

Abstract
1H detection can significantly improve solid‐state NMR spectral sensitivity and thereby allows studying more complex proteins. However, the common prerequisite for 1H detection is the introduction of exchangeable protons in otherwise deuterated proteins, which has thus far significantly hampered studies of partly water‐inaccessible proteins, such as membrane proteins. Herein, we present an approach that enables high‐resolution 1H‐detected solid‐state NMR (ssNMR) studies of water‐inaccessible proteins, and that even works in highly complex environments such as cellular surfaces. In particular, the method was applied to study the K+ channel KcsA in liposomes and in situ in native bacterial cell membranes. We used our data for a dynamic analysis, and we show that the selectivity filter, which is responsible for ion conduction and highly conserved in K+ channels, undergoes pronounced molecular motion. We expect this approach to open new avenues for biomolecular ssNMR.
Keywords: in-cell NMR spectroscopy, membrane proteins, protein dynamics, proton detection, solid-state NMR spectroscopy
Proton (1H) detection can greatly increase spectral sensitivity in solid‐state NMR (ssNMR) spectroscopy and thereby enables the study of complex proteins.1 1H‐detected ssNMR experiments usually require a stark dilution of the 1H network to diminish line‐broadening dipolar 1H–1H couplings, which can be achieved by protein expression in deuterated solvents.2 Whereas such high degrees of deuteration can provide excellent 1H resolution, they necessitate a proton/deuterium (H/D) back‐exchange step to incorporate amino protons (HN). Consequently, with these labeling techniques, water‐inaccessible protein regions remain invisible for 1H detection unless unfolding and refolding protocols are used, which are tedious, not broadly applicable, and limited to in vitro preparations. Moreover, deuterated solvents can reduce or prevent protein expression, especially in mammalian cells, and are generally associated with high costs for low‐yield proteins. A remedy to these problems would be the use of fully protonated proteins;3 however, such systems often provide limited spectral resolution. Even at very fast magic angle spinning (MAS) frequencies (>100 kHz),4 the resolution in fully protonated samples is governed by residual 1H–1H couplings. Moreover, the sensitivity loss that is due to the small sample volumes required for >100 kHz MAS may not be compensated for in heterogeneous systems such as cellular samples.
These issues have thus far critically limited the application of 1H detection for the study of water‐inaccessible protein regions, which is a major obstacle for ssNMR spectroscopy as binding pockets and active sites alike can be buried deep inside protein cores, far away from the bulk water. It is a particularly severe problem for the study of membrane proteins, whose transmembrane (TM) parts are critical for their function and do not undergo exchange in protonated buffers.2c, 3c, 5 This prompted us to develop a two‐step approach that is generally applicable in vitro and in situ, works already at moderate MAS frequencies of 60 kHz, and provides well‐resolved 1H‐detected NMR spectra of water‐inaccessible protein regions. In the first step, we enhance the 1H resolution by employing a novel labeling scheme, dubbed inverse fractional deuteration (iFD), which is based on protonated solvents (100 % H2O) and fully deuterated [13C]glucose in the growth medium. In the second step, we wash the iFD‐labeled protein with deuterated buffers (100 % D2O), which further markedly improves the spectral resolution. We first studied the iFD labeling approach on ubiquitin and then, in combination with the D2O wash, used it to assign the TM part of the ion channel KcsA in lipid bilayers. Based on our assignments, we present a site‐resolved study of the dynamics of the TM part, which is crucial to fully understand ion channel gating. Remarkably, our approach even provides sufficient spectral quality for the site‐resolved analysis of a membrane protein in situ, that is, directly in a native bacterial cell membrane.
iFD labeling is the first of two consecutive steps to improve the resolution of water‐inaccessible protons, especially for HN and Hα, which are most important for backbone assignments. 1H‐detected 2D NH and CH spectra of iFD‐labeled [13C,15N]ubiquitin in aqueous (100 % H2O) buffers, acquired at 60 kHz MAS and 950 MHz 1H frequency using MISSISSIPI water suppression7 and PISSARRO decoupling,8 are shown in Figures 1 A, B. These spectra are very well resolved and feature an average linewidth of 0.1–0.2 ppm for HN and Hα at 950 MHz, which is excellent considering the high 1H density of 80 % in iFD proteins, and constitutes a substantial average linewidth improvement of about 30–35 % compared to fully protonated ubiquitin (see Figure 1 C and the Supporting Information, Table S1; see also Section S2 for a quantitative 1H population analysis and a discussion of the 1H linewidth improvement). Moreover, as both Hα and, obviously, HN are recruited from the protonated solvent during protein expression, their 1H levels are close to 100 % in iFD ubiquitin. We also observed a very good resolution of around 0.1 ppm for many side‐chain 1H (Figure S3), although the resolution of certain methyl groups was compromised by isotopologue effects. Hence, owing to the high 1H density and 1H resolution in iFD ubiquitin, the backbone and many side‐chain protons could be readily assigned (Figure 1 D). We first correlated backbone HN that had been previously assigned2f and Hα protons using 3D CαNH and NCαHα experiments. We then used a 3D CCH experiment to detect side‐chain 1H, which were connected to backbone HN via the Hα protons (see the Supporting Information for experimental details).
Figure 1.
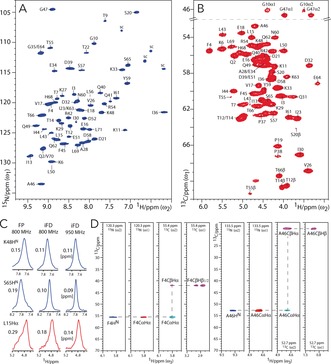
Dipolar 1H‐detected ssNMR experiments in iFD ubiquitin measured at 950 MHz and 60 kHz MAS. A) 2D NH spectrum (blue). B) CαHα region of a 2D CH spectrum (red). C) t1 cross‐sections extracted from 2D NH (blue) and CH (red) spectra of fully protonated (FP) and iFD ubiquitin. D) 1H‐detected side‐chain assignments. Strip plots are shown for 3D CαNH (blue), 3D NCαHα (red), and 3D CCH (cyan and magenta for negative and positive signals, respectively) experiments. DREAM6 13C–13C transfer was used for the 3D CCH experiment.
To further improve the 1H resolution of water‐inaccessible protein regions, we combined iFD labeling with a D2O wash. This was demonstrated with the K+ channel KcsA, a well‐accepted model for ion channel gating.9 Figure 2 A shows a 2D NH spectrum of iFD/[13C,15N]‐labeled KcsA, reconstituted in E. coli lipids and protonated buffers, acquired at 60 kHz MAS and 800 MHz. This spectrum, which shows the HN signals of the entire channel, is already of superior resolution compared to our earlier results with fully protonated KcsA,3c and readily allows identifying previously assigned water‐exposed signals (red, Figure 2 A).2f To exclusively select the TM part, we incubated iFD‐labeled KcsA in D2O and acquired a 2D NH spectrum, which featured a stark enhancement in spectral quality with well‐resolved signals as narrow as 0.13 ppm (Figures 2 B, C). The resolution improvement is partly due to the virtually complete disappearance of the water‐exposed residues, which reduces spectral congestion (see Figure S4 for a spectral overlay). Remarkably, the removal of water protons (and other exchangeable 1H) further narrows the linewidth of the TM HN by 25 % on average compared to the spectrum shown in Figure 2 A (Figure 2 D and Table S5). The latter has not been observed before in ssNMR spectroscopy, and implies that residual dipolar couplings to water protons can contribute to the 1H linewidth in highly protonated membrane proteins, even at 60 kHz MAS and a sample temperature of 35 °C. Such couplings are presumably substantial in KcsA owing to the presence of large internal water‐filled cavities and buried water.3c
Figure 2.
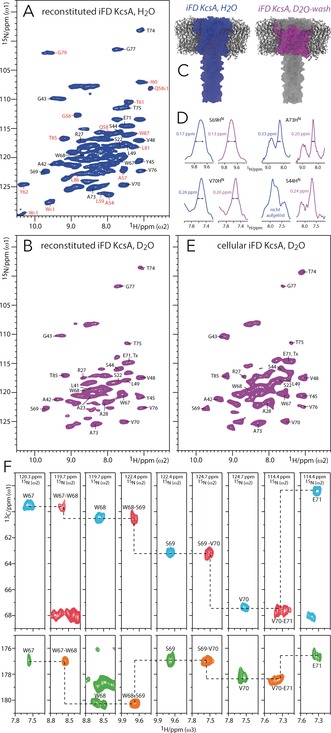
1H detection of water‐inaccessible regions in the ion channel KcsA. All data were recorded at 60 kHz MAS and 800 MHz. 2D NH spectra of reconstituted iFD KcsA A) before (blue) and B) after a D2O wash (magenta), and of E) cellular D2O‐incubated iFD KcsA, measured in native cell membranes (magenta). Assignments in red and black were obtained with FD KcsA2f and iFD KcsA, respectively. C) Color coding of regions that feature detectable HN. All HN are present in iFD KcsA whereas only the TM ones remain after the D2O wash. D) t1 cross‐sections extracted from 2D NH spectra of iFD KcsA before (blue) and after (magenta) a D2O wash. F) 1H‐detected assignments of residues W67–E71 of the pore helix. Strip plots are shown for 3D CαNH (cyan), Cα(CO)NH (red), CONH (orange), and CO(Cα)NH (green) experiments.
Notably, the high spectral quality conferred by our two‐step approach even allows measuring the TM part of KcsA directly in a native cell membrane.10 Cellular membranes are much more complex than reconstituted lipid bilayers in terms of the lipid and protein composition, and this can influence membrane protein function. However, adequate spectral quality is a challenge in cellular ssNMR studies owing to the low concentration of the target protein. Cellular iFD KcsA was expressed in the E. coli inner membrane using rifampicin to ensure selective 13C,15N‐labeling of KcsA and reduce the endogenous spectral background.11 The outer membrane was removed to increase the amount of KcsA in the sample, which was then incubated in deuterated solvent for three days prior to the ssNMR experiments. We obtained a surprisingly well‐resolved in situ 2D NH spectrum (Figure 2 E), which exhibited the clear spectral fingerprint of closed‐conductive KcsA9c without any discernible background signal (Figure S6). This is remarkable as the KcsA concentration in situ was about seven times lower than in vitro (see Section S10 in the Supporting Information). We could readily annotate the in situ spectrum based on our in vitro assignments (see below). To the best of our knowledge, this is the first time that a few nanomoles of a membrane protein (that is, ca. 3 nanomoles of KcsA, corresponding to ca. 175 μg of protein) could be assigned in situ with 1H‐detected ssNMR spectroscopy. Cellular KcsA showed only small chemical shift perturbations (CSPs) compared to reconstituted KcsA (Figure S11), implying that E. coli lipids are good membrane mimics. Yet, we observed a few marked differences in situ, which we discuss below, together with the assignments and relaxation studies performed in vitro. In addition, many signals in the in situ spectrum are strongly broadened by up to 100 % (Figure S7), which arises from inhomogeneous line broadening owing to the cellular heterogeneity. This type of broadening is independent of the MAS frequency and likely impacts on sensitivity when using smaller (<1.3 mm) rotor diameters.
We could assign approximately half of the resonances in the spectrum in Figure 2 B based on 3D CαNH, Cα(CO)NH, CONH, and CO(Cα)NH experiments (Figure 2 F), supplemented with 3D NCαCX and 2D N(CO)CX spectra to identify residue types, and supported by available 13C and 15N chemical‐shift data.9c, 12 These assignments include a number of functionally essential structural elements that we can access here for the first time with 1H detection.
The selectivity filter, responsible for the stringent selection of K+ over Na+ ions, comprises the 75‐TVGYG‐79 signature sequence, which is common to all K+ channels.9a Whereas only the exchangeable Y78 and G79 residues were detectable in a former study,2f here we could also assign residues T74–G77, providing access to the entire filter. Moreover, we assigned the pore helix L66–E71, a critical element for C‐type inactivation,9a as well as large stretches of the TM1 (transmembrane 1) helix including residues L41–L49, which precede the lipid‐sensitive turret.9c These signals could be largely retrieved in cellular KcsA. However, we could not unambiguously assign residues of the inner TM2 (transmembrane 2) helix, which was partly due to spectral overlap. Yet, while certain TM2 residues (such as V93–V95) were identified in the spectrum in Figure 2 B, our data suggest that the TM2 helix partly exchanges with D2O, especially C‐terminal of the flexible hinge G104.9a This hypothesis was corroborated by a 2D N(CO)CX experiment in D2O with short initial 1H–15N CP times,5 in which G104 and G116 could not be observed (Figure S8), and it is in line with studies in micelles.13 It was also corroborated by the observation of only about 50 signals in the 3D CαNH spectrum although we should observe approximately 65 resonances if all TM HN were retained in D2O. H/D exchange within the membrane is hence a curse and a blessing at the same time. Whereas it complicates assignments owing to missing sequential contacts, it can give insight into protein flexibility and membrane topology.
We used our assignments to perform a detailed analysis of the motion in the TM part of KcsA. Understanding this motion is of high relevance for the ion channel function, which depends on dynamic changes of membrane‐embedded structural elements.9a, 14 It is also of particular interest to decipher the motion of the filter as its flexibility is assumed to be related to a ubiquitous ion channel regulatory mechanism known as modal gating.9b NMR relaxation is a powerful method to study protein dynamics,15 and many of our assigned HN signals are well‐resolved (Figure 2 B), which is a prerequisite for site‐resolved analysis. It has previously been shown that 15N transverse rotation frame relaxation (T1ρ), which is a sensitive reporter of motion on the nano‐ to microsecond timescale, can be quantitatively extracted in fully protonated samples at higher (>50 kHz) MAS frequencies,16 at which coherent contributions to the magnetization decay are largely suppressed. We first measured the bulk 15N T1ρ as a function of the spinlock amplitude (Figure S9). In agreement with measurements in GB1 and membrane protein ASR,16, 17 we found a relaxation plateau for spinlock fields from about 10 to 25 kHz, yielding a bulk 15N T1ρ of approximately 100 ms for the TM part of KcsA. The site‐resolved 15N T1ρ relaxation rates (R1ρ) show a rather uniform dynamic regime for the lipid‐confined TM part, which is in line with the measurements in micelles18 (Figures 3 A, B).
Figure 3.
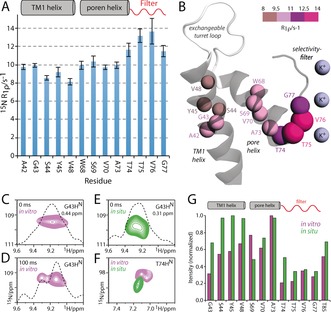
Dynamics recorded for the TM part of D2O‐washed iFD KcsA. All data were acquired at 60 kHz MAS and 800 MHz using a 15N spinlock field of 20 kHz. A) 15N R1ρ relaxation rates. B) Illustration of the 15N R1ρ rates on the KcsA structure (PDB No. 1K4C). Analyzed residues are shown as color‐coded spheres whose size is proportional to R1ρ. C, D) 15N T1ρ experiments in vitro with a spinlock duration of C) 0 ms or D) 100 ms reveal two signals for G43HN. E) G43HN exhibits only one conformation in situ. F) T74 showed a 15N chemical shift perturbation of +0.8 ppm in situ. G) Signal‐to‐noise ratios of resolved residues in in vitro (magenta) and in situ (green) D2O‐washed KcsA.
However, the entire filter stretch T74–G77 remarkably features modestly, but clearly enhanced 15N R1ρ, which is indicative of slow and presumably collective molecular motion. This motion very likely corresponds to slow nanosecond to microsecond dynamics as residues T74 and T75 did not show increased fast nanosecond motion in a previous 15N T1 relaxation study.19 Such slow motions have not been observed before in membrane‐embedded KcsA and can be important for its function as the filter backbone is immediately involved in ion conductance. Interestingly, flicker transitions,9b a form of modal gating represented by rapid channel opening and closure, also occur on the microsecond timescale, and flickering is indeed thought to be related to dynamic rearrangements around V76. Increased filter dynamics were also corroborated by reduced signal intensities in dipolar experiments (Figure 3 G). The conformational flexibility is also reflected in the HN linewidth, which is consistently larger for the filter than for the pore helix (Table S5). The TM1 helix residues A42–L49 are least subjected to slow motion and feature slightly smaller R1ρ rates than the pore helix. However, the G43HN resonance, which is by far the broadest of all HN (0.44 ppm), is split into two signals with different relaxation decays (Figures 3 C, D). As the G43 chemical shift is sensitive to the filter ion‐binding mode,12 this splitting may hence also be related to the conformational flexibility of the filter. Intriguingly, unlike almost all other resonances, the G43HN resonance is much narrower (0.31 ppm) in situ, most likely because only one conformation is present (Figure 3 E). This finding suggests that the KcsA conformational dynamics are, at least locally, altered in native bacterial membranes compared to liposomes.
While the spectral sensitivity inhibits site‐resolved relaxation studies in cellular KcsA, an analysis of signal intensities in dipolar experiments showed the lowest intensities for residues T74–G77. This result strongly suggests that the enhanced filter dynamics are maintained in the native membrane (Figure 3 G). In fact, the conformational flexibility of the filter seems to be further enhanced in situ as the filter signals are much reduced compared to those of the TM1 helix. Remarkably, also the relative intensities of the filter residues are changed, especially for T74, which displays the lowest intensity of all filter residues in vitro and by far the highest sensitivity in situ. This suggests that the dynamics of the filter are changed in cellular KcsA, which is further corroborated by a relatively strong 15N CSP of +0.8 ppm for T74 in situ (Figures 3 F and S11). A potential reason for this apparently different behavior of the filter may be the altered lipid composition in the bacterial membrane, which is known to influence the filter,9c or differential clustering of KcsA channels.20 Against the backdrops presented here, it is of great importance to explore how molecular motion modulates channel function. Whereas our data clearly show enhanced filter dynamics, further extensive measurements will be necessary to fully understand the timescales and amplitudes of these dynamics.
In conclusion, we have introduced an approach that enables the 1H detection of water‐inaccessible proteins with high sensitivity and resolution. We expect our method to be especially powerful for the study of membrane proteins and amyloid fibrils, which both exhibit domains that are highly resistant to hydrogen/deuterium exchange. Moreover, our method allows for the first time the use of 1H detection for the detailed analysis of a membrane protein in native cell membranes. This constitutes an important advance in ssNMR spectroscopy as membrane protein function often critically depends on the native environment.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge support by the NWO (projects 723.014.003, 700.10.443, 184.032.207). Experiments at the 950 MHz instrument were supported by uNMR‐NL, an NWO‐funded National Roadmap Large‐Scale Facility of the Netherlands.
J. Medeiros-Silva, D. Mance, M. Daniëls, S. Jekhmane, K. Houben, M. Baldus, M. Weingarth, Angew. Chem. Int. Ed. 2016, 55, 13606.
References
- 1. Ishii Y., Tycko R., J. Magn. Reson. 2000, 142, 199–204. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Chevelkov V., Rehbein K., Diehl A., Reif B., Angew. Chem. Int. Ed. 2006, 45, 3878–3881; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 3963–3966; [Google Scholar]
- 2b. Schanda P., Meier B. H., Ernst M., J. Am. Chem. Soc. 2010, 132, 15957–15967; [DOI] [PubMed] [Google Scholar]
- 2c. Linser R., Dasari M., Hiller M., Higman V., Fink U., Lopez del Amo J. M., Markovic S., Handel L., Kessler B., Schmieder P., Oesterhelt D., Oschkinat H., Reif B., Angew. Chem. Int. Ed. 2011, 50, 4508–4512; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4601–4605; [Google Scholar]
- 2d. Ward M. E., Shi L., Lake E., Krishnamurthy S., Hutchins H., Brown L. S., Ladizhansky V., J. Am. Chem. Soc. 2011, 133, 17434–17443; [DOI] [PubMed] [Google Scholar]
- 2e. Chevelkov V., Habenstein B., Loquet A., Giller K., Becker S., Lange A., J. Magn. Reson. 2014, 242, 180–188; [DOI] [PubMed] [Google Scholar]
- 2f. Mance D., Sinnige T., Kaplan M., Narasimhan S., Daniels M., Houben K., Baldus M., Weingarth M., Angew. Chem. Int. Ed. 2015, 54, 15799–15803; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 16025–16029; [Google Scholar]
- 2g. Dannatt H. R., Felletti M., Jehle S., Wang Y., Emsley L., Dixon N. E., Lesage A., Pintacuda G., Angew. Chem. Int. Ed. 2016, 55, 6638–6641; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6750–6753; [Google Scholar]
- 2h. Sinnige T., Daniels M., Baldus M., Weingarth M., J. Am. Chem. Soc. 2014, 136, 4452–4455. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Zhou D. H., Shah G., Cormos M., Mullen C., Sandoz D., Rienstra C. M., J. Am. Chem. Soc. 2007, 129, 11791–11801; [DOI] [PubMed] [Google Scholar]
- 3b. Marchetti A., Jehle S., Felletti M., Knight M. J., Wang Y., Xu Z. Q., Park A. Y., Otting G., Lesage A., Emsley L., Dixon N. E., Pintacuda G., Angew. Chem. Int. Ed. 2012, 51, 10756–10759; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10914–10917; [Google Scholar]
- 3c. Weingarth M., van der Cruijsen E. A., Ostmeyer J., Lievestro S., Roux B., Baldus M., J. Am. Chem. Soc. 2014, 136, 2000–2007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Wang S., Parthasarathy S., Xiao Y., Nishiyama Y., Long F., Matsuda I., Endo Y., Nemoto T., Yamauchi K., Asakura T., Takeda M., Terauchi T., Kainosho M., Ishii Y., Chem. Commun. 2015, 51, 15055–15058; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Xiang S., Biernat J., Mandelkow E., Becker S., Linser R., Chem. Commun. 2016, 52, 4002–4005. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Agarwal V., Penzel S., Szekely K., Cadalbert R., Testori E., Oss A., Past J., Samoson A., Ernst M., Bockmann A., Meier B. H., Angew. Chem. Int. Ed. 2014, 53, 12253–12256; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12450–12453; [Google Scholar]
- 4b. Mroue K. H., Nishiyama Y., Kumar Pandey M., Gong B., McNerny E., Kohn D. H., Morris M. D., Ramamoorthy A., Sci. Rep. 2015, 5, 11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi L. C., Kawamura I., Jung K. H., Brown L. S., Ladizhansky V., Angew. Chem. Int. Ed. 2011, 50, 1302–1305; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1338–1341. [Google Scholar]
- 6. Verel R., Baldus M., Ernst M., Meier B. H., Chem. Phys. Lett. 1998, 287, 421–428. [Google Scholar]
- 7. Zhou D. H., Rienstra C. M., J. Magn. Reson. 2008, 192, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weingarth M., Bodenhausen G., Tekely P., J. Magn. Reson. 2009, 199, 238–241. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Cuello L. G., Jogini V., Cortes D. M., Perozo E., Nature 2010, 466, 203–208; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Chakrapani S., Cordero-Morales J. F., Jogini V., Pan A. C., Cortes D. M., Roux B., Perozo E., Nat. Struct. Mol. Biol. 2011, 18, 67–74; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. van der Cruijsen E. A., Nand D., Weingarth M., Prokofyev A., Hornig S., Cukkemane A. A., Bonvin A. M., Becker S., Hulse R. E., Perozo E., Pongs O., Baldus M., Proc. Natl. Acad. Sci. USA 2013, 110, 13008–13013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Renault M., Tommassen-van Boxtel R., Bos M. P., Post J. A., Tommassen J., Baldus M., Proc. Natl. Acad. Sci. USA 2012, 109, 4863–4868; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Schanda P., Triboulet S., Laguri C., Bougault C. M., Ayala I., Callon M., Arthur M., Simorre J. P., J. Am. Chem. Soc. 2014, 136, 17852–17860; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Shahid S. A., Nagaraj M., Chauhan N., Franks T. W., Bardiaux B., Habeck M., Orwick-Rydmark M., Linke D., van Rossum B. J., Angew. Chem. Int. Ed. 2015, 54, 12602–12606; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12792–12797. [Google Scholar]
- 11. Baker L. A., Daniels M., van der Cruijsen E. A. W., Folkers G. E., Baldus M., J. Biomol. NMR 2015, 62, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wylie B. J., Bhate M. P., McDermott A. E., Proc. Natl. Acad. Sci. USA 2014, 111, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chill J. H., Louis J. M., Miller C., Bax A., Protein Sci. 2006, 15, 684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeuchi K., Takahashi H., Kawano S., Shimada I., J. Biol. Chem. 2007, 282, 15179–15186. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Lamley J. M., Oster C., Stevens R. A., Lewandowski J. R., Angew. Chem. Int. Ed. 2015, 54, 15374–15378; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15594–15598; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Ma P., Xue Y., Coquelle N., Haller J. D., Yuwen T., Ayala I., Mikhailovskii O., Willbold D., Colletier J. P., Skrynnikov N. R., Schanda P., Nat. Commun. 2015, 6, 8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lewandowski J. R., Sass H. J., Grzesiek S., Blackledge M., Emsley L., J. Am. Chem. Soc. 2011, 133, 16762–16765. [DOI] [PubMed] [Google Scholar]
- 17. Good D. B., Wang S. L., Ward M. E., Struppe J., Brown L. S., Lewandowski J. R., Ladizhansky V., J. Am. Chem. Soc. 2014, 136, 2833–2842. [DOI] [PubMed] [Google Scholar]
- 18. Chill J. H., Louis J. M., Baber J. L., Bax A., J. Biomol. NMR 2006, 36, 123–136. [DOI] [PubMed] [Google Scholar]
- 19. Ader C., Pongs O., Becker S., Baldus M., Biochim. Biophys. Acta Biomembr. 2010, 1798, 286–290. [DOI] [PubMed] [Google Scholar]
- 20. Sumino A., Yamamoto D., Iwamoto M., Dewa T., Oiki S., J. Phys. Chem. Lett. 2014, 5, 578–584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary