

Abstract
The formation of unusual multilayered parallel lamellae‐in‐lamellae in symmetric supramolecular double‐comb diblock copolymers is presented. While keeping the concentration of surfactant fixed, the number of internal layers was found to increase with molecular weight M up to 34 for the largest block copolymer. The number of internal structures n was established to scale as M 0.67 and therefore enables easy design of such structures with great precision.
Keywords: block copolymers, morphology, supramolecular self-assembly
Whereas linear diblock copolymers are known to form only four different stable structures upon self‐assembly (namely spheres, cylinders, lamellae, and gyroids),1, 2, 3 careful design of the macromolecular architecture or including additional blocks can enrich the phase behavior significantly.4 For instance, binary AB multiblock copolymers5 and A(AB)3 miktoarm star polymers6 were recently demonstrated to give parallel lamellae‐in‐lamellae or extremely asymmetric lamellae. Incorporation of a third component in ABC triblock terpolymers gives rise to an even higher morphological complexity.7 However, preparation of more complex polymeric architectures does not automatically imply formation of more complex structures, since reduced mobility and confinement of the junction points can hamper proper phase separation.
For real‐life technological applications, a synthetically less‐challenging approach is required. A supramolecular route8, 9, 10 enables researchers to produce comb‐shaped copolymers by simply combining regular linear block copolymers (BCPs) and small amphiphilic molecules, thereby avoiding such complex preparation methods.11, 12 Supramolecular complexes based on poly(4‐vinylpyridine) and 3‐pentadecylphenol (P4VP(3‐PDP)x, with x representing the number of 3‐PDP molecules per monomer) are among the most studied comb‐shaped systems. Since by now their phase behavior is very well understood13, 14 and materials are widely commercially available, several unique functional materials originating from such complexes have been reported. Examples include temperature‐responsive photonic crystals,15 CdSe/PbS nanoparticle assemblies,16 and highly ordered Au nanocomposites in thin films17, 18, 19 or solution.20 In polystyrene‐containing P4VP(3‐PDP)x‐b‐PS comb–coil diblock copolymers, the supramolecular nature of the complex also provides an easy route for creating ordered porous structures by simple dissolution of 3‐PDP. Such templates have for instance been refilled with metal or carbon for the preparation of actuating materials21 or nanoporous cathodes,22 respectively. From a fundamental point of view these comb–coil systems are surprisingly interesting as well, as simultaneous self‐assembly of both the comb (3‐PDP/P4VP) and diblock (P4VP(3‐PDP)x‐b‐PS) led to hierarchical structure formation.23 By changing the concentration of the alkylphenol or the composition of the parent PS‐b‐P4VP copolymer, self‐assembly gave access to multiple classical morphologies with an additional internal structure.24 Only highly sophisticated and synthetically challenging macromolecular architectures were previously found to give rise to such exceptional phase behavior.
Double‐comb (or bottlebrush) diblock copolymers could be realized by introducing a second hydrogen‐bond‐accepting block.25 By using a single, symmetric P4VP‐b‐PAPI (P4PA) diblock copolymer, several unique morphologies were observed in [poly(4‐vinylpyridine)‐block‐poly(N‐acryloylpiperidine)](3‐nonadecylphenol)x complexes as a function of the comb density x.26 Besides that, both the double parallel (x=0.5) and perpendicular alignment (x=1.0) of the internal layers with respect to the large lamellar BCP structure were in excellent agreement with our previous theoretical work.27 Interestingly though, the parallel morphology demonstrated multiple internal layers. Although not considered theoretically, to see whether this approach allows us to design materials with any number of internal layers, four additional lamella‐forming P4VP‐b‐PAPI BCPs were prepared and complexed to 3‐NDP (x=0.5; Scheme 1). Additional multiblock‐like structures28, 29 appeared upon self‐assembly using this simple and elegant route, with the number of internal layers increasing up to 34 per long period.
Scheme 1.
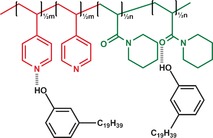
Representation of a P4PA(3‐NDP)0.5 supramolecular double‐comb diblock copolymer.
Symmetric (that is, f P4VP≈0.5) P4VP‐b‐PAPI diblock copolymers were synthesized via RAFT polymerization by starting from a P4VP macro chain transfer agent (Supporting Information, Tables S1, S2).30 The symmetric composition caused all P4PAyk‐z diblocks (y denotes the total molecular weight in kg mol−1, z the mass fraction P4VP in wt %) to self‐assemble into the expected lamellar structure, with its size increasing with molecular weight (Figures 1 a–e). Since perfect orientation of the lamellar nanostructure with respect to the viewer's eye can hardly ever be guaranteed, true domain spacings were obtained from SAXS.31, 32 Despite the weak scattering of these P4VP‐b‐PAPI diblocks owing to a low electron density contrast,30 the first order scattering maxima still allowed abstraction of their periodicity (Figure 1 f). Sizes ranged from 22.6 up to 146 nm (Supporting Information, Table S3).
Figure 1.
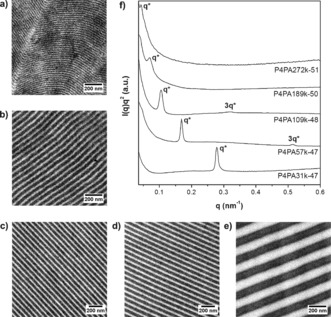
a)–e) Bright‐field TEM images of iodine‐stained P4PA diblock copolymers. P4VP appears dark. P4PA31k‐47 (a), P4PA57k‐47 (b), P4PA109k‐48 (c), P4PA189k‐50 (d), and P4PA272k‐51 (e). f) Room‐temperature Lorentz‐corrected SAXS patterns of neat P4PA diblock copolymers.
When mixed with 3‐NDP surfactants,33 TEM images of all five stained P4PA(3‐NDP)0.5 supramolecular complexes show parallel alignment of the small structure (Figures 2 a–e). Whereas the large length scale (BCP level) is indeed expected to increase with molecular weight, surprisingly though an increase of the number of small, internal layers was observed as well. For instance, only two dark P4VP lamellae can be observed in P4PA31k‐47, whereas P4PA272k‐51 displays an astonishing eight P4VP layers (Supporting Information, Table S4).
Figure 2.

a)–e) Stained and f)–j) unstained TEM images of P4PA(3‐NDP)0.5 double‐comb diblock copolymers. P4VP appears dark in the stained images (iodine). P4PA31k‐47 (a, f), P4PA57k‐47 (b, g), P4PA109k‐48 (c, h), P4PA189k‐50 (d, i), and P4PA272k‐51 (e, j).
According to room‐temperature SAXS patterns illustrated in Figure 3 a, regardless of the molecular weight, the small structure remains identical in all five complexes, that is, 4.4 to 4.5 nm (q≈1.4 nm−1). Multiple scattering events with integer‐valued ratios between the first‐ and higher‐order scattering maxima (q*, 2q*, 3q*,…, nq*) confirm the presence of large lamellae, starting at a spacing of 17.5 nm for P4PA31k‐47(3‐NDP)0.5 and going up to 77.0 nm for P4PA272k‐51(3‐NDP)0.5 (Supporting Information, Table S4 and Figure S1). Scherrer grain‐size analysis34 (Supporting Information, Table S5 and Figure S2) and TEM images recorded at a lower magnification (Supporting Information, Figure S3) support the high degree of ordering in all five complexes.
Figure 3.
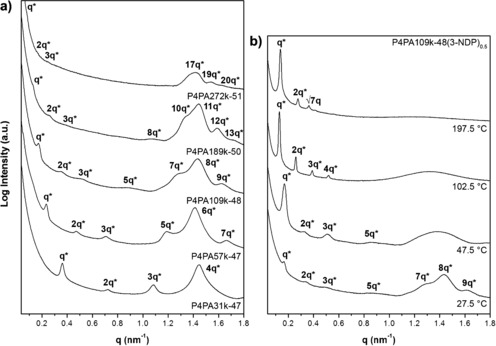
Room temperature SAXS intensity profiles of P4PA(3‐NDP)0.5 double‐combs (a). Temperature‐dependent SAXS profiles of P4PA109k‐48(3‐NDP)0.5 recorded at different stages of its self‐assembly (b).
A large lamellar morphology implies approximately equal distribution of 3‐NDP over both P4VP and PAPI. The diffraction patterns enable estimation of the number of layers inside the bright PAPI phase. Electron micrographs of unstained P4PA(3‐NDP)0.5 sections presented in Figures 2 f–j confirm the self‐assembly of 3‐NDP inside PAPI. For the low‐molecular‐weight complexes (P4PA31k‐47, P4PA57k‐47, and P4PA109k‐48), the predictions from SAXS are in excellent agreement with the local information provided by TEM, while the number of layers that can be observed in P4PA189k‐50 and P4PA272k‐51 fluctuate around these values (Supporting Information, Table S4). Although SAXS only provides average information, as a result of their higher molecular weights and broader distributions, local defects as can be seen in Figures 2 i and 2 j are expected to appear more often in larger BCP lamellae.
Besides DSC (Supporting Information, Figure S4), thermal properties of all x=0.5 double‐combs were also investigated by temperature‐resolved SAXS. A heating scan of P4PA109k‐48(3‐NDP)0.5 is displayed in the Supporting Information, Figure S5, with the most important phase transitions being highlighted in Figure 3 b. As the large P4PA lamellar morphology (d L=36.8 nm) is a superposition of the parallel, internal small structure (d S=4.4 nm), this explains the enhanced peak intensity of the 8th‐order scattering maximum (d L≈8d S) at room temperature. Upon melting around 35 °C, the small structure indeed disappeared and only a large lamellar BCP structure continued to exist. At elevated temperatures the complex simply followed phase behavior as observed before in PS‐b‐P4VP(3‐PDP)x coil–comb systems, with the alkylphenol only acting as plasticizer.35 Enhanced scattering and an increase of the long period were observed around 100 °C (d L=47.6 nm). Increased intensity of the second‐order peak suggests an asymmetric distribution of 3‐NDP over both P4VP and PAPI phases. Instead of the integer multiples of q* characteristic for lamellae, on further heating finally an additional peak appeared at q*, indicating the formation of a cylindrical structure due to migration of more 3‐NDP to the PAPI phase. Preference of 3‐NDP for the acrylamide block was observed before in P4PA57k‐47(3‐NDP)0.3.26 Similar phase behavior was observed in all other P4PA(3‐NDP)0.5 complexes (Supporting Information, Figure S6). A disordered melt was only found for the lowest‐molecular‐weight double‐comb P4PA31k‐47(3‐NDP)0.5 at higher temperatures (Supporting Information, Figure S6 a).
Since the small structure disappears upon melting of the complex, the ability of the aliphatic tails of 3‐NDP to crystallize seems to play a very important role. Therefore, we are convinced that microphase separation of the side chains is only beneficial upon crystallization, similar to PAPI(3‐NDP)1.0 homopolymer complexes.26
In the melt (55 °C), just after disappearance of the small structure, the size of the large lamellae is reduced significantly compared to the neat P4PA diblock copolymers, up to 42 %. In this state, the 3‐NDP surfactants simply act as a nonselective solvent, giving rise to contraction of the polymer chains36 with the long period d L scaling as Mn 0⋅67 (Figure 4). Maybe fortuitously, this value equals the well‐known exponent characteristic for the strong segregation regime. Upon crystallization of the side chains, the backbone largely maintains its random‐walk nature, which was present for T>T m, only leading to a slight reduction of d L owing to increased density of the crystalline comb layers (Supporting Information, Table S5). Because the size of the small structure d S remains identical in all complexes (ca. 4.5 nm), d L just above the crystallization temperature determines the number of small structures n per long period. As a result, n is found to scale with the molecular weight in the same way, that is, n=d L/d S∝Mn 0⋅67. This relationship therefore allows preparation of multiblock‐like structures with any desired number of internal layers 2n.
Figure 4.
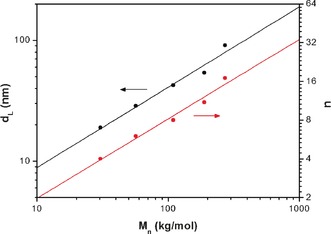
Molecular weights of P4PA in P4PA(3‐NDP)0.5 double‐combs plotted against the large length scale d L at T≈60 °C (T>T m) and number of small repeating structures n (=d L/d S). Both parameters (d L and n) scale as Mn 0.67.
Although a multilayered structure requires the BCP to pass through several crystalline amphiphilic layers (Scheme 2), in comparison to a single layer morphology such an alignment is entropically highly favorable. Furthermore, observation of a 4.5 nm d S implies formation of comb layers containing two interdigitating amphiphiles pointing in opposite directions, similar to x=1.0 comb copolymers.37 We believe that the combination of these two factors is responsible for the self‐assembly into multilayered parallel lamellae‐in‐lamellae with a fixed small feature size.
Scheme 2.
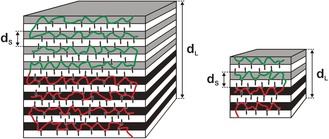
Representation of the 16‐layered (left) and 8‐layered structures (right) as characterized in P4PA109k‐48(3‐NDP)0.5 (n=d L/d S=8) and P4PA31k‐47(3‐NDP)0.5 (n=4) double‐comb diblock copolymers, respectively.
In summary, rather complex double‐comb diblock copolymers were realized via a supramolecular approach, using relatively simple components. Multiblock‐like, multilayered morphologies appeared in all P4PA(3‐NDP)0.5 supramolecular complexes. The established scaling behavior of the number of layers with molecular weight provides direct control over the internal structure and thus enables straightforward design of parallel morphologies with any number of internal lamellae.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work is supported by NanoNextNL, a micro and nanotechnology consortium of the Government of the Netherlands and 130 partners. Aalto University Nanomicroscopy Center (Aalto‐NMC) facilities were used for this research project. Beam time on the Dutch–Belgian Beamline (DUBBLE) of ESRF (Grenoble, France) has kindly been made available by NWO, and we would like to thank Wim Bras, Giuseppe Portale, and Daniel Hermida‐Merino for their experimental assistance.
A. H. Hofman, M. Reza, J. Ruokolainen, G. ten Brinke, K. Loos, Angew. Chem. Int. Ed. 2016, 55, 13081.
Contributor Information
Prof. Gerrit ten Brinke, Email: g.ten.brinke@rug.nl
Prof. Katja Loos, Email: k.u.loos@rug.nl.
References
- 1. Bates F. S., Science 1991, 251, 898–905. [DOI] [PubMed] [Google Scholar]
- 2. Khandpur A. K., Förster S., Bates S., Hamley I. W., Ryan A. J., Bras W., Almdal K., Mortensen K., Macromolecules 1995, 28, 8796–8806. [Google Scholar]
- 3. Lo T., Chao C., Ho R., Georgopanos P., Avgeropoulos A., Thomas E. L., Macromolecules 2013, 46, 7513–7524. [Google Scholar]
- 4. Bates F. S., Hillmyer M. A., Lodge T. P., Bates C. M., Delaney K. T., Fredrickson G. H., Science 2012, 336, 434–440. [DOI] [PubMed] [Google Scholar]
- 5. Faber M., Voet V. S. D., ten Brinke G., Loos K., Soft Matter 2012, 8, 4479. [Google Scholar]
- 6. Shi W., Hamilton A. L., Delaney K. T., Fredrickson G. H., Kramer E. J., Ntaras C., Avgeropoulos A., Lynd N. A., J. Am. Chem. Soc. 2015, 137, 6160–6163. [DOI] [PubMed] [Google Scholar]
- 7. Löbling T. I., Hiekkataipale P., Hanisch A., Bennet F., Schmalz H., Ikkala O., Gröschel A. H., Müller A. H. E., Polymer 2015, 72, 479–489. [Google Scholar]
- 8. Ahmed R., Patra S. K., Hamley I. W., Manners I., Faul C. F. J., J. Am. Chem. Soc. 2013, 135, 2455–2458. [DOI] [PubMed] [Google Scholar]
- 9. Houbenov N., Haataja J. S., Iatrou H., Hadjichristidis N., Ruokolainen J., Faul C. F. J., Ikkala O., Angew. Chem. Int. Ed. 2011, 50, 2516–2520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2564–2568. [Google Scholar]
- 10. Houbenov N., Milani R., Poutanen M., Haataja J., Dichiarante V., Sainio J., Ruokolainen J., Resnati G., Metrangolo P., Ikkala O., Nat. Commun. 2014, 5, 4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Runge M. B., Bowden N. B., J. Am. Chem. Soc. 2007, 129, 10551–10560. [DOI] [PubMed] [Google Scholar]
- 12. Miyake G. M., Piunova V. A., Weitekamp R. A., Grubbs R. H., Angew. Chem. Int. Ed. 2012, 51, 11246–11248; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11408–11410. [Google Scholar]
- 13. Ruokolainen J., ten Brinke G., Ikkala O., Torkkeli M., Serimaa R., Macromolecules 1996, 29, 3409–3415. [Google Scholar]
- 14. Ruokolainen J., Saariaho M., Ikkala O., ten Brinke G., Thomas E. L., Torkkeli M., Serimaa R., Macromolecules 1999, 32, 1152–1158. [Google Scholar]
- 15. Valkama S., Kosonen H., Ruokolainen J., Haatainen T., Torkkeli M., Serimaa R., ten Brinke G., Ikkala O., Nat. Mater. 2004, 3, 872–876. [DOI] [PubMed] [Google Scholar]
- 16. Zhao Y., Thorkelsson K., Mastroianni A. J., Schilling T., Luther J. M., Rancatore B. J., Matsunaga K., Jinnai H., Wu Y., Poulsen D., Fréchet J. M. J., Alivisatos A. P., Xu T., Nat. Mater. 2009, 8, 979–985. [DOI] [PubMed] [Google Scholar]
- 17. Kao J., Thorkelsson K., Bai P., Zhang Z., Xu T., Nat. Commun. 2014, 5, 4053. [DOI] [PubMed] [Google Scholar]
- 18. Chen X., Perepichka I. I., Bazuin C. G., ACS Appl. Mater. Interfaces 2014, 6, 18360–18367. [DOI] [PubMed] [Google Scholar]
- 19. Kao J., Xu T., J. Am. Chem. Soc. 2015, 137, 6356–6365. [DOI] [PubMed] [Google Scholar]
- 20. Liang R., Xu J., Li W., Liao Y., Wang K., You J., Zhu J., Jiang W., Macromolecules 2015, 48, 256–263. [Google Scholar]
- 21. Vukovic I., Punzhin S., Vukovic Z., Onck P., De Hosson J. T. M., ten Brinke G., Loos K., ACS Nano 2011, 5, 6339–6348. [DOI] [PubMed] [Google Scholar]
- 22. Choudhury S., Agrawal M., Formanek P., Jehnichen D., Fischer D., Krause B., Albrecht V., Stamm M., Ionov L., ACS Nano 2015, 9, 6147–6157. [DOI] [PubMed] [Google Scholar]
- 23. Ruokolainen J., Mäkinen R., Torkkeli M., Mäkelä T., Serimaa R., ten Brinke G., Ikkala O., Science 1998, 280, 557–560. [DOI] [PubMed] [Google Scholar]
- 24. Ruokolainen J., ten Brinke G., Ikkala O., Adv. Mater. 1999, 11, 777–780. [Google Scholar]
- 25. Faber M., Hofman A. H., Polushkin E., Alberda van Ekenstein G., Seitsonen J., Ruokolainen J., Loos K., ten Brinke G., Macromolecules 2013, 46, 500–517. [Google Scholar]
- 26. Hofman A. H., Reza M., Ruokolainen J., ten Brinke G., Loos K., Macromolecules 2014, 47, 5913–5925. [Google Scholar]
- 27. Markov V., Subbotin A., ten Brinke G., Phys. Rev. E 2011, 84, 041807. [DOI] [PubMed] [Google Scholar]
- 28. Masuda J., Takano A., Nagata Y., Noro A., Matsushita Y., Phys. Rev. Lett. 2006, 97, 098301. [DOI] [PubMed] [Google Scholar]
- 29. Masuda J., Takano A., Suzuki J., Nagata Y., Noro A., Hayashida K., Matsushita Y., Macromolecules 2007, 40, 4023–4027. [Google Scholar]
- 30. Hofman A. H., Alberda van Ekenstein G. O. R., Woortman A. J. J., ten Brinke G., Loos K., Polym. Chem. 2015, 6, 7015–7026. [Google Scholar]
- 31. Borsboom M., Bras W., Cerjak I., Detollenaere D., Glastra van Loon D., Goedtkindt P., Konijnenburg M., Lassing P., Levine Y. K., Munneke B., Oversluizen M., van Tol R., Vlieg E., J. Synchrotron Radiat. 1998, 5, 518–520. [DOI] [PubMed] [Google Scholar]
- 32. Bras W., Dolbnya I. P., Detollenaere D., van Tol R., Malfois M., Greaves G. N., Ryan A. J., Heeley E., J. Appl. Crystallogr. 2003, 36, 791–794. [Google Scholar]
- 33. Hofman A. H., Chen Y., ten Brinke G., Loos K., Macromolecules 2015, 48, 1554–1562. [Google Scholar]
- 34. Smilgies D. M., J. Appl. Crystallogr. 2009, 42, 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Valkama S., Ruotsalainen T., Nykänen A., Laiho A., Kosonen H., ten Brinke G., Ikkala O., Ruokolainen J., Macromolecules 2006, 39, 9327–9336. [Google Scholar]
- 36. Hashimoto T., Shibayama M., Kawai H., Macromolecules 1983, 16, 1093–1101. [Google Scholar]
- 37. Luyten M. C., Alberda van Ekenstein G. O. R., ten Brinke G., Ruokolainen J., Ikkala O., Torkkeli M., Serimaa R., Macromolecules 1999, 32, 4404–4410. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary