

Abstract
Investigation of the amnesic disorder Korsakoff Syndrome (KS) has been vital in elucidating the critical brain regions involved in learning and memory. Although the thalamus and mammillary bodies are the primary sites of neuropathology in KS, functional deactivation of the hippocampus and certain cortical regions also contributes to the chronic cognitive dysfunction reported in KS. The rodent pyrithiamine-induced thiamine deficiency (PTD) model has been used to study the extent of hippocampal and cortical neuroadaptations in KS. In the PTD model, the hippocampus, frontal and retrosplenial cortical regions display loss of cholinergic innervation, decreases in behaviorally stimulated acetylcholine release and reductions in neurotrophins. While PTD treatment results in significant impairment in measures of spatial learning and memory, other cognitive processes are left intact and may be recruited to improve cognitive outcome. In addition, behavioral recovery can be stimulated in the PTD model by increasing acetylcholine levels in the medial septum, hippocampus and frontal cortex, but not in the retrosplenial cortex. These data indicate that although the hippocampus and frontal cortex are involved in the pathogenesis of KS, these regions retain neuroplasticity and may be critical targets for improving cognitive outcome in KS.
Keywords: Korsakoff syndrome, Thiamine deficiency, Rodent, Thalamus, Hippocampus, Cortex, Memory
Thiamine Deficiency Protocols to Model Korsakoff Syndrome in Experimental Animals
Korsakoff Syndrome (KS), a disorder associated with thiamine deficiency (TD), is most commonly seen in alcoholics but is also reported in patients with bulimia, gastrointestinal distress and AIDS (Mair 1994; Kopelman et al. 2009). The study of individuals with KS has been critical in establishing the distinction between declarative and procedural memory systems (Squire 1980). Like temporal lobe amnesics, KS patients show poor performance in tests assessing content memory, but unlike temporal lobe amnesics, they also display impairments in contextual memory (e.g., spatial or temporal relationships between items or events), possibly attributable to alcoholism-related frontal lobe compromise (see Van Tilborg et al. 2011). Neuroimaging studies demonstrate that the most consistent neuropathology in KS is damage to the diencephalon, particularly to the thalamus and mammillary bodies (Kopelman et al. 2009; Sullivan and Pfefferbaum 2009). Indeed, one of the best predictors of memory loss in alcohol-related KS is pathology of the anterior thalamus (Harding et al. 2000). Damage to the hippocampus is not always observed in KS (Reed et al. 2003; Squire et al. 1990; but see Sullivan and Marsh 2003). However, given that the thalamus is connected to the hippocampus and other limbic regions, the functional distinction between temporal lobe amnesia and diencephalic amnesia has been questioned (Aggleton and Brown 1999; Squire 1982).
In addition to impairments in hippocampal-dependent tasks, KS patients perform poorly in tasks dependent on frontal cortical systems (Dirksen et al. 2006; Gansler et al. 2000; Oscar-Berman and Evert 1997; Oscar-Berman et al. 2004). Volumetric MRI analysis of KS brains reveals extensive frontal lobe atrophy (Reed et al. 2003; Christie et al. 1988), including white matter pathology (Fama et al. 2004). The somewhat diffuse pattern of neuropathology in KS patients has made it difficult to determine which brain regions should be targeted for therapeutic intervention.
Whereas preclinical site-specific lesion models have elucidated the relative contributions of different thalamic regions to learning and memory (Aggleton et al. 2011; Mitchell and Dalrymple-Alford 2005, 2006; Savage et al. 2011), preclinical TD models have increased our understanding of the mechanisms associated with TD-induced neurodegeneration. Over the past three decades, TD models have helped identify key neuropathological features of Wernicke’s Encephalopathy (WE) and KS, specific neurochemical and neuropathological consequences of TD and potential therapeutic approaches (Hazell and Butterworth 2009; Vetreno et al. 2011a, b; 2012).
Several experimental protocols have been used to induce TD in animals: a. a thiamine-deficient diet; b. a thiamine-deficient diet in conjunction with chronic alcohol treatment; and c. a thiamine-deficient diet combined with systemic injections of pyrithiamine, which inhibits thiamine pyrophosphorylation. All three approaches have revealed that TD results in limbic circuit dysfunction in thalamic, hippocampal and associated cortical regions—particularly the frontal cortex. However, the different protocols lead to varying degrees of behavioral dysfunction and neuropathology.
Thiamine-Deficient Diet
Thiamine plays a key role in the maintenance of normal cellular function. The brain is particularly sensitive to TD because of its high oxidative metabolism. Thiamine is a necessary cofactor in the production of transketolase, pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase (αKGDH), all of which are required for normal cellular function (Hazell and Butterworth 2009). A thiamine-deficient diet alone requires weeks to months to induce TD-associated neurological symptoms (Troncoso et al. 1981; Witt 1985); however, just 9 days on a TD diet reveals in rodents reduced hippocampal neurogenesis (Zhao et al. 2008). Furthermore, after 14 days there is evidence for a decrease in the number of cholinergic neurons in the forebrain (Zhao et al. 2008) and after 30 days levels of acetylcholinesterase (AChE) and cholinergic fibers are reduced in the cortex and hippocampus (Pires et al. 2001; 2005; Nakagawasai et al. 2000; 2001). At this time deficits in spatial (Pires et al. 2005) and nonspatial memory are observed (Nakagawasai et al. 2000; 2001); performance on the forced swim test is compromised; and the incidence of muricide increases (Nakagawasai et al. 2001). Increasing cholinergic tone in these TD-depleted regions by administration of AChE inhibitors (AChE-Is), muscarinic agonists or herbal compounds that increase the level of acetylcholine (ACh) has been shown to improve or restore cognitive abilities in TD mice (Nakagawasai 2005). A limitation of the TD diet protocol is that it does not result in lesions to the thalamus or mammillary bodies (Troncoso et al. 1981; Witt 1985). Thus, the TD diet better models the WE phase than the chronic KS phase of Wernicke-Korsakoff Syndrome (WKS).
Thiamine Deficiency and Chronic Ethanol Treatment
A handful of studies have assessed the interaction of TD with chronic ethanol exposure in rodents. In such protocols, animals are chronically exposed to ethanol and concurrently receive a diet deficient in thiamine with or without daily injections of pyrithiamine (Ciccia and Langlais 2000; Homewood et al. 1997; Pires et al. 2005). Generally, TD—whether produced via a TD diet alone or in combination with pyrithiamine—is interrupted before the animal displays extreme neurological signs (Pfefferbaum et al. 2007; He et al. 2007). It appears that the clinical stages associated with TD in the rodent (i.e., decreased grooming followed by ataxia and, subsequently, impaired righting reflex) progress at a faster rate when rats are also exposed to ethanol (Zimitat et al. 1990). Whether ethanol and TD have synergistic effects on pathology depends on the quantified measure. Ventricular size and pathology in the thalamus and hippocampus, as quantified using magnetic resonance imaging, did not show synergism (Pfefferbaum et al. 2007). By contrast, the corpus callosum was thinner and had a higher percentage of small fibers in the ethanol+TD group than in either of the thiamine- or ethanol-alone groups (He et al. 2007). Similarly, ethanol+TD induced a greater impairment in stimulated (in-vitro) cortical ACh release than either TD or ethanol alone (Pires et al. 2001). Kril and Homewood (1993) used a model of prolonged (28 weeks) ethanol exposure in which animals were subject to bouts of mild TD at various time points in the ethanol exposure protocol: early (after 4 weeks), in the middle (after 15 weeks) or late (after 26 weeks). TD at 4 weeks, but not 26 weeks, potentiated loss of neurons in frontal regions, suggesting a critical temporal window when TD synergistically interacts with ethanol toxicity.
Cerebellar Purkinje cells within the vermis are reduced in size and number in patients with alcohol-related KS, relative to uncomplicated alcoholics (Baker et al. 1999; Philips et al. 1987). Both in vitro and in vitro models support the hypothesis that TD alters the cerebellum: when TD is induced by either amprolium (an inhibitor of thiamine transport; Ke et al. 2009) or pyrithiamine (Pannunzio et al. 2000), cell death occurs in cultured cerebellar granule neurons. Furthermore, the addition of ethanol to the cell culture media potentiated TD-induced cell loss (Ke et al. 2009). A loss of Purkinje cells and shrinkage of the granular layers of the vermis have been observed after both PTD treatment and long-term exposure (20 months) to ethanol (Irle and Markowitsch 1983). However, the extent of cerebellar neuropathology following TD and/or chronic ethanol exposure has not been quantified and whether this damage contributes to cognitive or motor learning dysfunction in animal models of alcohol-related neuropathology has not been determined.
Behavioral measures likewise do not consistently show an interaction between ethanol exposure and bouts of TD. TD causes significant impairments in spatial learning over and above the deficits seen following exposure to ethanol alone (Pires et al. 2005) with a nonsignificant trend for intensified behavioral deficits in TD+chronic ethanol exposure. Importantly, TD and chronic ethanol produced differential cognitive dysfunction: chronic ethanol alone increased perseverative errors and impaired working memory on both delayed non-matching- and matching-to-sample tasks (DNMTS, DMTS), whereas TD alone only impaired working memory on the DMTS task (Ciccia and Langlais 2000). Reversal learning was compromised to the greatest extent in ethanol+TD rats, suggesting that cognitive flexibility may be one behavioral measure that is sensitive to the synergistic interaction between TD+ethanol.
In summary, given the limited number of studies and the variations in treatment parameters (e.g., ethanol concentration, exposure duration, extent of TD) among studies using the TD+ethanol exposure paradigm, the results are fairly inconclusive (see Vetreno et al. 2011a). When structures such as the hippocampus or thalamus are functionally or structurally assessed using in vivo models, no significant evidence exists for a synergistic interaction between TD and ethanol. The lack of synergistic effects between TD and other factors such as advanced age or ethanol toxicity is most likely due to the fact that severe TD disrupts the thalamic-hippocampal circuit to a point that further dysfunction would not be compatible with survival (see Pitkin and Savage 2001). However, when TD is mild to moderate, ethanol does exacerbate both white matter and gray matter pathology in forebrain regions. Furthermore, the in vitro data suggest that the cerebellum is sensitive to the compounded effects of TD+ethanol; however, this claim needs to be supported by in vivo evidence. Additional research is needed to determine optimal treatment parameters as well as the most sensitive neuropathology and behavior markers to use in the evaluation of the combined effects of ethanol exposure and TD.
Pyrithiamine-Induced Thiamine Deficiency (PTD)
When a TD diet is combined with systemic injections of pyrithiamine, the progression of the neurological sequelae of TD is accelerated: Within 15 to 18 days, rodents display a stereotyped progression of neurological symptoms that have been mapped to specific pathological changes in anatomy and neurochemistry (Zhang et al. 1995; Hazell and Butterworth 2009). Lesion progression and neurochemical alterations in the thalamus and mammillary bodies (outlined in Fig. 1) occur along a relatively well-established time course. Neurochemical dysfunction is also observed in the hippocampus, and there is evidence for degeneration in key limbic system fiber tracts and limbic cortical regions (Anzalone et al. 2010; Langlais and Savage 1995). The permanent neurological features of PTD in rodents mimic the persistent brain pathology and behavioral dysfunction described in humans with KS (Witt 1985; Vetreno et al. 2011a, b; 2012).
Fig. 1.
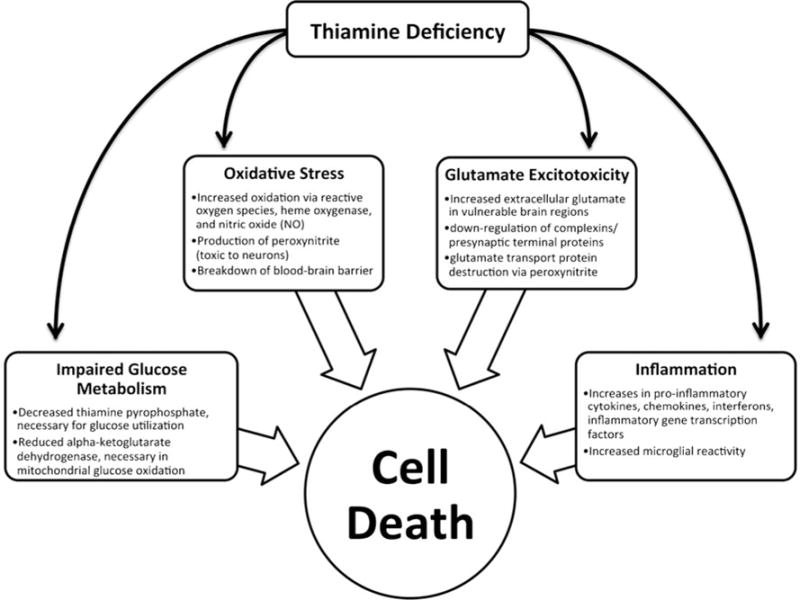
Schematic showing possible mediating mechanisms of cell death in thiamine deficiency. The information in this figure is a summary of Hazell and Butterworth (2009)
In the following sections, the proposed mechanisms of neuropathology, neurodegeneration and cognitive impairment in the PTD model will be outlined. Additionally, recent neuropharmacological data revealing the critical involvement of the hippocampus and frontal cortex in TD, and their potential roles as targets for therapeutic intervention, will be discussed.
Brain Changes Associated with the PTD-Induced Thiamine-Deficient State
Mechanisms of Cell Death in the PTD Model
Thiamine is critical for normal cellular functioning due to its key role in cellular respiration; TD can result in cell death. Possible, interrelated mechanisms underlying TD-induced neurodegeneration include impaired glucose metabolism, oxidative stress, glutamate excitotoxicity and inflammation (Hazell and Butterworth 2009; see Fig. 1). TD can reduce levels of thiamine pyrophosphate (TPP), which is a coenzyme and catalyst for a number of reactions involved in glucose metabolism (Héroux and Butterworth 1995; Vetreno et al. 2011a; Todd and Butterworth 1999). For example, an enzymatic derivative in the citric acid cycle, αKGDH, is decreased in TD (Butterworth and Héroux 1989). As αKGDH is critical in the oxidation of glucose, diminished levels have been associated with cell death (Gibson et al. 1984). The TD brain also shows evidence for a number of markers of oxidative stress, including peroxynitrite, a strong oxidizing free radical (Hazell and Butterworth 2009). Peroxynitrite can alter glutamate transport proteins, thereby inhibiting their function (Volterra et al. 1994) leading to increased concentrations of extracellular glutamate. Indeed, brain regions vulnerable to TD demonstrate high levels of extracellular glutamate (Hazell et al. 1993; Langlais and Zhang 1993), and lesion profile in the thalamus is consistent with glutamate excitotoxicity (Armstrong-James et al. 1988; Zhang et al. 1995). TD is also associated with increases in a number of indices of inflammation such as pro-inflammatory cytokines, chemokines and interferons (Hazell and Butterworth 2009). Hazell and Butterworth (2009) have suggested that activation of inflammatory markers may underlie the regional selectivity of thalamic damage in TD. Furthermore, increases in an inflammatory mediator, nitric oxide, are associated with decreased levels of αKGDH (Gibson et al. 1999). Thus, it is likely that many such mechanisms interact synergistically to induce cell death in TD. Parsing the unique and interactive effects of these mechanisms may result in a more thorough understanding of the pathology associated with TD.
Progress of Brain Pathology in the PTD Model
The diencephalon is the most studied brain region in TD due to the characteristic lesions observed in the thalamus and mammillary bodies and the association between these structures and the behavioral disturbances observed in KS (Langlais et al. 1996a, b). Although the thalamus is the first region to display neuronal degeneration in PTD models (Watanabe 1978; Armstrong-James et al. 1988), the severity and the specificity of the damage can vary among PTD-treated rats.
In the PTD model, the first symptom of TD is weight loss that begins typically on day 10. This is followed by loss of the righting reflex (days 12–13), severe ataxia (days 13–16) and dystonic posturing and convulsions prior to or during the excitotoxic event (Zhang et al. 1995). The timing of thiamine repletion after an acute TD episode is critical in determining the extent of neuropathology (Zhang et al. 1995; See Fig. 2). There is little evidence for persistent behavioral impairments if rats are treated with thiamine prior to experiencing seizures (Pitkin and Savage 2001).
Fig. 2.
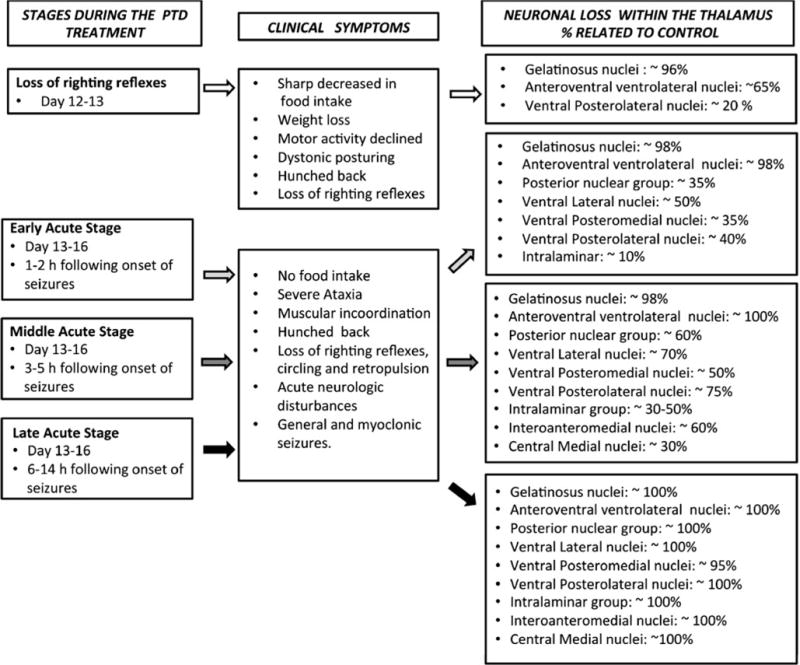
Summary of the progression of neuropathology within the thalamus as a function of PTD treatment (based on experimental data from Zhang et al. 1995). Although whether the seizure is a symptom or propagator of glutamate excitotoxicity is debated, it can be used as a time marker for progression of brain pathology
At the early acute stage (EAS) of the PTD model, degenerative changes occur in the gelatinosus and anteroventral ventrolateral nuclei of the thalamus, both of which undergo rapid neuronal loss (see Zhang et al. 1995). Within 1 to 2 h following seizure onset, the brains of PTD-treated rodents exhibit lesions in the ventral and posterior nuclear groups of the thalamus that are characterized by severe neuronal loss and pale neuropil. At the middle acute stage (MAS), 3 to 5 h following seizure onset, similar lesions are observed in the central medial, interoanteromedial and paracentral nuclei of the thalamus. Within the rostral intralaminar nuclei, neuronal loss appears complete (see Fig. 3-d). In the late acute stage (LAS), six to 14 h following onset of seizures, such lesions are present throughout the entire extent of the thalamus—that is, in the: i. anterior group (anteroventral, ventrolateral and anterior medial); ii. ventral nuclear group (ventral posterolateral, ventral posteromedial, ventral lateral and gelatinosus); iii. intralaminar group (central medial, paracentral and centrolateral); and iv. posterior group (parafascicular, posterior, medial geniculate and medial-lateral mammillary body). Damage includes neuronal loss, vascular proliferation, mild gliosis, spongiform changes and tissue loss.
Fig. 3.
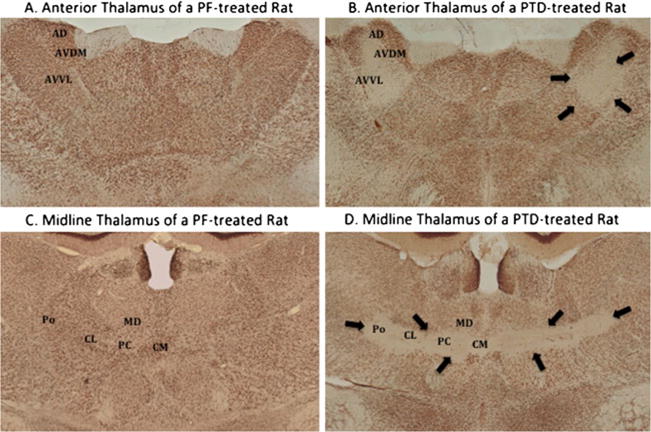
Thalamic sections stained with a nuclear specific stain (NeuN) the anterior (a & b) and medial thalamus (c & d). In the anterior thalamus (a & b), PTD rats show selective lesioning to the anteroventral ventrolateral (AVVL) area; however, there is comparative sparing of the anterodorsal (AD) and anteroventral ventrolateral (AVVL) nuclei. There is complete cell loss in the midline thalamus (D), specifically in the intralaminar nuclei (central medial [CM], paracentral [PC] and centrolateral [CL] nuclei) as well as the posterior (Po) nucleus. However, there is relative sparing of the medial dorsal (MD) nucleus, with the exception of the ventral portion. Arrows outline the regions of cell loss
Chronic Sequelae of PTD Treatment
Brain Pathology and Neurochemical Adaptations in the PTD Model
Diencephalon
During the recovery phase, 2 to 3 weeks following acute PTD, the third ventricle is enlarged. There is evidence for considerable neuronal loss (50–90%) in the anterior thalamic nuclear group (AT) and in particular in the anteroventral nucleus (Langlais and Savage 1995), which can be visualized clearly with immunohistochemical staining for the neuron-specific nuclear protein ([NeuN], see Fig. 3-b). Lesions and consequent gliotic scarring are observed in the intralaminar region (IML) including the central medial, paracentral, parafascicular, central-lateral (central part) and posterior medial (medial part) nuclei (Armstrong-James et al. 1988; Langlais and Mair 1990; Langlais et al. 1996b). The mammillary bodies (MB) are atrophied (Witt 1985), likely because of neuronal loss (>50%) in the medial nucleus (Langlais et al. 1996a, b). There is sparing of the lateral nucleus of the MB (Langlais and Savage 1995).
Damage to three diencephalic nuclei (AT, IML, MB) is proposed to underlie the learning and memory impairments observed in PTD-treated rats (Burk and Mair 2001; Mair et al. 1998; Savage et al. 1997; 1998; 2011; Vann 2010). Animals exposed to PTD are commonly categorized into two groups: those that have extensive cell loss and gliotic scarring throughout the internal medullary lamina region (IML-lesioned) and those that have some cell loss and gliosis within the IML, but no gliotic scarring (IML-spared). It was originally thought that an extensive midline lesion was required in order to observe significant behavioral impairment (Langlais et al. 1992; Mair et al. 1991a; Mair 1994), but moderate thalamic pathology (i.e., IML-spared) has also been associated with mild but functionally significant impairment (Langlais and Savage 1995). Indeed, while IML-lesioned PTD animals demonstrate a significant decrease in spontaneous alternation and impaired learning of both the initial spatial non-matching-to-position (NMTP) rule and the reversal to matching-to-position (MTP) rule, IML-spared PTD rats are only impaired in the acquisition of NMTP. Furthermore, PTD rats with moderate thalamic cell loss perform worse than pair-fed controls on the operant delayed MTP task (Savage et al. 1999). Since IML-lesioned and IML-spared PTD rats have the same degree of neuronal loss in the anterior thalamus (Langlais and Savage 1995), it may be that the anterior thalamus is responsible for some working memory impairment observed in PTD rats (Savage et al. 2011).
Long-term neurochemical alterations persist in the thalamus even after thiamine repletion, including significant reductions in overall GABA and glutamate levels in the midbrain thalamus (Langlais et al. 1988). Héroux and Butterworth (1988) found decreases in thalamic GABA levels 3 days after recovery from TD in the PTD model, and similar reductions have been observed 9 weeks after recovery (Langlais et al. 1988). However, the GABAergic and glutamatergic systems are not the only ones with proposed roles in the dementia associated with KS and PTD. Significant increases in serotonin and its metabolite have also been observed in the thalamus of PTD-treated rats (Langlais et al. 1988), and thalamic serotonergic dysfunction has been associated with impaired acquisition on a spatial task (Vigil et al. 2010). By contrast, Mair et al. (1985) reported no change in norepinephrine and dopamine concentrations in the medial thalamus of PTD-treated rats. Overall, it appears that the TD-induced neuropathology in the diencephalon leads to alterations in several neurochemical systems.
Hippocampus
Early reports examining the cytostructural integrity of the hippocampus following PTD treatment reported no gross neuropathology (Langlais et al. 1992; Mair et al. 1991b). However, recent in vivo MRI animal studies have revealed a hyperintense tissue response in the hippocampus (in addition to the thalamus and mammillary bodies) after PTD treatment that persisted for at least 1 month post treatment (Pfefferbaum et al. 2007). Moreover, our laboratory has demonstrated altered hippocampal cytogenesis in the PTD model (Vetreno et al. 2011). PTD treatment increased progenitor cell proliferation and survival immediately following, but not during, PTD treatment; however, this compensatory response did not translate into spared neurogenesis. On the contrary, neurogenesis was reduced at this early phase of recovery and at 28 days post treatment, at which time astrocytogenesis also increased. These data advocate either of two positions: i. damage to the thalamus alters hippocampal neurogenesis, or ii. hippocampal function is altered by another mechanism. Although the reason for the persistent decrease in hippocampal neurogenesis after PTD is unknown, we now have evidence of a compromised neurotrophic environment in the hippocampus of PTD-treated animals (see Fig. 4), including a chronic reduction in brain-derived neurotrophic factor (BDNF) protein expression (Vetreno et al. 2011a, b). BDNF is known to play a critical role in neurogenesis by enhancing the survival and maintenance of newly generated neurons (Cotman and Berchtold 2002; Lee et al. 2002; Nonner et al. 1996; Sairanen et al. 2005). Furthermore, degeneration of the major limbic system fiber tracts—including the fimbria/fornix and mammillothalamic tract that connect the thalamus, mammillary bodies and hippocampus—has been shown in the PTD model and likely leads to a loss of stimulation (Langlais and Zhang 1997).
Fig. 4.
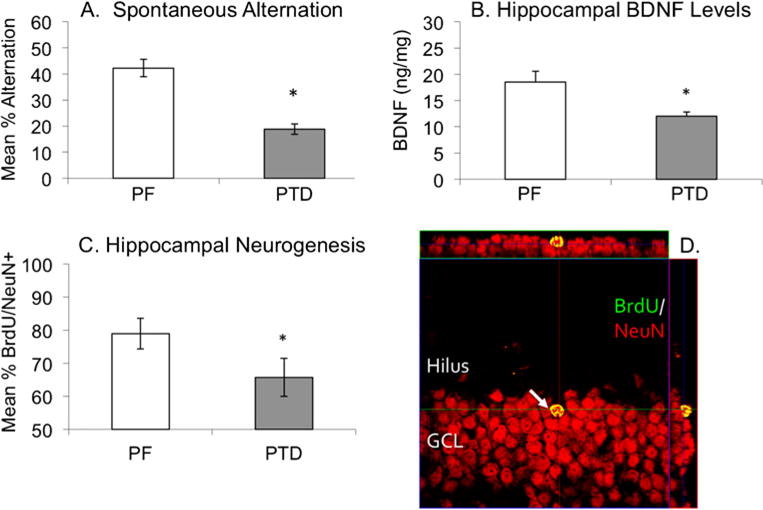
Confocal microscopy image (with orthogonal views) of Bromodeoxyuridine+cells (BrdU+; green) in a PTD brain co-localized with the cells immune positive for the neuronal marker NeuN (Neuronal Nuclei; red) in the subgranular zone (SGZ) of the dentate gyrus. PTD treatment produces a significant deficit in spontaneous alternation (a), chronically decreases hippocampal brain-derived neurotrophic factor (BNDF; b) and reduces hippocampal neurogenesis (c). Co-localized labels of BrdU/NeuN appear yellow and can be confirmed in all three projections (d)
Studies from our laboratory (Anzalone et al. 2010; Roland and Savage 2007; Roland et al. 2008; Savage et al. 2003, 2007; Vetreno et al. 2008) have also consistently documented a functional impairment in hippocampal ACh release in PTD rats (see Fig. 5). While basal levels of ACh are unaffected by PTD treatment, dysfunctional ACh efflux is observed when rats are cognitively challenged, reflecting a selective activity-dependent hippocampal impairment after TD. This is supported by our recent data demonstrating that following maze training PTD rats have fewer hippocampal cells in the dentate gyrus and CA 3 region that stain positive for the activity-dependent immediate early gene c-Fos (Fig. 5).
Fig. 5.
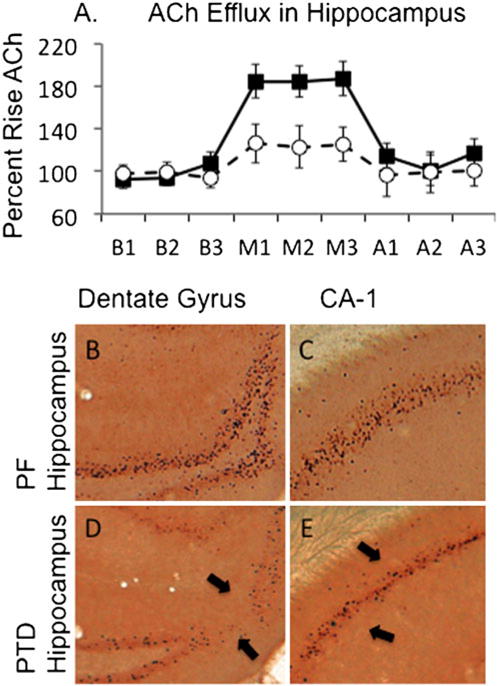
Behaviorally relevant acetylcholine (ACh) efflux in decreased in PTD rats (open circles) relative to control pair-fed (PF; closed squares) rats (a). Note that baseline levels (B1–B3) or after-maze levels (A1–A3) of ACh efflux are not different across the groups; the difference emerges only when the rats are tested for spontaneous alternation on the plus maze (M1–M3). In addition, relative to PF rats (b, c), behaviorally activated levels (induced by spontaneous alternation testing) of the early immediate gene c-Fos are decreased in PTD rats (d, e). The arrows point to the reduction of c-Fos positive cells with in the dentate gryrus and CA1 sector
In summary, it appears that, despite the absence of gross lesions or loss of mature neurons in the hippocampus following PTD treatment, the internal milieu of the hippocampus is altered by TD in such a way that it is unable to respond adequately to behavioral demands.
Cortex
Although structural abnormalities in the cortex following PTD have been documented, only recently has this pathology been directly implicated in the behavioral consequences of PTD. Prior to the MAS in PTD, there is extensive degeneration of axons in layers III through IV of the frontal and parietal cortices and moderate degeneration in layers IV through VI of the frontal, parietal, cingulate, temporal, retrosplenial, occipital and granular insular cortices (Langlais and Zhang 1997). Le Roch et al. (1987) similarly demonstrated neuronal degeneration in laminae III and V of the neocortex following PTD treatment. In addition, an electron microscopy study revealed splitting of myelin sheaths and shrinkage of cortical neurons within the cerebral cortex of PTD rats (Takahashi et al. 1988). Finally, a study by Langlais and Savage (1995) showed that PTD rats with significant IML lesions display thinning of the frontal and parietal cortices as well as the corpus callosum. Thus, both cortical gray and white matter are significantly disrupted by PTD treatment.
Cholinergic dysfunction within the cortex is evident in rats exposed to PTD treatment: both AChE activity and in vitro ACh release are decreased in the cerebral cortex of PTD rats and the level of cortical AChE activity was found to correlate with poor spatial memory performance (Pires et al. 2005). Furthermore, stereological analysis of AChE fiber density revealed a 25% reduction in the medial frontal cortex and an 18% reduction in the retrosplenial cortex of PTD treated rats compared with pair-fed controls (Anzalone et al. 2010). This reduction in AChE fiber density was associated with shrinkage of midline thalamic tissue. Interestingly, the frontal and prefrontal cortical areas display the greatest reductions in behaviorally activated ACh efflux after PTD treatment (see Fig. 6). In addition, spontaneous alternation performance correlates with ACh efflux in the medial frontal cortex but not in the retrosplenial cortex, suggesting that depressed cholinergic activity in the frontal cortex contributes to the behavioral impairment observed in the PTD model.
Fig. 6.
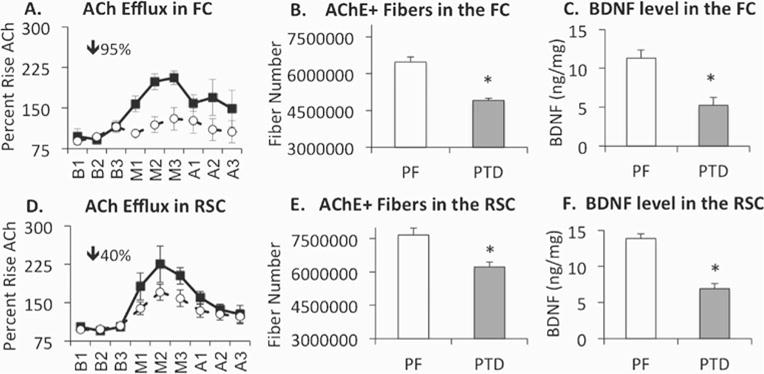
In both the frontal (FC) and retrosplenial (RSC) cortices, PTD treatment induces decreases in ACh efflux (a & d; open circles=PTD; closed squares=PF), number of acetylcholinesterase (AChE) fibers (b & e) and brain-derived neurotrophic factor (BDNF) signaling (c & f). It should be noted that the impairment of ACh efflux induced by PTD treatment is greater in the frontal cortex than in the retrosplenial cortex, despite a similar loss in AChE fibers and BDNF across the two cortical regions
The cortical glutamatergic system is also altered by PTD. In a study by Carvalho et al. (2006), TD, but not chronic ethanol consumption, in rats impaired performance on the Morris water maze and decreased in vitro glutamate uptake in the prefrontal cortex. Thus, it appears likely that the severity of cortical dysfunction and behavioral deficits in PTD is modulated through both cholinergic and glutamatergic systems.
Examination of the literature suggests a few possible mechanisms by which cortical alterations may contribute to cognitive decline in the PTD model and in human KS. First, reduced cholinergic innervation of the cortex from the basal forebrain may contribute to diminished cortical ACh functioning (Arendt et al. 1983; Anzalone et al. 2010; Harper and Kril 1993). Second, damage to key thalamic regions by PTD treatment may alter both ACh-dependent feedback synapses within cortical circuits and the release of glutamate from thalamocortical afferents (Savage 2012). Third, given reduced levels of BDNF in the frontal cortex of PTD rats (Vetreno et al. 2011a), decreased neurotrophin signaling may result in a broad decrease in cortical plasticity. A considerable body of evidence suggests that functional deactivation in the cortex may be a critical element in the devastating behavioral consequences of TD.
Cognitive Dysfunction in the PTD Model
An array of PTD studies has exposed behavioral impairments that are remarkably similar to those of KS patients. Rats recovered from PTD are impaired on tests of spatial memory, including delayed alternation (Mair et al. 1985), spontaneous alternation (Savage et al. 2003), spatial matching- and non-matching-to-sample (Mair et al. 1991a, b; Robinson and Mair 1992) and acquisition of a water maze task (Langlais et al. 1992; Pires et al. 2005). Deficits are also observed on olfactory- and auditory-based matching- and non-matching-to-sample tasks (Mair et al. 1991a, b) and avoidance tasks (Langlais et al. 1992).
Observations of impaired spatial navigation learning assessed by the Morris water maze (Langlais et al. 1992; Pires et al. 2005; Carvalho et al. 2006) have revealed that recovered PTD rats have difficulty shifting strategies due to perseveration of a previous learned behavior. Nevertheless, the animals were eventually able to master this task, unlike other tasks in which a significant proportion of PTD-treated rats displayed persistent learning/memory deficits such as spontaneous alternation (Savage et al. 2003), shock avoidance (active and passive) or spatial delayed alternation (Mair et al. 1985). These variations in the effects of PTD treatment are likely a product of differences in task demands. For example, successful performance on a delayed alternation task requires the animal to learn a win-shift rule relies heavily on working memory; by contrast, the demands on working memory are minimized in a water maze task in which the location of a hidden platform does not change across trials (Langlais et al. 1992). The foregoing studies suggest that PTD-induced neuropathology primarily affects the performance of tasks that have heavy demands on episodic working memory. Episodic memory, a type of explicit memory for a past experience of an event and/or location, in rodents is subserved by the diencephalic-temporal lobe axis (see Aggleton 2008), which the AT and MB are critical components. It is presumed that the severe damage that occurs to those diencephalic regions and the associated reduced activation of the retrosplenial cortex after PTD treatment is the reason why such severe memory deficits persist on such tasks.
While an impaired capacity for learning is an established consequence of PTD, the effects of TD on retrograde memory are still unclear. In one of the few studies to assess retrograde memory in PTD-treated rats, subjects displayed normal retention of a hidden-platform water maze task that was initially learned over a two-week period just prior to the commencement of PTD treatment (Langlais et al. 1992). These results were echoed by another study in which neither ethanol nor TD nor a combination of both induced retrograde amnesia (Pires et al. 2005). On the other hand, deficiencies in retrograde memory were suggested by a study in which PTD animals were unable to reacquire a spatial delayed alternation task that they had learned prior to treatment (Mair et al. 1985). The manifestation of retrograde memory loss may thus be dependent on task type, lesion intervals, and other factors.
Three hypotheses on the localization of the neural dysfunction responsible for the cognitive impairments reported in TD and KS have been formulated (see Béracochéa 2005). The first posits that the relevant functional damage is restricted to the diencephalon. Lesion studies have indicated that the midline thalamic region, in particular the IML, is critical for memory performance (see Mair 1994). A second hypothesis is that TD affects temporal lobe structures, including the hippocampus, and that diencephalic amnesia arises from disrupted connections between the diencephalon and the medial temporal lobe (Béracochéa 2005; Savage et al. 2003). Evidence suggests that even discrete diencephalic damage alters the activation of other regions, in particular the hippocampus. Our laboratory has demonstrated that the hippocampus is functionally impaired when PTD-treated rats perform a spontaneous alternation task (Savage et al. 2003) or learn a NMTP task (Roland and Savage 2007). A third hypothesis is that functional impairments resulting from TD are widespread and encompass multiple cortical areas (Paller et al. 1997). In vivo studies have demonstrated that under certain behavioral demands, cortical regions are not fully activated in PTD rats. Anzalone et al. (2010) showed that behaviorally evoked ACh efflux is blunted not only in the hippocampus but also in two critical cortical regions (the medial frontal and retrosplenial cortices) in the PTD model; another study reported a significant correlation between cortical ACh activity and distance from the platform in the Morris water maze (Pires et al. 2005). Such data support the theory that although there are no overt lesions in the hippocampus, retrosplenial cortex or frontal cortex in the PTD model, these regions have reduced activity-dependent neural response (neurotransmitter release and c-Fos immunoreactivity). Such “functional lesions” contribute to the cognitive impairments observed with diencephalic damage.
Spared and Rescued Cognitive Functions in the PTD Model
Spared Cognitive Functions
As with KS patients, PTD-treated rats show preserved and even enhanced performance on some learning and memory tasks, including simple discrimination (Mair et al. 1988) and response tasks (Vetreno et al. 2008) and some types of Pavlovian associative tasks. Such tasks are known to be independent of hippocampal function. For example, PTD-treated rats learning a left–right discrimination task demonstrated an accelerated release of ACh in the striatum and a trend toward accelerated learning relative to control rats (Vetreno et al. 2008). Similarly, no learning or memory deficits were observed when PTD rats were trained using a Pavlovian procedure pairing unique rewards (rat chow vs. chocolate milk) with to-be-remembered locations (left vs. right) on either a maze or operant delayed MTP task (Langlais and Savage 1995; Savage and Guarino 2010). Pairing unique rewards with specific locations enabled PTD-treated rats to learn the maze task at the same rate as control rats; without such pairing, PTD rats require about twice as many trials to learn this task (Langlais and Savage 1995). On the operant delayed MTP task, PTD rats trained with unique rewards actually outperformed control rats (Savage and Guarino 2010). Thus, training protocols that incorporate Pavlovian-based learning appear to “immunize” PTD-treated animals against memory impairment, suggesting that these cognitive demands recruit neural circuits involving structures (such as the amygdala) that are resistant to PTD-induced pathology (Savage and Ramos 2009). Clearly, these data have implications for the development of behavioral strategies for individuals with KS (see Hochhalter et al. 2001).
Pharmacological Promotion of Cognitive Recovery
Several lines of evidence suggest that the learning and memory deficits seen in the PTD model are a consequence of neurochemical dysfunction, particularly of the cholinergic and glutamatergic systems. Our laboratory has demonstrated a 25% to 30% loss of choline acetyltransferase (ChAT)–positive neurons in the medial septum/diagonal band of PTD-treated rats (Pitkin and Savage 2001, 2004; Roland and Savage 2009) and reduced cholinergic innervation of the hippocampus and cortex (Anzalone et al. 2010). This neuropathology likely contributes to the diminution of functional ACh efflux in the hippocampus and cortex that correlates with poor performance on spatial tasks (Vetreno et al. 2011a). Similar results have been found for rats fed only a TD diet; in vitro stimulation studies have revealed less ACh release from the hippocampal and frontal cortical tissue of TD rats compared with controls (Pires et al. 2005).
Whereas drugs that enhance cholinergic activity, such as AChE-Is, have had limited success in improving cognitive functioning in KS patients (Cochrane et al. 2005; Iga et al. 2001), site-specific targeted drugs that increase ACh function have resulted in significant or complete recovery of cognitive functioning in the PTD model (see Fig. 7). Two cholinergic targets affected by PTD treatment, the hippocampus and cortex, have been the focus of several recent studies. Systemic or intrahippocampal injections of the AChE-I physostigmine completely eliminated PTD-associated behavioral impairment (Roland et al. 2008). Similarly, decreasing GABA inhibition of the septohippocampal pathway by intraseptal infusion of bicuculline (a GABAA antagonist) increased hippocampal ACh release and resulted in complete behavioral recovery in PTD animals (Roland and Savage 2009). Although both systemic and intraseptal administration of the AChE-I tacrine dose-dependently improved spontaneous alternation behavior in PTD-treated rats, only systemic administration completely eliminated the behavioral deficit (Roland et al. 2010), suggesting that areas outside of the septohippocampal pathway are also involved in pharmacologically induced recovery.
Fig. 7.
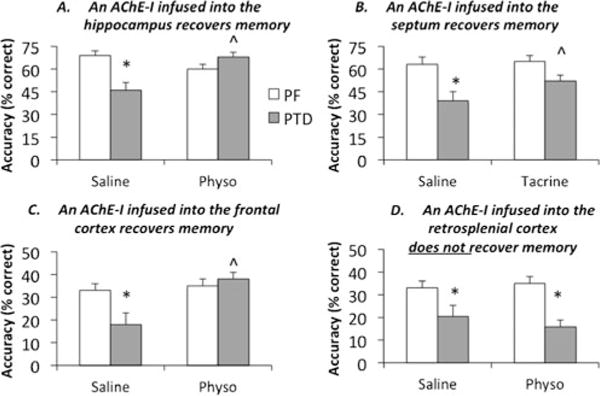
Acetylcholinesterase inhibitors (AChE-Is) acutely infused into the hippocampus (a: 40 ng, ∂=0.97), medial septum (b: 5 μg, ∂=0.99) and medial frontal cortex (c: 1 μg, ∂=3.14) all significantly increased spontaneous alternation performance in PTD rats. However, infusion of the AChE-I physostigmine into the retrosplenial cortex did not improve cognitive outcome (d: 1 μg), although it increased ACh levels. *Significant (P<.01) difference between PF and PTD rats in the saline condition; ˆSignificant (P<.05) drug recovery in PTD rats. For comparisons across studies, Cohen’s effect size (∂) is reported. Dose response curves for all AChE-Is were conducted for behavior and ACh efflux. Only the maximum effective dose is shown for each drug condition
Given significant reductions in ACh efflux in the limbic cortical regions of PTD rats during cognitive challenges, we recently sought to determine whether cognitive abilities could be restored by increasing cholinergic tone in the frontal or retrosplenial cortex. Interestingly, increased ACh levels in the frontal cortex, but not in the retrosplenial cortex, were associated with recovered spatial memory in PTD rats (Savage 2012). Thus, not all “functionally lesioned” structures are appropriate targets for therapeutic intervention. Because we believe that intact circuitry is necessary for cholinergic stimulation to be effective in brain-damaged subjects, we feel that regions that are “functionally” lesioned (septum, hippocampus, frontal cortex) rather than “structurally” lesioned (thalamus, mammillary bodies) by PTD may be the most credible targets for neurochemical modulation to improve behavioral outcomes.
Enrichment
Recently, environmental enrichment has been suggested as a natural promoter of neural plasticity that can aid the recovery of cognitive performance in both animal models of memory dysfunction and humans in the early stages of memory loss (Heyn et al. 2004). Although there are no published studies assessing the effects of environmental enrichment or exercise on animals with TD- or PTD-induced brain pathology or cognitive dysfunction, there is evidence that exercise prior to binge alcohol exposure (Leasure and Nixon 2010) or during chronic alcohol exposure (Crews et al. 2004) does protect against alcohol-induced hippocampal cell loss.
In addition, environmental enrichment has been shown to have beneficial behavioral effects in animals following neurotoxin-induced lesions to the anterior thalamic nuclei (ATN), an area of critical damage in PTD and KS. Loukavenko et al. (2007) showed that rats recovering from ATN lesions exhibited better performance when exposed to enriched housing, and a subsequent study suggested that enrichment improves the ability of AT-lesioned rats to update spatial maps (Wolff et al. 2008).
Given the loss of neurogenesis and BDNF in the PTD brain, investigation of the effects of environmental enrichment and exercise on brain and behavioral function in PTD-treated animals is of considerable interest. Indeed, voluntary exercise following traumatic brain injury in rats has been shown to reduce learning impairments on the Morris water maze task and to lead to upregulation of BDNF (Griesbach et al. 2004), and a more recent study demonstrated that the ameliorative effects of exercise on memory following brain injury are dependent on BDNF activation (Griesbach et al. 2009). These data, combined with evidence that voluntary exercise increases neurogenesis (specifically precursor cell proliferation in the adult hippocampus; Fabel et al. 2009), encourage investigation into enrichment as a means of diminishing the behavioral deficits in the PTD model. An understanding of the mechanisms involved may lead to valuable insights into therapeutic remedies for KS, and research in our laboratory aimed at revealing these mechanisms is ongoing.
Conclusions
The PTD model of KS has provided a means to a more thorough understanding of the brain structures involved in learning and memory and has implicated not only diencephalic structures but the hippocampus and frontal cortex in the cognitive deficits associated with PTD and KS. However, the PTD brain also demonstrates functional plasticity and an ability to compensate for memory deficiencies when task demands permit (for example, through the use of spared striatal- and amygdalar-based circuits). Finally, our work has shown that although the functional integration of the limbic system (thalamus, hippocampus and cortex) is compromised with PTD, the hippocampus and frontal cortex are able to respond when cholinergic tone is increased via direct pharmacological manipulation, and cognitive function may be thus restored.
The application of models of human pathological conditions not only permits insight into the neuroanatomical and neurochemical underpinnings of cognitive dysfunction, but also provides a glimpse into the plastic properties of the brain that remain despite insult. Greater understanding of the mechanisms of plasticity that are critical for learning and memory and for cognitive adaptation will aid in the development of treatments for memory disorders in humans.
Acknowledgments
We would like to thank Ryan Vetreno for assistance with photographs of the brain. This research was funded by grants NINDS 054272 and ARRA NINDS 054272-S1 to LMS.
Abbreviations
- ACh
Acetylcholine
- AChE
Acetylcholinesterase
- AChE-I
Acetylcholinesterase inhibitor
- AT
Anterior thalamus
- ATN
Anterior thalamic nuclei
- BDNF
Brain-derived neurotrophic factor
- ChAT
Choline acetyltransferase
- DNMTS
Delayed non-matching-to-sample
- DMTS
Delayed matching-to-sample
- EAS
Early acute stage
- IML
Intralaminar
- KS
Korsakoff syndrome
- LAS
Late acute stage
- MAS
Middle acute stage
- MB
Mammillary bodies
- MTP
Matching-To-Position
- MTS
Matching-To-Sample
- NeuN
Neuronal-Specific nuclear protein
- NMTP
Non-Matching-To-Position
- NMTS
Non-Matching-To-Sample
- PTD
Pyrithiamine-induced thiamine deficiency
- TD
Thiamine deficiency
- TPP
Thiamine pyrophosphate
- WE
Wernicke’s encephalopathy
- WKS
Wernicke-Korsakoff syndrome
Contributor Information
Lisa M. Savage, Email: lsavage@binghamton.edu, Behavioral Neuroscience Program, Department of Psychology, State University of New York at Binghamton, Binghamton, NY 13902, USA.
Joseph M. Hall, Behavioral Neuroscience Program, Department of Psychology, State University of New York at Binghamton, Binghamton, NY 13902, USA
Leticia S. Resende, Laboratorio de Neurociencia e Comportamento, Universidade Federal de Minas Gerais, Belo Horizonte 31270-010, Brazil
References
- Aggleton JP. Understanding anterograde amnesia: disconnections and hidden lesions. The Quarterly Journal of Experimental Psychology. 2008;61:1441–1471. doi: 10.1080/17470210802215335. [DOI] [PubMed] [Google Scholar]
- Aggleton JP, Brown MW. Episodic memory, amnesia, and the hippocampal-anterior thalamic axis. Behavioral Brain Research. 1999;22:425–489. [PubMed] [Google Scholar]
- Aggleton JP, Dumont JR, Warburton EC. Unraveling the contributions of the diencephalon to recognition memory: a review. Learning & Memory. 2011;18:384–400. doi: 10.1101/lm.1884611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone S, Vetreno RP, Ramos RL, Savage LM. Cortical cholinergic abnormalities contribute to the amnesic state induced by pyrithiamine-induced thiamine deficiency in the rat. European Journal of Neuroscience. 2010;32:847–858. doi: 10.1111/j.1460-9568.2010.07358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korskaoff’s disease. Acta Neuropathologica. 1983;61:101–108. doi: 10.1007/BF00697388. [DOI] [PubMed] [Google Scholar]
- Armstrong-James M, Ross DT, Chen F, Ebner FF. The effect of thiamine deficiency on the structure and physiology of the rat forebrain. Metabolic Brain Disease. 1988;3:91–124. doi: 10.1007/BF01001012. [DOI] [PubMed] [Google Scholar]
- Baker KG, Harding AJ, Halliday GM, Kril JJ, Harper CG. Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke’s encephalopathy. Neuroscience. 1999;91:429–438. doi: 10.1016/s0306-4522(98)90664-9. [DOI] [PubMed] [Google Scholar]
- Béracochéa D. Interaction between emotion and memory: importance of mammillary body damage in a mouse model of the alcoholic Korsakoff syndrome. Neural Plasticity. 2005;12:275–287. doi: 10.1155/NP.2005.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk JA, Mair RG. Effects of intralaminar thalamic lesions on sensory attention and motor intention in the rat: a comparison with lesions involving frontal cortex and hippocampus. Behavioural Brain Research. 2001;123:49–63. doi: 10.1016/s0166-4328(01)00202-9. [DOI] [PubMed] [Google Scholar]
- Butterworth RF, Héroux M. Effect of pyrithiamine treatment and subsequent thiamine rehabilitation on regional cerebral amino acids and thiamine-dependent enzymes. Journal of Neurochemistry. 1989;52:1079–1084. doi: 10.1111/j.1471-4159.1989.tb01850.x. [DOI] [PubMed] [Google Scholar]
- Carvalho FM, Pereira SRC, Pires RGW, Ferraz VP, Romano-Silva MA, Oliveira-Silva IF, Ribeiro AM. Thiamine deficiency decreases glutamate uptake in the prefrontal cortex and impairs performance in a water maze test. Pharmacology Biochemistry and Behavior. 2006;83:481–489. doi: 10.1016/j.pbb.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Christie JE, Kean DM, Douglas RH, Engleman HM, St Clair D, Blackburn IM. Magnetic resonance imaging in pre-senile dementia of the Alzheimer-type, multi-infarct dementia and Korsakoff’s syndrome. Psychological Medicine. 1988;18:319–329. doi: 10.1017/s0033291700007868. [DOI] [PubMed] [Google Scholar]
- Ciccia RM, Langlais PJ. An examination of the synergistic interaction of ethanol and thiamine deficiency in the development of neurological signs and long-term cognitive and memory impairments. Alcoholism, Clinical and Experimental Research. 2000;24:622–634. [PubMed] [Google Scholar]
- Cochrane M, Cochrane A, Jauhar P, Ashton E. Acetylcholinesterase inhibitors for the treatment of Wernicke-Korsakoff syndrome— three further cases show response to donepezil. Alcohol and Alcoholism. 2005;40:151–154. doi: 10.1093/alcalc/agh127. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends in Neuroscience. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- Crews FT, Nixon K, Wilkie ME. Exercise reverses ethanol inhibition of neural stem cell proliferation. Alcohol. 2004;33:63–71. doi: 10.1016/j.alcohol.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Dirksen CL, Howard JA, Cronin-Golomb A, Oscar-Berman M. Patterns of prefrontal dysfunction in alcoholics with and without Korsakoff’s syndrome, patients with Parkinson’s disease, and patients with rupture and repair of the anterior communicating artery. Neuropsychiatric Disease and Treatment. 2006;2:327–339. doi: 10.2147/nedt.2006.2.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabel K, Wolf SA, Ehninger D, Babu H, Leal-Galicia P, Kemperman G. Additive effects of physical exercise and environmental enrichment on adult hippocampal neurogenesis in mice. Frontiers in Neuroscience. 2009;3:1–7. doi: 10.3389/neuro.22.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fama R, Marsh L, Sullivan EV. Dissociation of remote and anterograde memory impairment and neural correlates in alcoholic Korsakoff syndrome. Journal of International Neuropsychological Society. 2004;10:427–441. doi: 10.1017/S135561770410310X. [DOI] [PubMed] [Google Scholar]
- Gansler DA, Harris GJ, Oscar-Berman M, Streeter C, Lewis RF, Ahmed I, Achong D. Hypoperfusion of inferior frontal brain regions in abstinent alcoholics: a pilot SPECT study. Journal of Studies on Alcohol. 2000;61:32–37. doi: 10.15288/jsa.2000.61.32. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Ksiezak-Reding H, Sheu KFR, Mykytyn V, Blass JP. Correlation of enzymatic metabolic and behavioral deficits in thiamine deficiency and its reversal. Neurochemistry Research. 1984;9:803–814. doi: 10.1007/BF00965667. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Park LCH, Sheu KFR, Blass JP, Calingasan NY. The α-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochemistry International. 1999;36:97–112. doi: 10.1016/s0197-0186(99)00114-x. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Molteni R, Wu A, Gomez-Pinilla F. Voluntary exercise following traumatic brain injury: brain-derived neurotrophic factor upregulation and recovery of function. Neuroscience. 2004;125:129–139. doi: 10.1016/j.neuroscience.2004.01.030. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Gomez-Pinilla F. Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain Research. 2009;1228:105–115. doi: 10.1016/j.brainres.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AJ, Halliday G, Caine D, Kril JJ. Degeneration of anterior thalamic nuclei differentiates alcoholics with amnesia. Brain. 2000;123:141–154. doi: 10.1093/brain/123.1.141. [DOI] [PubMed] [Google Scholar]
- Harper CG, Kril JJ. Neuropathological changes in alcoholics. Research Monograph: Alcohol-induced Brain Damage. 1993;22:39–70. [Google Scholar]
- Hazell AS, Butterworth RF. Update of cell damage mechanisms in thiamine deficiency: focus on oxidative stress, excitotoxicity and inflammation. Alcohol and Alcoholism. 2009;44:141–147. doi: 10.1093/alcalc/agn120. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Butterworth RF, Hakim AM. Cerebral vulnerability is associated with selective increase in extracellular glutamate concentration in experimental thiamine deficiency. Journal of Neurochemistry. 1993;61:1155–1158. doi: 10.1111/j.1471-4159.1993.tb03635.x. [DOI] [PubMed] [Google Scholar]
- He X, Sullivan EV, Stankovic RK, Harper CG, Pfefferbaum A. Interaction of thiamine deficiency and voluntary alcohol consumption disrupts rat corpus callosum ultrastructure. Neuropsychopharmacology. 2007;32:2207–2216. doi: 10.1038/sj.npp.1301332. [DOI] [PubMed] [Google Scholar]
- Héroux M, Butterworth RF. Reversible alterations of cerebral α–aminobutyric acid in pyrithiamine-treated rats: implications for the pathogenesis Wernicke’s encephalopathy. Journal of Neurochemistry. 1988;51:1221–1226. doi: 10.1111/j.1471-4159.1988.tb03090.x. [DOI] [PubMed] [Google Scholar]
- Héroux M, Butterworth RF. Regional alterations of thiamine phosphate esters and of thiamine diphosphate-dependent enzymes in relation to function in experimental Wernicke’s encephalopathy. Neurochemical Research. 1995;20:87–93. doi: 10.1007/BF00995157. [DOI] [PubMed] [Google Scholar]
- Heyn P, Abreau BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Archives of Physical Medicine and Rehabilitation. 2004;85:1694–1704. doi: 10.1016/j.apmr.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Hochhalter AK, Sweeney WA, Savage LM, Bakke BL, Overmier JB. Using animal models to address the memory deficits of Wernicke-Korsakoff syndrome. In: Carroll ME, Overmier JB, editors. Animal research and human health: Advancing human welfare through behavioral science. Washington, DC: American Psychological Association; 2001. pp. 281–292. [Google Scholar]
- Homewood J, Bond NW, Mackenzie A. The effects of single and repeated episodes of thiamin deficiency on memory in alcohol-consuming rats. Science. 1997;14:81–91. doi: 10.1016/s0741-8329(96)00111-5. [DOI] [PubMed] [Google Scholar]
- Iga JI, Araki M, Ishimoto Y, Ohmori T. A case of Korsakoff’s syndrome improved by high doses of donepezil. Alcohol and Alcoholism. 2001;36:553–555. doi: 10.1093/alcalc/36.6.553. [DOI] [PubMed] [Google Scholar]
- Irle E, Markowitsch HJ. Widespread neuroanatomical damage and learning deficits following chronic alcohol consumption or vitamin-B1 (thiamine) deficiency in rats. Behavioural Brain Research. 1983;9:277–294. doi: 10.1016/0166-4328(83)90133-x. [DOI] [PubMed] [Google Scholar]
- Ke ZJ, Wang X, Fan Z, Luo J. Ethanol promotes thiamine deficiency-induced neuronal death: involvement of double-stranded RNA-activated protein kinase. Alcohol Clinical Experimental Research. 2009;33:1097–1103. doi: 10.1111/j.1530-0277.2009.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelman MD, Thomson AD, Guerrini I, Marshall EJ. The Korskakoff syndrome: clinical aspects, psychology and treatment. Alcohol and Alcoholism. 2009;44:148–154. doi: 10.1093/alcalc/agn118. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Homewood J. Neuronal changes in the cerebral cortex of the rat following alcohol treatment and thiamine deficiency. Journal of Neuropathology and Experimental Neuropathology. 1993;52:586–593. doi: 10.1097/00005072-199311000-00005. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Mair RG. Protective effects of the glutamate antagonist MK-801 on pyrithiamine-induced lesions and amino acid changes in rat brain. Journal of Neuroscience. 1990;10:1664–1674. doi: 10.1523/JNEUROSCI.10-05-01664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX. Extracellular glutamate is increased in thalamus during thiamine deficiency-induced lesions and is blocked by MK-801. Journal of Neurochemistry. 1993;61:2175–2182. doi: 10.1111/j.1471-4159.1993.tb07457.x. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Savage LM. Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks that correlate with tissue loss in diencephalon, cortex and white matter. Behavioural Brain Research. 1995;68:75–89. doi: 10.1016/0166-4328(94)00162-9. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX. Cortical and subcortical white matter damage without Wernicke’s encephalopathy after recovery from thiamine deficiency in the rat. Alcoholism, Clinical and Experimental Research. 1997;21:434–443. doi: 10.1111/j.1530-0277.1997.tb03788.x. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Mair RG, Anderson CD, McEntee WJ. Long-lasting changes during thiamine deficiency-induced lesions and amino acid changes in the rat brain. Neurochemistry Research. 1988;13:1199–1206. doi: 10.1007/BF00971639. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Mandel RJ, Mair RG. Diencephalic lesions, learning impairments, and intact retrograde memory following acute thiamine deficiency in the rat. Behavioural Brain Research. 1992;48:177–185. doi: 10.1016/s0166-4328(05)80155-x. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Anderson G, Guo SX, Bondy SC. Increased cerebral free radical production during thiamine deficiency. Metabolic Brain Disease. 1996a;12:137–143. [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX, Savage LM. Neuropathology of thiamine deficiency: an update on the comparative analysis of human disorders and experimental models. Metabolic Brain Disease. 1996b;11:19–35. doi: 10.1007/BF02080929. [DOI] [PubMed] [Google Scholar]
- Lee J, Duan W, Mattson MP. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. Journal of Neurochemistry. 2002;82:1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x. [DOI] [PubMed] [Google Scholar]
- Le Roch K, Riche D, Sara SJ. Persistence of habituation deficits after neurological recovery from severe thiamine deprivation. Behavioural Brain Research. 1987;26:37–46. doi: 10.1016/0166-4328(87)90014-3. [DOI] [PubMed] [Google Scholar]
- Leasure JL, Nixon K. Exercise neuroprotection in a rat model of binge alcohol consumption. Alcoholism, Clinical and Experimental Research. 2010;34:404–414. doi: 10.1111/j.1530-0277.2009.01105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukavenko EA, Ottley MC, Morgan JP, Wolff M, Dairymple-Alford JC. Towards therapy to relieve memory impairments after anterior thalamic lesions: improved spatial working memory after immediate and delayed postoperative enrichment. European Journal of Neuroscience. 2007;26:3267–3276. doi: 10.1111/j.1460-9568.2007.05879.x. [DOI] [PubMed] [Google Scholar]
- Mair RG. On the role of thalamic pathology in diencephalic amnesia. Reviews in Neuroscience. 1994;5:105–140. doi: 10.1515/revneuro.1994.5.2.105. [DOI] [PubMed] [Google Scholar]
- Mair RG, Anderson CD, Langlais PJ, McEntee WJ. Thiamine deficiency depletes cortical norepinephrine and impairs learning processes in the rat. Brain Research. 1985;360:273–284. doi: 10.1016/0006-8993(85)91243-0. [DOI] [PubMed] [Google Scholar]
- Mair RG, Anderson CD, Langlais PJ, McEntee WJ. Behavioral impairments, brain lesions, and monoaminergic activity in the rat following recovery from a bout of thiamine deficiency. Behavioural Brain Research. 1988;27:223–239. doi: 10.1016/0166-4328(88)90119-2. [DOI] [PubMed] [Google Scholar]
- Mair RG, Knoth RL, Rabchenuk SA, Langlais PJ. Impairment of olfactory, auditory, and spatial serial reversal learning in rats recovered from pyrithiamine-induced thiamine deficiency. Behavioral Neuroscience. 1991a;105:360–374. [PubMed] [Google Scholar]
- Mair RG, Otto TA, Knoth RL, Rabchenuk SA, Langlais PJ. Analysis of aversively conditioned learning and memory in rats recovered from pyrithiamine-induced thiamine deficiency. Behavioral Neuroscience. 1991b;105:351–359. [PubMed] [Google Scholar]
- Mair RG, Burk JA, Porter MC. Lesions of the frontal cortex, hippocampus, and intralaminar thalamic nuclei have distinct effects on remembering in rats. Behavioral Neuroscience. 1998;112:772–792. doi: 10.1037//0735-7044.112.4.772. [DOI] [PubMed] [Google Scholar]
- Mitchell AS, Dalrymple-Alford JC. Dissociable memory effects after medial thalamus lesions in the rat. European Journal of Neuroscience. 2005;22:973–985. doi: 10.1111/j.1460-9568.2005.04199.x. [DOI] [PubMed] [Google Scholar]
- Mitchell AS, Dalrymple-Alford JC. Lateral and anterior thalamic lesions impair independent memory systems. Learning & Memory. 2006;13:388–396. doi: 10.1101/lm.122206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawasai O. Behavioral and neurochemical alterations following thiamine deficiency in rodents: relationship to functions of cholinergic neurons. The Pharmaceutical Society of Japan. 2005;125:549–554. doi: 10.1248/yakushi.125.549. [DOI] [PubMed] [Google Scholar]
- Nakagawasai O, Tadano T, Hozumi S, Tan-No K, Niijima F, Kisara K. Immunohistochemical estimation of brain choline acetyltransferase and somatostatin related to the impairment of avoidance learning induced by thiamine deficiency. Brain Research Bulletin. 2000;52:189–196. doi: 10.1016/s0361-9230(00)00248-3. [DOI] [PubMed] [Google Scholar]
- Nakagawasai O, Tadano T, Hozumi S, Taniguchi R, Yamadera F, Tan-No F, Niijima H, Arai H, Yasuhara H, Kinemuchi H, Kisara K. Inolvement of muscarinic receptor on the impairment of avoidance learning in mice fed a thiamine-deficient diet. Biogenic Amines. 2001;16:199–210. [Google Scholar]
- Nonner D, Barrett EF, Barrett JN. Neurotrophin effects on survival and expression of cholinergic properties in cultured rat septal neurons under normal and stress conditions. The Journal of Neuroscience. 1996;16:6665–6675. doi: 10.1523/JNEUROSCI.16-21-06665.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oscar-Berman M, Evert D. Alcoholic Korsakoff’s syndrome. In: Nussbaum PD, editor. Handbook of neuropsychology and aging. New York: Plenum; 1997. pp. 201–215. [Google Scholar]
- Oscar-Berman M, Kirkley SM, Gansler DA, Couture A. Comparisons of Korsakoff and non-Korsakoff alcoholics on neuropsychological tests of prefrontal brain functioning. Alcoholism, Clinical and Experimental Research. 2004;28:667–675. doi: 10.1097/01.alc.0000122761.09179.b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paller KA, Acharya A, Richardson AP, Plaisant O, Shimamura AP, Reed BR, Jagust WJ. Functional neuroimaging of cortical dysfunction in alcoholic Korsakoff’s syndrome. Journal of Cognitive Neuroscience. 1997;9:277–293. doi: 10.1162/jocn.1997.9.2.277. [DOI] [PubMed] [Google Scholar]
- Pannunzio P, Hazell AS, Pannunzio M, Rao KV, Butterworth RF. Thiamine deficiency results in metabolic acidosis and energy failure in cerebellar granule cells: an in vitro model for the study of cell death mechanisms in Wernicke’s encephalopathy. Journal of Neuroscience Research. 2000;62:286–292. doi: 10.1002/1097-4547(20001015)62:2<286::AID-JNR13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Adalsteinsson E, Bell RL, Sullivan EV. Development and resolution of brain lesions caused by pyrithiamine- and dietary-induced thiamine deficiency and alcohol exposure in the alcohol-preferring rat: a longitudinal magnetic resonance imaging and spectroscopy study. Neuropsychopharmacology. 2007;32:1159–1177. doi: 10.1038/sj.npp.1301107. [DOI] [PubMed] [Google Scholar]
- Philips SC, Harper C, Kril J. A quantitative histological study of the cerebellar vermis in alcoholic patients. Brain. 1987;110:301–314. doi: 10.1093/brain/110.2.301. [DOI] [PubMed] [Google Scholar]
- Pires RGW, Pereira SRC, Pittella JEH, Franco GC, Ferreira CLM, Fernandes PA, Ribeiro AM. The contribution of mild thiamine deficiency and ethanol consumption to central cholinergic dysfunction and rats’ open-field performance impairment. Pharmacology Biochemistry and Behavior. 2001;70:227–235. doi: 10.1016/s0091-3057(01)00593-7. [DOI] [PubMed] [Google Scholar]
- Pires RGW, Pereira SRC, Oliveira-Silva I, Franco GC, Ribeiro AM. Cholinergic parameters and the retrieval of learned and re-learned spatial information: a study using a model of Wernicke-Korsakoff Syndrome. Behavioural Brain Research. 2005;162:11–21. doi: 10.1016/j.bbr.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Aging potentiates the acute and chronic neurological symptoms of pyrithiamine-induced thiamine deficiency in the rodent. Behavioural Brain Research. 2001;119:167–177. doi: 10.1016/s0166-4328(00)00350-8. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Age-related vulnerability to diencephalic amnesia produced by thiamine deficiency: the role of time of insult. Behavioural Brain Research. 2004;148:93–105. doi: 10.1016/s0166-4328(03)00208-0. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Lasserson D, Marsden P, Stanhope N, Stevens T, Bello F, Kingsley D, Colchester A, Kopelman MD. FDG-PET findings in the Wernicke-Korsakoff syndrome. Cortex. 2003;39:1027–1045. doi: 10.1016/s0010-9452(08)70876-1. [DOI] [PubMed] [Google Scholar]
- Robinson JK, Mair RG. MK-801 prevents brain lesions and delayed-nonmatching-to-sample deficits produced by pyrithiamine-induced encephalopathy in rats. Behavioral Neuroscience. 1992;106:623–633. [PubMed] [Google Scholar]
- Roland JJ, Savage LM. Blunted hippocampal, but not striatal, acetylcholine efflux parallels learning impairment in diencephalic-lesioned rats. Neurobiology of Learning and Memory. 2007;87:123–132. doi: 10.1016/j.nlm.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. The role of cholinergic and GABAergic medial septal/diagonal band cell populations in the emergence of diencephalic amnesia. Neuroscience. 2009;160:32–41. doi: 10.1016/j.neuroscience.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Mark K, Vetreno RP, Savage LM. Increasing hippocampal acetylcholine levels enhance behavioral performance in an animal model of diencephalic amnesia. Brain Research. 2008;1234:116–127. doi: 10.1016/j.brainres.2008.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Levinson M, Vetreno RP, Savage LM. Differential effects of systemic and intraseptal administration of the acetylcholinesterase inhibitor tacrine on the recovery of spatial behavior in an animal model of diencephalic amnesia. European Journal of Pharmacology. 2010;629:31–39. doi: 10.1016/j.ejphar.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sairanen M, Lucas G, Emfors P, Castren M, Castren E. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. The Journal of Neuroscience. 2005;25:1089–1094. doi: 10.1523/JNEUROSCI.3741-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM. Sustaining high acetylcholine levels in the frontal cortex, but not retrosplenial cortex, recovers spatial memory performance in a rodent model of diencephalic amnesia. Behavioral Neuroscience. 2012;126:226–236. doi: 10.1037/a0027257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Guarino S. Memory for reward location is enhanced even though acetylcholine efflux within the amygdala is impaired in rats with damage to the diencephalon produced by thiamine deficiency. Neurobiology of Learning and Memory. 2010;94:554–560. doi: 10.1016/j.nlm.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Ramos RL. Reward expectation alters learning and memory: the impact of the amygdala on appetitive-driven behaviors. Behavioural Brain Research. 2009;198:1–12. doi: 10.1016/j.bbr.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Castillo R, Langlais PJ. Effects of lesions of thalamic intralaminar and midline nuclei and internal meduallary lamina on spatial memory and object discrimination. Behavioral Neuroscience. 1998;112:1339–1352. doi: 10.1037//0735-7044.112.6.1339. [DOI] [PubMed] [Google Scholar]
- Savage LM, Pitkin SR, Knitowski KM. Rats exposed to acute pyrithiamine-induced thiamine deficiency are more sensitive to the amnestic effects of scopolamine and MK-801: examination of working memory, response selection, and reinforcement contingencies. Behavioural Brain Research. 1999;104:13–26. doi: 10.1016/s0166-4328(99)00049-2. [DOI] [PubMed] [Google Scholar]
- Savage LM, Chang Q, Gold PE. Diencephalic damage decreases hippocampal acetylcholine release during spontaneous alternation testing. Learning & Memory. 2003;10:242–246. doi: 10.1101/lm.60003. [DOI] [PubMed] [Google Scholar]
- Savage LM, Roland JJ, Klintsova A. Selective septohippocampal-but not forebrain amygdalar-cholinergic dysfunction in diencephalic amnesa. Brain Research. 2007;1139:210–219. doi: 10.1016/j.brainres.2006.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Hall JM, Vetreno RP. Anterior thalamic lesions alter both hippocampal-dependent behavior and hippocampal acetylcholine release in the rat. Learning & Memory. 2011;18:751–758. doi: 10.1101/lm.023887.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Sweet AJ, Castillo R, Langlais PJ. The effects of lesions to the thalamic lateral internal medullary lamina nuclei and posterior nuclei on learning, memory, and habituation in the rat. Behavioural Brain Research. 1997;82:133–147. doi: 10.1016/s0166-4328(97)80983-7. [DOI] [PubMed] [Google Scholar]
- Squire LR. Specifying the defect in human amnesia: storage, retrieval and semantics. Neuropsychologia. 1980;18:369–372. doi: 10.1016/0028-3932(80)90134-7. [DOI] [PubMed] [Google Scholar]
- Squire LR. The neuropsychology of human memory. Annual Review of Neuroscience. 1982;5:241–273. doi: 10.1146/annurev.ne.05.030182.001325. [DOI] [PubMed] [Google Scholar]
- Squire LR, Amaral DG, Press GA. Magnetic resonance imaging of the hippocampal formation and mammillary nuclei distinguish medial temporal lobe and diencephalic amnesia. Journal of Neuroscience. 1990;10:3106–3117. doi: 10.1523/JNEUROSCI.10-09-03106.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Marsh L. Hippocampal volume deficits in alcoholic Korsakoff’s syndrome. Neurology. 2003;61:1716–1719. doi: 10.1212/01.wnl.0000098940.31882.bb. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Neuroimaging of the Wernicke-Korsakoff syndrome. Alcohol and Alcoholism. 2009;44:155–165. doi: 10.1093/alcalc/agn103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Nakazawa S, Yoshino Y, Shimura T. Metabolic studies of the edematous cerebral cortex of the pyrithiamine-treated thiamine deficient rat. Brain Research. 1988;441:202–208. doi: 10.1016/0006-8993(88)91399-6. [DOI] [PubMed] [Google Scholar]
- Todd KG, Butterworth RF. Mechanisms of selective neuronal cell death due to thiamine deficiency. Annals of the New York Academy of Sciences. 1999;893:404–411. doi: 10.1111/j.1749-6632.1999.tb07866.x. [DOI] [PubMed] [Google Scholar]
- Troncoso JC, Johnston MV, Hess KM, Griffin JW, Price DL. Model of Wernicke’s encephalopathy. Archives of Neurology. 1981;38:350–354. doi: 10.1001/archneur.1981.00510060052007. [DOI] [PubMed] [Google Scholar]
- Vann SD. Re-evaluating the role of the mammillary bodies in memory. Neuropsychologia. 2010;48:2316–2327. doi: 10.1016/j.neuropsychologia.2009.10.019. [DOI] [PubMed] [Google Scholar]
- Van Tilborg IA, Kessels RP, Kruijt P, Wester AJ, Hulstijn W. Spatial and nonspatial implicit motor learning in Korsakoff’s amnesia: evidence for selective deficits. Experimental Brain Research. 2011;214:427–435. doi: 10.1007/s00221-011-2841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Anzalone SJ, Savage LM. Impaired, spared, and enhanced ACh efflux across the hippocampus and striatum in diencephalic amnesia is dependent on task demands. Neurobiology of Learning and Memory. 2008;90:237–244. doi: 10.1016/j.nlm.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Hall JM, Savage LM. Alcohol-related amnesia and dementia: animal models have revealed the contributions of different etiological factors on neuropathology, neurochemical dysfunction and cognitive impairment. Neurobiology of Learning and Memory. 2011a;96:596–608. doi: 10.1016/j.nlm.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Klintsova A, Savage LM. Stage-dependent alterations of progenitor cell proliferation and neurogenesis in an animal model of Wernicke-Korsakoff syndrome. Brain Research. 2011b;1391:132–146. doi: 10.1016/j.brainres.2011.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Ramos RL, Anzalone S, Savage LM. Brain and behavioral pathology in an animal model of Wernicke’s encephalopathy and Wernicke-Korsakoff syndrome. Brain Research. 2012;1436:178–192. doi: 10.1016/j.brainres.2011.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil AFB, Oliveira-silva IF, Ferreira LF, Pereira SRC, Ribeiro AM. Spatial memory deficits and thalamic serotonergic metabolite change in thiamine deficient rats. Behavioural Brain Research. 2010;210:140–142. doi: 10.1016/j.bbr.2010.02.019. [DOI] [PubMed] [Google Scholar]
- Volterra A, Trotti D, Tromba C, Floridi S, Racagni G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. The Journal of Neuroscience. 1994;14:2924–2932. doi: 10.1523/JNEUROSCI.14-05-02924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe I. Pyrithiamine-induced acute thiamine-deficient encephalopathy in the mouse. Experimental and Molecular Pathology. 1978;28:381–394. doi: 10.1016/0014-4800(78)90012-6. [DOI] [PubMed] [Google Scholar]
- Witt ED. Neuroanatomical consequences of thiamine deficiency: a comparative analysis. Alcohol and Alcoholism. 1985;20:201–221. [PubMed] [Google Scholar]
- Wolff M, Loukavenko EA, Will BE, Dalrymple-Alford JC. The extended hippocampal-diencephalic memory system: enriched housing promotes recovery of the flexible use of spatial representations after anterior thalamic lesions. Hippocampus. 2008;18:996–1007. doi: 10.1002/hipo.20457. [DOI] [PubMed] [Google Scholar]
- Zhang SX, Weilersbacher GS, Henderson SW, Corso T, Olney JW, Langlais PJ. Excitotoxic cytopathology, progression, and reversibility of thiamine deficiency-induced diencephalic lesions. Journal of Neuropathology and Experimental Neurology. 1995;54:255–267. doi: 10.1097/00005072-199503000-00012. [DOI] [PubMed] [Google Scholar]
- Zhao N, Zhong C, Wang Y, Zhao Y, Gong N, Zhou G, Xu T, Hong Z. Impaired hippocampal neurogenesis is involved in cognitive dysfunction induced by thiamine deficiency at early pre-pathological lesion stage. Neurobiology of Disease. 2008;29:176–185. doi: 10.1016/j.nbd.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Zimitat C, Kril J, Harper C, Nixon P. Progression of neurological disease in thiamine-deficient rats is enhanced by ethanol. Alcohol. 1990;7:493–501. doi: 10.1016/0741-8329(90)90038-e. [DOI] [PubMed] [Google Scholar]