactoglobulin18 and myoglobin19 reproduced from previous reports. The FTIR amide‐I band of β‐lactoglobulin shows the typical shape expected for a β‐sheet‐rich protein, while that of myoglobin is rather symmetrical and is found in a position typical for helical species. Clearly, in both cases, the match between the corresponding gas‐phase and condensed‐phase spectra in the amide‐I region is very good, thus giving further evidence that, for low charge states, the condensed‐phase structure is at least in part preserved after transfer to the gas phase.
IR spectroscopy directly probes local secondary structure and can differentiate between helices and β‐sheets. However, it does not provide direct information about the tertiary structure in which those secondary structure elements are embedded. IR spectroscopy is thus a good complement to IMS, which on its own is blind to structural details and only probes the overall shape and size. The combination of IR spectroscopy and IMS shown here gives a clear picture of the structural evolution of isolated proteins as their charge increases. Low charge states can retain the native structure. When Coulomb repulsion increases and charged side chains coordinate to the protein backbone,21 the native fold begins to lose stability, which is indicated by an increase in IMS cross‐section. When the original native structure is predominantly helical, IR spectroscopy shows that the unfolded species remain helical. This is to be expected, since extended helical structures can retain most of their hydrogen bonds while maximizing the distances between equal charges. However, the situation is different for native β‐sheet proteins. In these, the individual β‐sheet strands are embedded in a hydrogen‐bond network, and when an increase in charge causes unfolding, preservation of the β‐sheet strands would result in many disrupted hydrogen bonds. Hence, for an initial β‐sheet‐dominated structure, the unfolded protein will adopt a helix‐rich structure as well. Finally, at very high charges, Coulomb repulsion will cause helix unzipping in both cases.9
In summary, the presented data clearly show that for β‐lactoglobulin and myoglobin, the condensed‐phase secondary as well as tertiary structure can be conserved when solvent is completely removed. Methods based on gas‐phase mass spectrometry provide extremely high sensitivity. When mass spectrometry is combined with IMS, additionally high selectivity can be achieved through being able to select individual charge states, conformations, or aggregation states, which then can be investigated using spectroscopy. Such sensitivity and selectivity cannot be achieved using condensed‐phase methods. However, care must be taken to be sure the journey from the condensed phase to the solvent‐free gas phase is very gentle to avoid possible refolding and structure change.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge the expert assistance from the staff of the FHI free‐electron laser facility, in particular S. Gewinner and W. Schöllkopf. M.T.B. gratefully acknowledges the support of the National Science Foundation (USA) for support under grant CHE‐1301032 and support from the Alexander von Humboldt foundation.
J. Seo, W. Hoffmann, S. Warnke, M. T. Bowers, K. Pagel, G. von Helden, Angew. Chem. Int. Ed. 2016, 55, 14173.
References
- 1. Pandey A., Mann M., Nature 2000, 405, 837–846. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Heck A. J. R., Nat. Methods 2008, 5, 927–933; [DOI] [PubMed] [Google Scholar]
- 2b. Ruotolo B. T., Robinson C. V., Curr. Opin. Chem. Biol. 2006, 10, 402–408. [DOI] [PubMed] [Google Scholar]
- 3. Siuzdak G., Bothner B., Yeager M., Brugidou C., Fauquet C. M., Hoey K., Chang C. M., Chem. Biol. 1996, 3, 45–48. [DOI] [PubMed] [Google Scholar]
- 4. Engen J. R., Anal. Chem. 2009, 81, 7870–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oh H. B., Moon B., Mass Spectrom. Rev. 2015, 34, 116–132. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wyttenbach T., Bowers M. T., Annu. Rev. Phys. Chem. 2007, 58, 511–533; [DOI] [PubMed] [Google Scholar]
- 6b. Bohrer B. C., Mererbloom S. I., Koeniger S. L., Hilderbrand A. E., Clemmer D. E., Annu. Rev. Anal. Chem. 2008, 1, 293–327; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Lanucara F., Holman S. W., Gray C. J., Eyers C. E., Nat. Chem. 2014, 6, 281–294. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Simons J. P., Mol. Phys. 2009, 107, 2435–2458; [Google Scholar]
- 7b. Polfer N. C., Oomens J., Mass Spectrom. Rev. 2009, 28, 468–494; [DOI] [PubMed] [Google Scholar]
- 7c. Polfer N. C., Chem. Soc. Rev. 2011, 40, 2211–2221. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Shelimov K. B., Clemmer D. E., Hudgins R. R., Jarrold M. F., J. Am. Chem. Soc. 1997, 119, 2240–2248; [Google Scholar]
- 8b. Wyttenbach T., Bowers M. T., J. Phys. Chem. B 2011, 115, 12266–12275. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. González Flórez A. I., Mucha E., Ahn D.-S., Gewinner S., Schöllkopf W., Pagel K., von Helden G., Angew. Chem. Int. Ed. 2016, 55, 3295–3299; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3356–3360; [Google Scholar]
- 9b. Segev E., Wyttenbach T., Bowers M. T., Gerber R. B., Phys. Chem. Chem. Phys. 2008, 10, 3077. [DOI] [PubMed] [Google Scholar]
- 10. Jackson M., Mantsch H. H., Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [DOI] [PubMed] [Google Scholar]
- 11. Oomens J., Sartakov B. G., Meijer G., von Helden G., Int. J. Mass Spectrom. 2006, 254, 1–19. [Google Scholar]
- 12.
- 12a. Pierson N. A., Chen L., Valentine S. J., Russell D. H., Clemmer D. E., J. Am. Chem. Soc. 2011, 133, 13810–13813; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Shi L., Holliday A. E., Shi H., Zhu F., Ewing M. A., Russell D. H., Clemmer D. E., J. Am. Chem. Soc. 2014, 136, 12702–12711. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Oh H., Breuker K., Sze S. K., Ge Y., Carpenter B. K., McLafferty F. W., Proc. Natl. Acad. Sci. USA 2002, 99, 15863–15868; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Oomens J., Polfer N., Moore D. T., van der Meer L., Marshall A. G., Eyler J. R., Meijer G., von Helden G., Phys. Chem. Chem. Phys. 2005, 7, 1345–1348; [DOI] [PubMed] [Google Scholar]
- 13c. Fung Y. M., Besson T., Lemaire J., Maitre P., Zubarev R. A., Angew. Chem. Int. Ed. 2009, 48, 8340–8342; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 8490–8492; [Google Scholar]
- 13d. Pagel K., Kupser P., Bierau F., Polfer N. C., Steill J. D., Oomens J., Meijer G., Koksch B., von Helden G., Int. J. Mass Spectrom. 2009, 283, 161–168. [Google Scholar]
- 14.
- 14a. Liu L., Kitova E. N., Klassen J. S., J. Am. Soc. Mass Spectrom. 2011, 22, 310–318; [DOI] [PubMed] [Google Scholar]
- 14b. Gross D. S., Zhao Y., Williams E. R., J. Am. Soc. Mass Spectrom. 1997, 8, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shvartsburg A. A., Jarrold M. F., Chem. Phys. Lett. 1996, 261, 86–91. [Google Scholar]
- 16.
- 16a. Shelimov K. B., Jarrold M. F., J. Am. Chem. Soc. 1997, 119, 2987–2994; [Google Scholar]
- 16b. Schenk E. R., Almeida R., Miksovska J., Ridgeway M. E., Park M. A., Fernandez-Lima F., J. Am. Soc. Mass Spectrom. 2015, 26, 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schöllkopf W., Gewinner S., Junkes H., Paarmann A., von Helden G., Bluem H., Todd A. M. M., Proc. SPIE 2015, 9512, 95121L. [Google Scholar]
- 18. Casal H. L., Köhler U., Mantsch H. H., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1988, 957, 11–20. [DOI] [PubMed] [Google Scholar]
- 19. Meersman F., Smeller L., Heremans K., Biophys. J. 2002, 82, 2635–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kelly S. M., Jess T. J., Price N. C., BBA-Proteins Proteom. 2005, 1751, 119–139. [DOI] [PubMed] [Google Scholar]
- 21. Warnke S., von Helden G., Pagel K., J. Am. Chem. Soc. 2013, 135, 1177–1180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary