

Abstract
Novel methodology for O‐functionalization of carbohydrate derivatives has been established using bench‐stable and easily prepared iodonium(III) reagents. Both electron‐withdrawing and electron‐donating aryl groups were introduced under ambient conditions and without precautions to exclude air or moisture. Furthermore, the approach was extended both to full arylation of cyclodextrin, and to trifluoroethylation of carbohydrate derivatives. This is the first general approach to introduce traditionally non‐electrophilic groups into any of the OH groups around the sugar backbone. The methodology will be useful both in synthetic organic chemistry and biochemistry, as important functional groups can be incorporated under simple and robust reaction conditions in a fast and efficient manner.
Keywords: arylation, carbohydrates, electrophilic addition, fluorine, hypervalent compounds
Early geniuses of organic chemistry chose their interest wisely. Their topics initiated modern organic chemistry and are still fascinating for the scientists of the modern time. One of these great topics is the chemistry of carbohydrates, which is important in biochemistry, medicinal chemistry, and drug discovery, as well as in sustainable chemistry as a cheap and renewable energy source. However, the tremendous variety of carbohydrate structures complicates the development of general methodology for their derivatization. The functionalization of carbohydrates with non‐carbohydrate structures has an essential role in organic and medicinal chemistry.1 Such groups are commonly introduced by stepwise preparation of leaving groups and subsequent nucleophilic substitution (Scheme 1 a), and by either esterification or traditional Williamson etherfication of carbohydrate hydroxy groups with reactive primary electrophiles (e.g. allyl, propargyl, and benzyl halides, or α‐halocarbonyl compounds; Scheme 1 b).2 The first strategy leads to (sometimes undesired) inversion of carbohydrate carbon stereochemistry, while the last two are limited in structural scope or require subsequent transformations to reach structural diversity.2
Scheme 1.
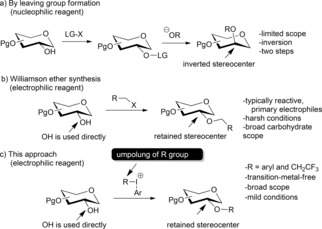
Approaches to introduce noncarbohydrate functional groups. LG=leaving group, Pg=protecting group.
Introduction of O‐linked aglycones in the glycosidic linkage has great importance in modern science,3 but the chemical and enzymatic instability of these molecules restricts their use in many applications. Methods for introduction of pharmaceutically interesting, non‐electrophilic functional groups to nonglycosidic carbohydrate hydroxy groups would broaden the chemical space and could improve the biological activities and pharmacokinetic profiles of the compounds. Such methods would indeed be valuable tools in the discovery and development of glycomimetic compounds targeting medically relevant proteins.1b,1c, 4
While the first synthesis of an O‐arylated carbohydrate (phenyl glycoside) was accomplished in 1879,5 there are no general solutions for introduction of conventionally non‐electrophilic groups to the carbohydrate hydroxy groups. Herein, we report a general methodology to functionalize carbohydrate hydroxy groups with either aryl or fluoroalkyl groups using electrophilic hypervalent iodine reagents (Scheme 1 c). The methodology is transition metal‐free, has unsurpassed efficiency and yields, features mild reaction conditions, and demonstrates very broad substrate scope.
Fluoroalkylated and aromatic compounds are important building blocks in modern pharmaceutical and fine‐chemical industries.6 Because of their low electrophilicity, there is no general method to introduce such functionalities to carbohydrate hydroxy groups, and only a handful of examples functionalized with such groups at O2−O6 have been reported. Carbohydrate hydroxy groups can be decorated with highly electron‐deficient aryl groups by SNAr reactions (Scheme 2 a).7 Arylation of secondary alcohols on the sugar backbone usually requires leaving‐group preparation, and results in epimerization.8 A phenyl group was introduced in the O3‐position with an organobismuth(V) reagent in low yield.9 Transition metal catalyzed alkyl–aryl C−O couplings are usually sensitive to steric hindrance and require high temperatures.10 This sensitivity could explain why transition metals have not been applied in carbohydrate OH functionalization, with the exception of a nickel/iridium‐catalyzed photoredox arylation of the primary OH of galactose (Scheme 2 b).11 Ritter and co‐workers presented an elegant OH−OH coupling strategy which was also exemplified on carbohydrates, but it did require an expensive carbene reagent and strictly anhydrous conditions (Scheme 2 c).12
Scheme 2.
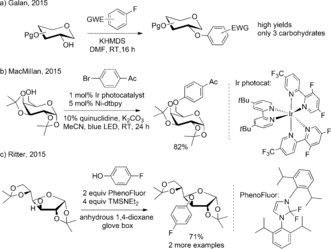
State of the art in carbohydrate O‐arylation. EWG=electron‐withdrawing group, DMF=N,N‐dimethylformamide, HMDS=hexamethyldisilazide, TMS=trimethylsilyl.
Recent developments in hypervalent iodine chemistry have enabled efficient synthesis of highly electrophilic, and bench‐stable iodonium reagents for the transfer of carbon ligands.13 While hypervalent iodine reagents have been utilized as oxidants in carbohydrate chemistry,14 they have never been applied as group‐transfer agents to derivatize carbohydrates. Such an approach could have several advantages: 1) direct O‐functionalization instead of leaving group/glycal preparation; 2) retention of stereochemistry; 3) no need for Lewis acid or transition‐metal catalyst; 4) robust, mild, and user‐friendly conditions; and 5) no need for excess carbohydrate derivatives.
We have previously demonstrated metal‐free O‐arylations with diaryliodonium salts,15 and set out to assess the feasibility of this approach. Carbohydrate derivatives 1 were treated with 4‐nitrophenyl(phenyl)iodonium triflate (2 a) to provide nitroarylated carbohydrates 3 (Table 1). The nitroaryl group was chosen as it can be used as a protecting group, as a handle for conjugation to other bioactive structures after reduction, or serve as a good substrate for enzymes in the glycosidic position.16 To our delight, the anomeric OH of mannofuranose (1 a) was smoothly arylated (entry 1). The use of simple alkali bases proved sufficient (entries 2–3), and even aqueous sodium hydroxide resulted in 95 % yield (entry 4). The method also allowed the arylation of fructose and glucose derivatives 1 b–d with both NaOH and tBuOK in excellent yields (entries 5–10). The practical benefit of extremely short reaction times with tBuOK led us to choose this base for subsequent experiments.
Table 1.
Screening of the arylation of secondary hydroxy groups.[a]
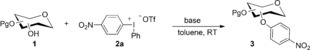
| Entry | 1 | Base | t | 3 | Yield [%][a] |
|---|---|---|---|---|---|
| 1[b] |
|
tBuOK | 20 min | 3 a | 95 |
| 2 | KOH | 3 h | 96 | ||
| 3 | NaOH | 3 h | 95 | ||
| 4 | NaOH(aq) [c] | 3 h | 93 | ||
| 5 |
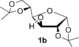
|
NaOH | 2 h | 3 b | quant |
| 6[b] | tBuOK | 40 min | 92 | ||
| 7 |
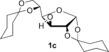
|
NaOH | 5 h | 3 c | 89 |
| 8[b] | tBuOK | 10 min | 97 | ||
| 9 |
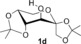
|
NaOH | 3 h | 3 d | 99 |
| 10[b] | tBuOK | 10 min | 99 |
[a] Reaction conditions: sugar 1 (0.2 mmol), salt 2 a (1.5 equiv) and base (1.5 equiv), toluene (1 mL), RT. Yield of isolated product. [b] 0.4 mmol scale. [c] 0.3 mmol NaOH in 100 μL water. Tf=trifluoromethanesulfonyl.
To investigate the scope of the reaction, a set of furanose and pyranose structures 1 were subsequently converted into the corresponding nitrophenylated derivatives (Scheme 3). Protected glucosamine and allose derivatives were both O3‐arylated to provide 3 e,f. A series of galactose derivatives were also subjected to the reaction conditions. The O6‐unprotected galactose gave 3 h in good yield within a 30 minute reaction time, and the O3‐product 3 i was obtained in even shorter time. Noteworthy is that modification of the O3 of galactosides is an important approach towards selective ligands for galectin proteins.17
Scheme 3.
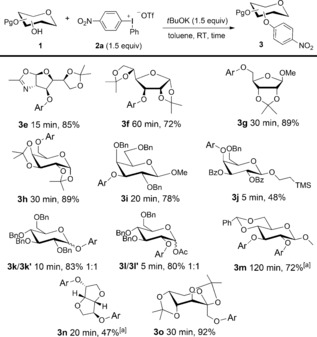
Scope of the nitroarylation reaction. Yield of isolated product given. [a] 3.0 equiv of base and 2 a. Bz=benzoyl.
Furthermore, the typically unreactive axial O4 of a mixed benzoyl‐ and benzyl‐protected galactoside was also nitroarylated to 3 j in a very fast reaction, albeit in moderate yield (Scheme 3). Arylation of the glycosidic OH in anomeric mixtures of substrates provided the products 3 k,l in 1:1 anomeric ratios. Diarylation of a glucose diol was also conducted, thus resulting in the corresponding diether 3 m. Isosorbide is one of the promising sustainable resources,18 and both the more accessible and the hydrogen bonded hydroxy group of isosorbide were arylated in a fast reaction to provide 3 n. Attempts to selectively monoarylate diols failed, and might be explained by the poor solubility of the diol compared to the monoarylated alcohol, thus leading to faster arylation of the latter. Synthesis of similar compounds in an SNAr manner required heating at 150 °C for 4 hours.18c Topiramate is an effective drug in the treatment of epilepsy and bipolar disorder.19 The sugar‐component of this drug was nitroarylated to 3 o in the same short reaction time and high efficacy.
Having established efficient nitroarylation conditions, the transfer of various other aryl groups was investigated next.20 As isopropylidene glucofuranose can be easily deprotected and isomerized into glucopyranose, this was chosen as a model substrate (Figure 1). Aryl groups with electron‐withdrawing substituents were easily installed to yield 3 p,q, and even a sterically demanding ortho‐methoxycarbonyl aryl group could be transferred to give 3 r at a slightly elevated temperature. The incorporation of a phenyl ring (3 s), aryl groups with electron‐donating alkyl substituents (3 t–u), and the sterically congested mesityl group (3 v) were also possible in good yields.
Figure 1.
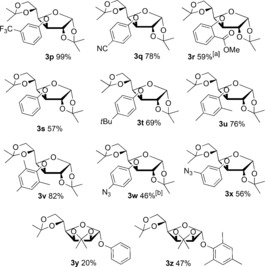
Scope with respect to the aryl groups. Reaction conditions: sugar 1 (1 equiv), salt 2 (2 equiv), and tBuOK (2 equiv) in toluene (4 mL) at RT for 2–4 h. Yield of isolated product given. [a] At 50 °C. [b] 56 % with TMP salt; see the Supporting Information.
The use of azido groups has a great impact on carbohydrate chemistry, even more so since click chemistry has revolutionized the field of biochemistry.21 In addition, azidophenyl groups are important groups in photolabeling biochemistry. To the best of our knowledge, azido‐substituted diaryliodonium salts have never been applied to azidophenylate organic molecules.22 Importantly, both para‐ and meta‐azidophenylated glucopyranose derivatives 3 w,x could be obtained under mild reaction conditions (Figure 1). Arylation of the glycosidic OH of mannose to give 3 y,z proved more difficult than the O3 of glucose, thus highlighting that this method is complementary to existing methods to introduce aryl groups to carbohydrates.
The methodology was extended to the synthesis of more complex products, as exemplified with an arylation using a phenylalanine‐derived iodonium salt to generate the unprecedented glucose tyrosine glycopeptide‐type system 3 aa (Scheme 4 a). Under unoptimized reaction conditions, the reaction proceeded in moderate yield with recovered 1 b. Furthermore, a cyclodextrin derivative was sevenfold nitroarylated in to give 4 in 57 % yield (Scheme 4 b).
Scheme 4.
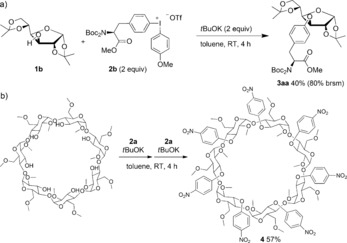
Advanced arylation of carbohydrates. Boc=tert‐butoxycarbonyl.
Fluorine‐containing functional groups have high impact in pharmaceutical and crop science. While trifluoromethylation is straightforward, the introduction of a trifluoroethyl moiety to hydroxy groups is not established despite their potential use in medicinal carbohydrate chemistry.23 The low electrophilicity of trifluoroethyl iodide and related pseudohalogenides prevents O‐functionalization with these reactants under mild reaction conditions.24 Inspired by our earlier findings, we envisioned the use of trifluoroethyl(mesityl)iodonium triflate (5), which is another class of hypervalent iodine reagent,25 for carbohydrate O‐functionalization (Scheme 5). To our delight, 5 was successfully used to trifluoroethylate a small series of mono‐unprotected sugar derivatives. The reactions took place with complete chemoselectivity, that is, without formation of any arylated byproducts, to give fluorinated carbohydrates 6 under mild and practical reaction conditions.
Scheme 5.
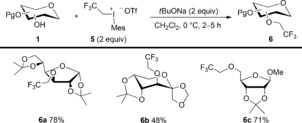
Fluoroalkylation of carbohydrates. Mes=mesityl.
In conclusion, novel methodology to O‐functionalize carbohydrate derivatives was developed using bench‐stable and easily prepared iodonium(III) reagents. To the best of our knowledge, this is the first general approach to introduce traditionally non‐electrophilic (aryl and trifluoroethyl) groups onto a variety of different carbohydrate hydroxy groups, thus attaining products which have previously been unobtainable or required long synthetic sequences. The strategy was applied to introduce both electron‐withdrawing and electron‐donating aryl groups under ambient conditions and without precautions to avoid air and moisture. Furthermore, the reaction could be extended both to full arylation of cyclodextrin, and to trifluoroethylation of carbohydrate derivatives.
We anticipate that this methodology will receive great interest in both organic chemistry and biochemistry, as important functional groups can be implemented under simple and robust reaction conditions. Application of the methodology towards synthesis and evaluation of novel bioactive compounds are underway in our laboratories.
Experimental Section
The carbohydrate derivative 1 (0.04–0.4 mmol) was stirred in a 20 mL screw‐cap vial in toluene (no dry solvent needed; 0.4–4 mL) for 3 minutes. A mixture of 4‐nitrophenyl(phenyl) iodonium triflate (2 a; 1.5 equiv/OH) and potassium tert‐butoxide (1.5 equiv/OH) was added, without taking precautions to exclude air, over a period of 2–3 minutes, while the mixture turned yellow. The reaction was stirred for 5–60 min, followed by TLC. Upon completion, the mixture was transferred to a round‐bottom flask with EtOAc (3×10 mL) and Celite was added. The volatiles were removed under reduced pressure, and the residue was subjected to column chromatography to provide the product 3.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful to Dr. Johan Olsson and Prof. Thisbe Lindhorst for initial discussions. Olle Engkvist Byggmästare foundation is acknowledged for project funding and GLT's postdoctoral scholarship.
G. L. Tolnai, U. J. Nilsson, B. Olofsson, Angew. Chem. Int. Ed. 2016, 55, 11226.
Contributor Information
Dr. Gergely L. Tolnai, http://www.organ.su.se/bo/
Prof. Berit Olofsson, Email: berit.olofsson@su.se.
References
- 1.
- 1a. Dalziel M., Crispin M., Scanlan C. N., Zitzmann N., Dwek R. A., Science 2014, 343, 1235681; [DOI] [PubMed] [Google Scholar]
- 1b. Hudak J. E., Bertozzi C. R., Chem. Biol. 2014, 21, 16–37; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Ernst B., Magnani J. L., Nat. Rev. Drug Discovery 2009, 8, 661–677; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Pieters R. J., ChemBioChem 2006, 7, 721–728. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Jacobsson M., Malmberg J., Ellervik U., Carbohydr. Res. 2006, 341, 1266–1281; [DOI] [PubMed] [Google Scholar]
- 2b. Jensen K. J., J. Chem. Soc. Perkin Trans. 1 2002, 2219–2233; For recent examples, see [Google Scholar]
- 2c. Issa J. P., Bennett C. S., J. Am. Chem. Soc. 2014, 136, 5740–5744 and refererences therein. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Burke H. M., Gunnlaugsson T., Scanlan E. M., Chem. Commun. 2015, 51, 10576–10588; [DOI] [PubMed] [Google Scholar]
- 3b. Pershagen E., Borbas K. E., Angew. Chem. Int. Ed. 2015, 54, 1787–1790; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1807–1810; [Google Scholar]
- 3c. Dirks-Hofmeister M. E., Verhaeghe T., De Winter K., Desmet T., Angew. Chem. Int. Ed. 2015, 54, 9289–9292; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9421–9424; [Google Scholar]
- 3d. Ichikawa Y., Kamiya M., Obata F., Miura M., Terai T., Komatsu T., Ueno T., Hanaoka K., Nagano T., Urano Y., Angew. Chem. Int. Ed. 2014, 53, 6772–6775; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6890–6893. [Google Scholar]
- 4. Fernández-Tejada A., Cañada F. J., Jiménez-Barbero J., Chem. Eur. J. 2015, 21, 10616–10628. [DOI] [PubMed] [Google Scholar]
- 5. Michael A., J. Am. Chem. Soc. 1879, 1, 305–312. [Google Scholar]
- 6.All top-selling small-molecule drugs contain aromatic or fluorinated groups. See: McGrath N. A., Brichacek M., Njardarson J. T., J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar]
- 7.
- 7a. Henderson A. S., Medina S., Bower J. F., Galan M. C., Org. Lett. 2015, 17, 4846–4849; [DOI] [PubMed] [Google Scholar]
- 7b. Huchel U., Schmidt C., Schmidt R. R., Eur. J. Org. Chem. 1998, 1998, 1353–1360; [Google Scholar]
- 7c. Huchel U., Schmidt C., Schmidt R. R., Tetrahedron Lett. 1995, 36, 9457–9460; [Google Scholar]
- 7d. Sharma S. K., Corrales G., Penadés S., Tetrahedron Lett. 1995, 36, 5627–5630. [Google Scholar]
- 8. Das Adhikary N., Chattopadhyay P., Eur. J. Org. Chem. 2011, 7346–7354. [DOI] [PubMed] [Google Scholar]
- 9. Barton D. H. R., Finet J.-P., Motherwell W. B., Pichon C., J. Chem. Soc. Perkin Trans. 1 1987, 251–259. [Google Scholar]
- 10.
- 10a. Torraca K. E., Huang X., Parrish C. A., Buchwald S. L., J. Am. Chem. Soc. 2001, 123, 10770–10771; [DOI] [PubMed] [Google Scholar]
- 10b. Bhadra S., Dzik W. I., Goossen L. J., J. Am. Chem. Soc. 2012, 134, 9938–9941; [DOI] [PubMed] [Google Scholar]
- 10c. Tolnai G. L., Pethő B., Králl P., Novák Z., Adv. Synth. Catal. 2014, 356, 125–129; [Google Scholar]
- 10d. Bhadra S., Dzik W. I., Gooßen L. J., Angew. Chem. Int. Ed. 2013, 52, 2959–2962; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3031–3035. [Google Scholar]
- 11. Terrett J. A., Cuthbertson J. D., Shurtleff V. W., MacMillan D. W. C., Nature 2015, 524, 330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen X., Neumann C. N., Kleinlein C., Goldberg N. W., Ritter T., Angew. Chem. Int. Ed. 2015, 54, 5662–5665; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5754–5757. Ligand price at Sigma–Aldrich, 360 €/mmol, April 2016. [Google Scholar]
- 13.
- 13a. Yoshimura A., Zhdankin V. V., Chem. Rev. 2016, 116, 3328–3435; [DOI] [PubMed] [Google Scholar]
- 13b. Aradi K., Tóth B. L., Tolnai G. L., Novák Z., Synlett 2016, 27, 1456–1485; [Google Scholar]
- 13c. Olofsson B., Top. Curr. Chem. 2015, 373, 135–166; [DOI] [PubMed] [Google Scholar]
- 13d. Merritt E. A., Olofsson B., Angew. Chem. Int. Ed. 2009, 48, 9052–9070; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9214–9234. [Google Scholar]
- 14.
- 14a. Kirschning A., Eur. J. Org. Chem. 1998, 1998, 2267–2274; [Google Scholar]
- 14b. Kajimoto T., Morimoto K., Ogawa R., Dohi T., Kita Y., Eur. J. Org. Chem. 2015, 2015, 2138–2142; [Google Scholar]
- 14c. Kunst E., Gallier F., Dujardin G., Kirschning A., Org. Biomol. Chem. 2008, 6, 893–898; [DOI] [PubMed] [Google Scholar]
- 14d. Kirschning A., J. Org. Chem. 1995, 60, 1228–1232. [Google Scholar]
- 15.
- 15a. Ghosh R., Stridfeldt E., Olofsson B., Chem. Eur. J. 2014, 20, 8888–8892; [DOI] [PubMed] [Google Scholar]
- 15b. Ghosh R., Lindstedt E., Jalalian N., Olofsson B., ChemistryOpen 2014, 3, 54–57; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Lindstedt E., Ghosh R., Olofsson B., Org Lett. 2013, 15, 6070–6073; [DOI] [PubMed] [Google Scholar]
- 15d. Jalalian N., Petersen T. B., Olofsson B., Chem. Eur. J. 2012, 18, 14140–14149; for other O-arylations of alcohols with diaryliodonium salts, see [DOI] [PubMed] [Google Scholar]
- 15e. Sundalam S. K., Stuart D. R., J. Org. Chem. 2015, 80, 6456–6466; [DOI] [PubMed] [Google Scholar]
- 15f. Lubinkowski J. J., Knapczyk J. W., Calderon J. L., Petit L. R., McEwen W. E., J. Org. Chem. 1975, 40, 3010–3015. [Google Scholar]
- 16.
- 16a. Zhu W., Wang Y., Li K., Gao J., Huang C.-H., Chen C.-C., Ko T.-P., Zhang Y., Guo R.-T., Oldfield E., J. Med. Chem. 2015, 58, 1215–1227; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Müller A., Kobarg H., Chandrasekaran V., Gronow J., Sönnichsen F. D., Lindhorst T. K., Chem. Eur. J. 2015, 21, 13723–13731; [DOI] [PubMed] [Google Scholar]
- 16c. Ramström O., Lohmann S., Bunyapaiboonsri T., Lehn J.-M., Chem. Eur. J. 2004, 10, 1711–1715. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Cumpstey I., Salomonsson E., Sundin A., Leffler H., Nilsson U. J., Chem. Eur. J. 2008, 14, 4233–4245; [DOI] [PubMed] [Google Scholar]
- 17b. Cumpstey I., Sundin A., Leffler H., Nilsson U. J., Angew. Chem. Int. Ed. 2005, 44, 5110–5112; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5240–5242; [Google Scholar]
- 17c. Sörme P., Arnoux P., Kahl-Knutsson B., Leffler H., Rini J. M., Nilsson U. J., J. Am. Chem. Soc. 2005, 127, 1737–1743; [DOI] [PubMed] [Google Scholar]
- 17d. Rajput V. K., Leffler H., Nilsson U. J., Mukhopadhyay B., Bioorg. Med. Chem. Lett. 2014, 24, 3516–3520. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Galbis J. A., García-Martín M. d. G., de Paz M. V., Galbis E., Chem. Rev. 2016, 116, 1600–1636; [DOI] [PubMed] [Google Scholar]
- 18b. Fenouillot F., Rousseau A., Colomines G., Saint-Loup R., Pascault J. P., Prog. Polym. Sci. 2010, 35, 578–622; [Google Scholar]
- 18c. Medimagh R., Mghirbi S., Saadaoui A., Fildier A., Desloir-Bonjour M., Raffin G., Kricheldorf H. R., Chatti S., Compt. Rend. Chim. 2013, 16, 1127–1139. [Google Scholar]
- 19.
- 19a. Arnone D., Ann. Gen. Psychiatry 2005, 4, 1–14; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Maryanoff B. E., Costanzo M. J., Nortey S. O., Greco M. N., Shank R. P., Schupsky J. J., Ortegon M. P., Vaught J. L., J. Med. Chem. 1998, 41, 1315–1343; [DOI] [PubMed] [Google Scholar]
- 19c. Maryanoff B. E., Nortey S. O., Gardocki J. F., Shank R. P., Dodgson S. P., J. Med. Chem. 1987, 30, 880–887. [DOI] [PubMed] [Google Scholar]
- 20.Diaryliodonium salts were synthesized according to:
- 20a. Bielawski M., Zhu M., Olofsson B., Adv. Synth. Catal. 2007, 349, 2610–2618; [Google Scholar]
- 20b. Bielawski M., Aili D., Olofsson B., J. Org. Chem. 2008, 73, 4602–4607; [DOI] [PubMed] [Google Scholar]
- 20c. Zhu M., Jalalian N., Olofsson B., Synlett 2008, 592–596. [Google Scholar]
- 21. Tiwari V. K., Mishra B. B., Mishra K. B., Mishra N., Singh A. S., Chen X., Chem. Rev. 2016, 116, 3086–3240. [DOI] [PubMed] [Google Scholar]
- 22. Chun J.-H., Pike V. W., Eur. J. Org. Chem. 2012, 2012, 4541–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Han J. B., Hao J. H., Zhang C. P., Qin H. L., Curr. Org. Chem. 2015, 19, 1554–1565; [Google Scholar]
- 23b. Le Mai Hoang K., Liu X.-W., Nat. Commun. 2014, 5, 5051; [DOI] [PubMed] [Google Scholar]
- 23c. Fröhlich R. F. G., Schrank E., Zangger K., Carbohydr. Res. 2012, 361, 100–104; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23d. Vuluga D., Legros J., Crousse B., Bonnet-Delpon D., Eur. J. Org. Chem. 2009, 2009, 3513–3518; [Google Scholar]
- 23e. Gueyrard D., Rollin P., Nga T. T. T., Ourévitch M., Bégué J.-P., Bonnet-Delpon D., Carbohydr. Res. 1999, 318, 171–179. [Google Scholar]
- 24.
- 24a. Yang Q., Njardarson J. T., Tetrahedron Lett. 2013, 54, 7080–7082; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Bodor N., Huang M.-J., C. Szántay, Jr. , Szántay C., Tetrahedron 1992, 48, 5823–5830. [Google Scholar]
- 25.
- 25a. Tóth B. L., Kovács S., Sályi G., Novák Z., Angew. Chem. Int. Ed. 2016, 55, 1988–1992; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2028–2032; [Google Scholar]
- 25b. Tolnai G. L., Székely A., Makó Z., Gáti T., Daru J., Bihari T., Stirling A., Novák Z., Chem. Commun. 2015, 51, 4488–4491. Similar reagents have been used as promotors in glycosylation reactions, but have not been utilized as group-transfer reagents. See: [DOI] [PubMed] [Google Scholar]
- 25c. Chu A.-H. A., Minciunescu A., Bennett C. S., Org. Lett. 2015, 17, 6262–6265; [DOI] [PubMed] [Google Scholar]
- 25d. Chu A.-H. A., Minciunescu A., Montanari V., Kumar K., Bennett C. S., Org. Lett. 2014, 16, 1780–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary