

Abstract
Six new polyketides, simplexolides A–E (1–5) and a furan ester, plakorfuran A (6), together with four known furanylidenic methyl esters (7–10) were isolated from the marine sponge Plakortis simplex. Compounds 1–5 feature a tetrahydrofuran ring opened seco-plakortone skeleton. These new structures, including relative configurations, were determined on the basis of extensive analysis of spectroscopic data. The absolute configurations of 1–6 were established by the modified Mosher's method, and the CD exciton chirality method. However, configurations of the remote stereocenters at C-8 in compounds 1–5 were not determined. Antifungal, cytotoxicity, antileismanial, and antimalarial activities of these poly-ketides were evaluated.
Keywords: Plakortis simplex, Simplexolide, Mosher's method, Antifungal, Cytotoxicity, Antimalarial, Absolute configuration
1. Introduction
Sponges of the genera Plakortis and Plakinastrella have been widely investigated for their biologically active polyketides.1–8 Apart from a large number of cyclic peroxides they also contain a group of γ-lactones, including bicyclic peroxylactones (plakortolides),9,10 bicyclic furanolactones (plakortones),8,11 N-alkylated lactones (amphiasterins),12 α,β-unsaturated lactones (butenolides),13 and some seco derivatives (seco-plakortolides).8 Plakortones A–F, featuring bicyclic furanolactones, are a class of ethyl branched (butyrate derived) polyketides from the genus Plakortis. They show interesting biological activities that have been the focus of extensive chemical and pharmaceutical studies. Plakortones A–D are cardiac sacroplasmic reticulum Ca2+-pumping ATPase activators, being active at micromolar concentrations;11 plakortones B–F exhibited moderate cytotoxicity against the murine fibro sarcoma cell line WEHI 164;14 plakortone G, a closely related analogof the γ-lactone isolated by Stierle and Faulkner,15 without a tetrahydrofuran moiety, showed antimalarial activity against both the D6 and W2 clones of Plasmodium falciparum.16 The absolute configurations of these plakortones were not confirmed during the initial isolation. The total syntheses of plakortones B, D, and E helped to establish their absolute configurations later.17–19 However, the absolute configurations of the other four plakortones A, C, F, and G still remain unknown. Recently, plakortones L, N, and P were isolated from the sponge of Plakinastrella clathrata.8 They have bicyclic furanolactone fragments like plakortones A–F, but with methyl rather than ethyl substituents.
During the course of our continuing search for new drug leads from marine sponges collected off the Xisha Islands in the South China Sea, besides the previously reported two unusual polyketides simplextones A and B,20 we have recently isolated and identified six new polyketides, simplexolides A–E (1–5) and plakorfuran A (6), together with four known furanylidenic methyl esters (2Z,6R,8R,9E) [3-ethyl-5-(2-ethyl-hex-3-enyl)-6-ethyl-5H-furan-2-ylidene]-acetic acid methyl ester (7),15 methyl (2Z,6R,8S)-4,6-diethyl-3,6-epoxy-8-methyldeca-2,4-dienoate (8),21 methyl (2Z,6R,8S)-3,6-epoxy-4,6,8-triethyldodeca-2,4-dienoate (9),21 and (2Z,6R,8R,9E)[3-ethyl-5-(2-ethyl-hex-3-enyl)-6-methyl-5H-furan-2-ylidene]-acetic acid methyl ester (10)22 from the marine sponge Plakortis simplex. The primary difference between compounds 1–5 and the previously isolated plakortones A–F from Plakortis spp. is the opening of the tetrahydrofuran ring in 1–5. Herein, we describe the isolation, structure elucidation, and initial biological evaluation of compounds 1–6.
2. Results and discussion
A sample of the sponge P. simplex, collected off the Xisha Islands in the South China Sea, was exhaustively extracted with MeOH. The extract was suspended in water and successively extracted with n-hexane, CH2Cl2, EtOAc, and n-BuOH. The CH2Cl2-soluble extract was subjected to repeated column chromatography followed by reversed-phase preparative HPLC to afford compounds 1–10 as colorless oils.
The molecular formula of compound 1 was assigned as C17H30O3 by its HRESIMS (m/z 305.2094, [M+Na]+) and NMR data (Tables 1 and 2), displaying three degrees of unsaturation. The IR absorptions indicated the presence of hydroxyl (3451 cm−1), double bond (1655 cm−1), and ester carbonyl (1763 cm−1) groups. Analysis of the 1H NMR data (Table 1) and HSQC spectrum revealed the presence of four methyls, seven methylenes, two sp3 methines, one of which was oxygenated, one sp2 methine, one oxygenated sp3 quaternary carbon, and two sp2 quaternary carbons. By interpretation of 1H–1H COSY correlations, it was possible to establish five partial structures of consecutive proton systems: H-2/H-3, H-7/H-8/Me-17, H-11/Me-12, H-13/Me-14, and H-15/Me-16 (Fig. 1). The HMBC correlations from Me-14 (δH 1.01) to C-4 (δC 93.3), from H-13a (δH 1.64) to C-3 (δC 72.4) and C-5 (δC 120.1), and from H-3 (δH 4.22) to C-1 (δC 174.9) indicated the attachment of an ethyl group at C-4. Moreover, the HMBC correlations from Me-16 (δH 1.03) to C-6 (δC 93.3) and from H-15 (δH 2.13) to C-5 (δC 120.1), C-6 (δC 148.6), and C-7 (δC 44.9) demonstrated the linkage of C-5, C-7, and C-15 via C-6. The HMBC correlations from Me-17 (δH 0.85) to C-9 (δC 36.5), from H-9a (δH 1.11) to C-11 (δC 22.9), and from Me-12 (δH 0.89) to C-10 (δC 29.2) defined the structure of the C-1 to C-12 portion of the molecule. The C-1 ester carbonyl was confirmed to form a γ-butyrolactone with the oxygenated C-4, which was severely downfield shifted at δC 93.3, to consume the remaining one degree of unsaturation.
Table 1. 1H NMR data of compounds 1–6 in CDCl3.
| No. | 1a | 2b | 3b | 4b | 5b | 6b |
|---|---|---|---|---|---|---|
| 2 | 2.59, dd (18.0, 1.2) | 2.42, d (17.5) | 2.43, d (17.5) | 2.42, d (17.5) | 2.46, d (17.5) | 4.80, s |
| 2.85, dd (18.0, 6.0) | 2.80, dd (17.5, 5.5) | 2.83, dd (17.5, 5.5) | 2.82, dd (17.5, 5.5) | 2.83, dd (17.5, 5.5) | ||
| 3 | 4.22, d (5.4) | 4.30, d (5.0) | 4.32, t (4.0) | 4.34, t (4.0) | 4.31, d (5.5) | |
| 5 | 5.09, s | 5.14, s | 5.04 s | 5.15 s | 5.03, s | 6.22, s |
| 7 | 1.87, dd (13.8, 8.4) | 2.11, dd (14.0, 9.5) | 1.95, m | 2.15, dd (14.5, 7.5) | 1.70, dd (13.5, 9.0) | 2.53 d (14.0) |
| 2.17, m | 2.21, dd (13.5, 7.0) | 2.23, dd (14.5, 7.5) | 2.13, dd (13.5, 5.5) | 2.36 d (14.0) | ||
| 8 | 1.62, m | 1.65, m | 1.21, m | 1.45, m | 1.58, m | |
| 9 | 1.11, m | 1.14, m | 1.41, m | 1.17, m | 1.10, m | 5.17, d (9.0) |
| 1.23, m | 1.28, m | 1.27, m | 1.27, m | |||
| 10 | 1.32, m | 1.30, m | 1.23, m | 1.28, m | 1.27, m | 4.24, dt (9.0, 6.5) |
| 11 | 1.30, m | 1.27, m | 1.24, m | 1.28, m | 1.27, m | 1.55, m 1.39, m |
| 12 | 0.89, t (7.2) | 0.90, t (7.5) | 0.84, t (7.5) | 0.99, t (7.5) | 0.89, t (7.5) | 0.86, t (7.5) |
| 13 | 1.64, dq (7.2, 7.2) | 1.89, m | 1.88, m | 1.92, m | 1.89, dq (15.0, 7.5) | 2.15, m |
| 1.72, dq (7.2, 7.2) | 1.99, m | 2.02, m | 2.02, m | 2.03, dq (15.0, 7.5) | ||
| 14 | 1.01, t (7.2) | 0.95, t (7.5) | 0.99, t (7.5) | 0.98, t (7.5) | 1.01, t (7.5) | 1.15, t (7.5) |
| 15 | 2.13, m | 2.04, m | 2.10, dq (7.5, 7.5) | 2.03, m | 2.03, dq (14.0, 7.0) | 1.88, m |
| 2.27, dq (7.5, 7.5) | 2.33, dq (14.0, 7.0) | 1.76, m | ||||
| 16 | 1.03, t (7.2) | 0.99, t (7.5) | 1.00, t (7.5) | 1.00, t (7.5) | 0.98, t (7.5) | 0.81, t (7.5) |
| 17 | 0.85. d (6.6) | 0.84. d (6.5) | 1.25, m | 1.20, m | 0.81. d (6.5) | 2.09, m |
| 18 | 0.89, t (7.5) | 0.85, t (7.5) | 0.97, t (7.5) | |||
| 19 | 3.68, s |
Recorded at 600 MHz.
Recorded at 500 MHz.
Table 2. 13 C NMR data of compounds 1–6 in CDCl3.
| No. | 1a | 2b | 3b | 4b | 5b | 6b |
|---|---|---|---|---|---|---|
| 1 | 174.9 qC | 175.7 qC | 175.1 qC | 175.0 qC | 175.6 qC | 166.8 qC |
| 2 | 37.6 CH2 | 38.8 CH2 | 38.8 CH2 | 38.8 CH2 | 38.9 CH2 | 84.0 CH |
| 3 | 72.4 CH | 74.4 CH | 74.6 CH | 74.6 CH | 74.5 CH | 171.4 qC |
| 4 | 93.3 qC | 92.8 qC | 92.3 qC | 92.2 qC | 92.7 qC | 140.3 qC |
| 5 | 120.1 CH | 123.4 CH | 124.0 CH | 123.5 CH | 124.0 CH | 139.4 CH |
| 6 | 148.6 qC | 147.3 qC | 147.3 qC | 147.7 qC | 147.0 qC | 97.4 qC |
| 7 | 44.9 CH2 | 37.5 CH2 | 41.4 CH2 | 35.1 CH2 | 44.7 CH2 | 43.6 CH2 |
| 8 | 30.8 CH | 30.6 CH | 32.6 CH | 37.0 CH | 30.9 CH | 139.0 qC |
| 9 | 36.5 CH2 | 37.1 CH2 | 36.8 CH2 | 33.1 CH2 | 36.8 CH2 | 132.9 CH |
| 10 | 29.2CH2 | 29.2 CH2 | 28.8 CH2 | 29.2 CH2 | 29.3 CH2 | 69.6 CH |
| 11 | 22.9 CH2 | 22.9 CH2 | 23.1 CH2 | 23.1 CH2 | 23.0 CH2 | 30.4 CH2 |
| 12 | 14.0 CH3 | 14.1 CH3 | 14.1 CH3 | 14.1 CH3 | 14.1 CH3 | 9.7 CH3 |
| 13 | 31.8 CH2 | 27.4 CH2 | 27.2 CH2 | 27.4 CH2 | 27.1 CH2 | 18.5 CH2 |
| 14 | 8.1 CH3 | 8.3 CH3 | 8.4 CH3 | 8.4 CH3 | 8.4 CH3 | 12.1 CH3 |
| 15 | 23.9 CH2 | 29.8 CH2 | 23.4 CH2 | 30.0 CH2 | 23.5 CH2 | 30.9 CH2 |
| 16 | 12.9 CH3 | 13.0 CH3 | 12.8 CH3 | 13.1 CH3 | 12.8 CH3 | 8.1 CH3 |
| 17 | 19.6 CH3 | 19.1 CH3 | 25.5 CH2 | 26.1 CH2 | 19.5 CH3 | 24.5 CH2 |
| 18 | 10.6 CH3 | 11.6 CH3 | 13.6 CH3 | |||
| 19 | 50.6 CH3 |
Recorded at 150 MHz.
Recorded at 125 MHz.
Fig. 1.

COSY (
 ), key HMBC (→), and selected NOE (
), key HMBC (→), and selected NOE (
 ) correlations of 1,2, and 6.
) correlations of 1,2, and 6.
The relative configuration of 1 was established on the basis of NOESY data (Fig. 1). The crucial NOE correlations between H-3 (δH 4.22) and H-13a (δH 1.64) suggested that these protons were oriented on the same face of the γ-butyrolactone moiety. The E-geometry of the Δ5,6 double bond was deduced from a NOESY correlation between H-5 (δH 5.09) and H-7a (δH 1.87). The absolute configuration of C-3 was determined by applying the modified Mosher's method to the secondary hydroxyl group.23,24 Compound 1 was reacted with (R)-(−)- and (S)-(+)- α -methoxy- α -tri-fluoromethylphenylacetic acid (MTPA) chlorides to give MTPA esters 1a and 1b, respectively. A consistent distribution of positive and negative Δδ values around C-3 allowed the assignment of S-configuration for C-3 (Fig. 2). On the basis of the previously determined relative configuration, a 3S,4S configuration was assigned to simplexolide A (1). However, configuration of the remote stereocenter at C-8 was not determined.
Fig. 2.

ΔδS–R values (ppm) for the MTPA derivatives of 1, 2, and 6 in CDCl3.
Simplexolide B (2) was clearly an isomer of compound 1 on the basis of the identical molecular formula of C17H30O3 obtained by HRESIMS (m/z 305.2095 [M+Na]+). Comparison of the NMR data between 2 and 1 (Table 1) suggested that they had the same planar structures except for the configuration of the double bond at C-5–C-6, which was assigned as Z geometry on the basis of a NOESY correlation between H-5 and H-15 (Fig. 1). The NOE correlation between H-3 and H-5 indicated the same orientation of these two protons. The absolute configuration at C-3 was determined to be S by the modified Mosher's method (Fig. 2), implying that 1 and 2 were epimeric at C-4. On the basis of above-mentioned analysis, a 3S,4R configuration was assigned to simplexolide B (2).
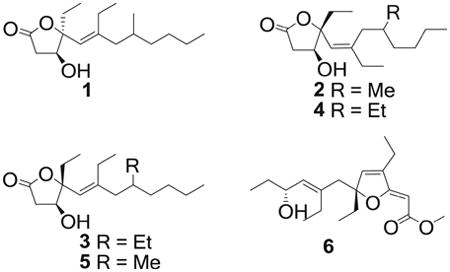
The molecular formula of compound 3 was established as C18H32O3 on the basis of HRESIMS (m/z 319.2247, [M+Na]+) and NMR data. The 1H and 13C NMR data indicated that 3 is a homolog of 2. The 1H NMR spectrum of 3 was almost identical to that of 2 except that a methyl group was replaced by an ethyl group at C-8 (Table 2). The geometry of the Δ5,6 double bond was assigned as E based on the NOESY correlation between H-5 and H-7. The crucial NOE correlation between H-3 and H-5 indicated that the relative configuration was assumed to be the same as 2. The CD spectrum of 3 displayed a positive Cotton effect at 196 nm, which was similar to that of 2 (Fig. 3), suggesting that the absolute configuration of 3 was the same as that of 2. Thus, a 3S,4R configuration was assigned to simplexolide C (3).
Fig. 3.
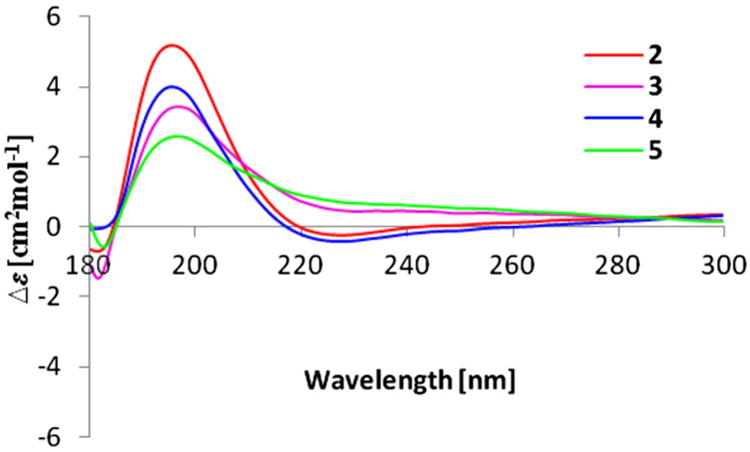
CD curves of compounds 2–5.
Compound 4 exhibited a quasi-molecular ion peak at m/z 319.2251 ([M+Na]+, calcd 319.2249), consistent with the molecular formula of C18H32O3. The 1H and 13C NMR data of 4 (Tables 1 and 2) were similar to those of 3. The geometry of the double bond at C-5 was assigned as Z, supported by the NOE correlation between H-5 and H-15. The CD spectrum of 4 revealed a pronounced positive Cotton effect with maxima observed at 196 nm (Fig. 3), and this closely matched the CD spectrum of 3. Therefore, the absolute configuration of simplexolide D (4) was assigned to be 3S,4R, consistent with those of 3.
The positive HRESIMS spectrum of simplexolide E (5) exhibited a pseudomolecular ion peak at m/z 305.2091 [M+Na]+, consistent with the molecular formula of C17H30O3, implying three degrees of unsaturation. Analysis of its NMR spectroscopic data revealed nearly identical structural features to those of 2, except for the geometry of the double bond at C-5, which was assigned as E on the basis of an NOE correlation between H-5 and H-7. The CD spectra of 5 showed a positive Cotton effect at 196 nm (Fig. 3), similar to that of 2, suggesting that the absolute configuration of 5 was 3S,4R.
The HRESIMS of compound 6 exhibited a pseudomolecular ion peak at m/z 345.2040 [M+Na]+ and established a molecular formula of C19H30O4, indicating five degrees of unsaturation. The 13C NMR (Table 2) displayed 19 carbon signals, which were identified by the assistance of the DEPT spectrum as 5 methyls, 5 methylenes, 3 sp2 methines, 1 oxygenated sp3 methine, and 5 quaternary carbons. The carbon resonances at δC 84.0 (C-6) and 171.4 (C-3) are identical as those for a furano α,β-unsaturated ester.14,15 Furthermore, an ester carbonyl was recognized as being present in 6 from its 13C NMR signal at C 166.8 (qC, C-1) and strong IR absorptions at 1752 cm−1. By interpretation of COSY correlations (Fig. 1), it was possible to establish the proton connections between H-15 and H-16, H-17 and H-18, H-9 and H-10, H-13 and H-14, and between H-11 and H-12. The connectivities of these partial structures were further established by the HMBC correlations (Fig. 1). The conjunction of C-10 and C-11 was elucidated on the basis of the HMBC correlation from H3-12 to C-10. Moreover, the HMBC correlations from H3-18 and H-10 to C-8 indicated the attachment between C-9 and C-17 via C-8. The HMBC correlations observed from H3-16 to C-6, and from H-15 and H-17 to C-7 indicated the connection of C-8 and C-15 through C-6 and C-7. The HMBC correlations of H3-19/C-1, H3-14 and H-2/C-4, H-5/C-3 and C-13, and H-15/C-5 further confirmed the existence of an unsaturated furan ester. With this assignment secured, the final oxymethine at C-10 had to be substituted with a hydroxyl group to satisfy the molecular formula. Finally, the E-geometry of the Δ8,9 double bond was deduced from a NOESY correlation between H-7 and H-9. The crucial NOE correlation between H-2 and H-13 indicated that the geometry of the Δ2,3 double bond is assumed to be Z (Fig. 1). This completed the assignment of the planar structure of plakorfuran A (6).
The absolute configuration at C-10 was determined to be R by applying a modified Mosher's ester method to the secondary hydroxyl group (Fig. 2). The absolute configuration at C-6 was determined by applying the CD exciton chirality method.25 The CD spectrum of 6 revealed a negative Cotton effect at λmax 285 nm (Δε −5.49) and a positive Cotton effect at λmax 205 nm (Δε 5.85) due to the transition interaction between two different chromophores of the unsaturated furan ester and the Δ8,9 double bond (Fig. 4), indicating a negative chirality for 6, thus concluding the absolute configuration as 6R,10R.
Fig. 4.
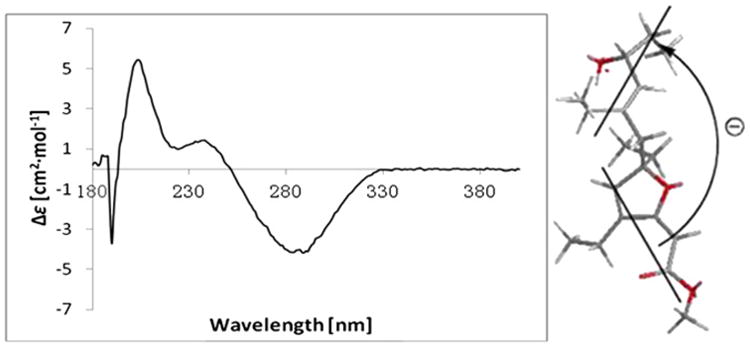
Experimental CD spectrum of 6. Bold lines denote the electric transition dipole of the chromophores for 6.
Simplexolides A–E (1–5) feature a previously unknown tetrahydrofuran opened plakortone skeleton. A possible biogenetic pathway is proposed as shown in Scheme 1. Acetate, propionate, and butyrate units are required to assemble the polyketide skeleton.26 The double bond at Δ3,4 is oxidized to an epoxide. With acyl carrier protein (ACP) domain releasing, the carboxylic acid cyclizes onto the opening epoxide togenerate a γ-lactone with the insertion of a hydroxyl.27 In this lactonization step, a pair of stereoisomers are formed at C-4 due to the nucleophilic attack of the hydroperoxy group onto C-4 of the epoxide group from both of the side.
Scheme 1.

Plausible biogenetic pathway for simplexolides A–E (1–5).
Compounds 1, 2, and 5–10 were tested for antifungal, cytotoxicity, antileismanial, and antimalarial activities (Table 3). Compounds 2, 5, and 7–10 showed weak to moderate antifungal activity against the fungi Cryptococcus neoformans. The cytotoxic activity of compounds 1 and 2 against five human cancer cell lines, HCT-116 (colon cancer), HeLa (cervical cancer), SW480 (colon cancer), QGY-7703 (hepatocarcinoma), and A549 (lung carcinoma) was also assayed. The results showed that compound 1 exhibited much stronger cytotoxicity against the five cancer cell lines, which was due to the stereochemistry influenced. Compounds 2 and 10 showed moderate antileismanial activity againt Leishmania donovani, while compounds 7–9 were a little weaker, and compounds 1, 5 and 6 were inactive. Furthermore, the antimalarial activity against chloroquine sensitive (D6, Sierra Leone) and resistant (W2, Indo China) strains of P. falciparum was tested. In this assay, only compound 10 displayed antimalarial activity against D6 and W2 with IC50 values of 2.0 and 2.0 μg/mL, respectively, and both with SI [IC50(fibroblast)/IC50(parasite)] of 2.4, while the other compounds were inactive.
Table 3. Biological activities of 1, 2 and 5–10.
| Compound | Antifungal IC50 (μg/ml)/MIC (μg/ml) | Cytotoxicity IC50 (μg/ml) | Antileismanial IC50 (μg/ml) | Antimalarial IC50 (μg/ml) | |||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| HCT-116 | HeLa | SW480 | QGY-7703 | A549 | D6 | W2 | |||
| 1 | – | 9.23 | 27.43 | 17.08 | 26.53 | 16.79 | >40 | >50 | >50 |
| 2 | 10.49/20 | 4.34 | 6.51 | 11.28 | 17.98 | 10.77 | 13.82 | >50 | >50 |
| 5 | 3.66 | >50 | – | – | – | – | >40 | >50 | >50 |
| 6 | – | >50 | – | – | – | – | >40 | >50 | >50 |
| 7 | 20.00 | – | – | – | – | – | 38.56 | >50 | >50 |
| 8 | 15.30 | – | – | – | – | – | 31.24 | >50 | >50 |
| 9 | 20.00 | – | – | – | – | – | 20.20 | >50 | >50 |
| 10 | 11.72 | – | – | – | – | – | 7.11 | 2.0 | 2.0 |
| Amphotericin B | 0.317 | – | – | – | – | – | 0.34 | – | – |
| Camptothecin | – | 3.22 | 2.43 | 8.26 | 1.41 | 0.81 | – | – | – |
| Pentamidine | – | – | – | – | – | – | 1.62 | – | – |
| Chloroquine | – | – | – | – | – | – | – | 0.06 | 0.83 |
3. Experimental section
3.1. General experimental procedures
Optical rotations were determined with a Perkin–Elmer 341 polarimeter equipped with a 1 mm cell. The CD spectra were obtained with a JASCO J-715 spectropolarimeter. IR spectra were recorded on a Bruker Vector 22 spectrometer using KBr pellets. The NMR experiments were conducted on Bruker AVANCE-600 and Bruker AMX-500 MHz instruments in CDCl3 with TMS as an internal standard. HRESIMS and ESIMS were obtained on a Q-Tof micro YA019 mass spectrometer. Reversed-phase HPLC was performed using Sunfire C18 (5 μm) and YMC-Pack Pro C18 RS (5 μm) columns with a Waters 1525/2998 liquid chromatograph. Column chromatography (CC) was carried out on silica gel 60 (200–300 mesh; Yantai, China), and Sephadex LH-20 (Pharmacia). TLC was carried out using HSGF 254 plates and visualized by spraying with anisaldehyde/H2SO4 reagent. Samples were weighed on a Shushi analytical balance.
3.2. Animal material
The sponge specimen was collected around Yongxing Island and seven connected islets in the South China Sea in June 2007, and were identified by Prof. Jin-He Li (Institute of Oceanology, Chinese Academy of Sciences, China). A voucher sample (No. B-3) was deposited in the Laboratory of Marine Drugs, Department of Pharmacy, Changzheng Hospital, Second Military Medical University, China.
3.3. Extraction and isolation
The air-dried and powdered sponge (2.0 kg, dry weight) was extracted with MeOH, and the crude extract was concentrated under reduced pressure at 45 °C to yield 500 g of residue. The residue was then extracted successively with n-hexane, CH2Cl2, EtOAc, and n-BuOH. The CH2Cl2 extract (41 g) was separated by vacuum liquid chromatography (VLC) on silica gel using CH2Cl2/MeOH as the eluent to give three fractions (Fr. A–C). The lower polarity part fraction A (CH2Cl2/MeOH 25:1) was subjected to VLC eluting with petroleum ether/EtOAc (PE) to give four subfractions (Fr. A1–A4). Fraction A3 was purified by reversed-phase preparative HPLC (Sunfire C18, 5 μm, 10×250 mm) to yield 31.7 mg of compound 1 (CH3OH/H2O 95:5, 2.0 mL/min, UV detection at 210 nm, tR=17.0 min), 22.1 mg of compound 6 (CH3CN/H2O 50:50, 2.0 mL/min, UV detection at 282 nm, tR=17.2 min), a mixture of compounds 3 and 4 (CH3CN/H2O 70:30, 2.0 mL/min, UV detection at 200 nm, tR=29.0 min), and a mixture of compounds 2 and 5 (CH3CN/H2O 70:30, 2.0 mL/min, UV detection at 200 nm, tR=24.4 min). The mixture of compounds 3 and 4 and the mixture of compounds 2 and 5 were further purified by reversed-phase preparative HPLC (YMC-Pack Pro C18 RS, 5 μm, 10×250 mm) to yield 21.3 mg of compound 3 (CH3CN/H2O 85:15, 2.0 mL/min, UV detection at 200 nm, tR=18.4 min), 1.8 mg of compound 5 (CH3CN/H2O 85:15, 2.0 mL/min, UV detection at 200 nm tR=17.4 min), 8.2 mg of compound 2 (CH3CN/H2O 85:15, 2.0 mL/min, UV detection at 200 nm, tR=66.9 min), and 52.1 mg of compound 5 (H3CN/H2O 85:15, 2.0 mL/min, UV detection at 200 nm, tR=62.6 min). Similarly, the four known compounds 7 (22.0 mg), 8 (15.1 mg), 9 (8.3 mg), and 10 (2.2 mg) were obtained from fraction A1.
3.3.1. Simplexolide A(1)
Colorless oil; (c 0.215, MeOH); 1R (KBr) νmax 3451, 2960, 2927, 2873, 1763, 1655, 1464, 1412, 1378, 1343, 1290, 1248, 1210, 1182, 1119, 1103, 1090, 1071, 1014, 980, 958, 926, 885, 800 cm−1; 1H NMR (CDCl3, 600 MHz) and 13C NMR (CDCl3,150 MHz) data, see Tables 1 and 2; HRESIMS m/z 305.2094 [M+Na]+ (calcd for C17H30O3Na, 305.2093). CD spectrum (c 1.1 mg/mL, CH3CN), 193 nm (Δε -0.65), 222 nm (Δε 0.84).
3.3.2. Simplexolide B (2)
Colorless oil; (c 0.120, MeOH); IR (KBr) νmax 3439, 2962, 2930, 2874, 1758, 1655, 1460, 1400, 1379, 1341, 1280, 1250, 1208, 1165, 1111, 1063, 1023, 972, 954, 916, 879, 799 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) data, see Tables 1 and 2; HRESIMS m/z 305.2095 [M+Na]+ (calcd for C17H30O3Na, 305.2093). CD spectrum (c 0.5 mg/mL, CH3CN), 196 nm (Δε+5.16).
3.3.3. Simplexolide C (3)
Colorless oil; (c 0.090, MeOH); IR (KBr) νmax 3444, 2962, 2928, 2874, 1758, 1655, 1464, 1399, 1379, 1341, 1287, 1251, 1203, 1164, 1113, 1099, 1022, 975, 951, 915, 879, 800 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) data, see Tables 1 and 2; HRESIMS m/z 319.2247 [M+Na]+ (calcd for C18H32O3Na, 319.2249). CD spectrum (c 0.5 mg/mL, CH3CN), 196 nm (Δε +3.43).
3.3.4. Simplexolide D (4)
Colorless oil; (c 0.130, MeOH); IR (KBr) νmax 3448, 2962, 2930, 2874, 1759, 1655, 1460, 1400, 1379, 1340, 1280, 1250, 1205, 1163, 1111, 1036, 1024, 975, 955, 914, 798cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) data, see Tables 1 and 2; HRESIMS m/z 319.2251 [M+Na]+ (calcd for C18H32O3Na, 319.2249). CD spectrum (c 0.9 mg/mL, CH3CN), 196 nm (Δε +3.99).
3.3.5. Simplexolide E (5)
Colorless oil; (c 0.170, MeOH); IR (KBr) νmax 3446, 2961, 2927, 2874, 1758, 1655, 1465, 1400, 1379, 1342, 1288, 1252, 1202, 1166, 1112, 1063, 1021, 974, 951, 916, 879, 800 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) data, see Tables 1 and 2; HRESIMS m/z 305.2091 [M+Na]+ (calcd for C17H30O3Na, 305.2093). CD spectrum (c 0.7 mg/mL, CH3CN), 196 nm (Δε + 2.57).
3.3.6. Plakorfuran A (6)
Colorless oil; (c 0.085, MeOH); 1H NMR(CDCl3, 500 MHz) and 13C NMR(CDCl3,125 MHz) data, see Tables 1 and 2; HRESIMS m/z 345.2040 [M+Na]+ (C19H30O4Na, calcd 345.2042). CD spectrum (c 0.85 mg/mL, CH3CN), 205 nm ((Δε 5.85), 239 nm ((Δε 1.70), 285 nm ((Δε −5.49).
3.4. Preparation of MTPA esters 1a and 1b
Simplexolide A (1; 1.0 mg and 0.8 mg, respectively) was reacted with R-(−)- or S-(+)-MTPACl (15 μL) in freshly distilled dry pyridine (500 μL) and stirred under N2 at room temperature for 18 h, respectively, and the solvent was removed in vacuo. The products were purified by mini-column chromatography on silica gel (200 mesh, petroleum ether/EtOAc, 1:1) to afford S-(−)- and R-(+)-MTPA esters 1a and 1b, respectively.
3.5. Preparation of MTPA esters 2a and 2b
Simplexolide C (3; 1 mg each) was similarly processed to give S-(−)- and R-(+)-MTPA esters 3a and 3b, respectively.
3.6. Preparation of MTPA esters 6a and 6b
Plakorfuran A (6; 1 mg each) was similarly processed to give S-(−)- and R-(+)-MTPA esters 6a and 6b, respectively.
3.7. Biological tests
Antifungal assay against C. neoformans was performed as described by Ikhlas A. Khan et al.28 Amphotericin B was used as the positive control. Cytotoxicity was determined against human cancer cell lines HCT-116 (colon cancer), HeLa (cervical cancer), SW480 (colon cancer), QGY-7703 (hepatocarcinoma), and A549 (lung carcinoma) using the MTT assay method. Camptothecin was used as the positive control. The experimental details of this assay were carried out according to a previously described procedure.29 Antimalarial activity was determined in vitro against chloroquine sensitive (D6, Sierra Leone) and resistant (W2, Indo China) strains of P. falciparum by measuring plasmodial LDH activity.30 Chloroquine was used as the positive control. In vitro antileishmanial activity was tested on a culture of L. donovani promastigotes. In a 96-well microplate assay, test compounds were diluted to an appropriate concentration and added to the Leishmania promastigotes culture (2×106 cell/mL). The plates were incubated at 26 °C for 72 h and growth of Leishmania promastigotes was determined by Alamar blue assay.31 Pentamidine and Amphotericin B were used as the standard antileishmanial agents. The growth inhibition curve was used to compute the IC50 value for each compound.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81072573), the Major Program of Modernization of Chinese Medicine (STCSM, 09dZ1975800), the NIH, NIAID, Division of AIDS (No. AI 27094), and the USDA Agricultural Research Service Specific Cooperative Agreement (No. 58-6408-2-0009). The authors thank Dr. Melissa R. Jacob and Ms. Marsha A. Wright for antifungal assay assistance, and Dr. Shabana Khan for antimalarial assay assistance.
Footnotes
Supplementary data: Supplementary data associated with this article can be found in the online version, at doi:10.1016/j.tet.2012.04.025. These data include MOL files and InChiKeys of the most important compounds described in this article.
References and notes
- 1.Higgs MD, Faulkner DJ. J Org Chem. 1978;43:3454–3457. [Google Scholar]
- 2.Braekman JC, Daloze D, De Groote S, Fernandes JB, Van Soest RWM. J Nat Prod. 1998;61:1038–1042. doi: 10.1021/np980096z. [DOI] [PubMed] [Google Scholar]
- 3.Hu JF, Gao HF, Kelly M, Hamann MT. Tetrahedron. 2001;57:9379–9383. [Google Scholar]
- 4.Kossuga MH, Nascimento AM, Reimao JQ, Tempone AG, Taniwaki NN, Veloso K, Ferreira AG, Cavalcanti BC, Pessoa C, Moraes MO, Mayer AMS, Hajdu E, Berlinck RGS. J Nat Prod. 2008;71:334–339. doi: 10.1021/np0705256. [DOI] [PubMed] [Google Scholar]
- 5.Feng Y, Davis RA, Sykes M, Avery VM, Camp D, Quinn RJ. J Nat Prod. 2010;73:716–719. doi: 10.1021/np900535z. [DOI] [PubMed] [Google Scholar]
- 6.Rahm F, Hayes P, Kitching W. Heterocycles. 2004;64:523–575. [Google Scholar]
- 7.Qureshi A, Salva J, Harper MK, Faulkner DJ. J Nat Prod. 1998;61:1539–1542. doi: 10.1021/np9802724. [DOI] [PubMed] [Google Scholar]
- 8.Yong KWL, De Voss JJ, Hooper JNA, Garson MJ. J Nat Prod. 2011;74:194–207. doi: 10.1021/np100620x. [DOI] [PubMed] [Google Scholar]
- 9.Rudi A, Afanii R, Gravalos LG, Aknin M, Gaydou E, Vacelet J, Kashman Y. J Nat Prod. 2003;66:682–685. doi: 10.1021/np020589a. [DOI] [PubMed] [Google Scholar]
- 10.Perry TL, Dickerson A, Khan AA, Kondru RK, Beratan DN, Wipf P, Kelly M, Hamann MT. Tetrahedron. 2001;57:1483–1487. [Google Scholar]
- 11.Patil AD, Freyer AJ, Bean MF, Carte BK, Westley JW, Johnson RK, Lahouratate P. Tetrahedron. 1996;52:377–394. [Google Scholar]
- 12.Zampella A, Giannini C, Debitus C, D'Auria MV. Tetrahedron. 2001;57:257–263. [Google Scholar]
- 13.De Guzman FS, Schmitz FJ. J Nat Prod. 1990;53:926–931. [Google Scholar]
- 14.Cafieri F, Fattorusso E, Taglialatela-Scafati O, Ianaro A. Tetrahedron. 1999;55:13831–13840. [Google Scholar]
- 15.Stierle DB, Faulkner DJ. J Org Chem. 1980;45:3396–3401. [Google Scholar]
- 16.Gochfeld DJ, Hamann MT. J Nat Prod. 2001;64:1477–1479. doi: 10.1021/np010216u. [DOI] [PubMed] [Google Scholar]
- 17.Xie XG, Wu XW, Lee HK, Peng XS, Wong HN. Chem —Eur J. 2010;16:6933–6941. doi: 10.1002/chem.201000189. [DOI] [PubMed] [Google Scholar]
- 18.Hayes PY, Kitching W. J Am Chem Soc. 2002;124:9718–9719. doi: 10.1021/ja026728s. [DOI] [PubMed] [Google Scholar]
- 19.Akiyama M, Isoda Y, Nishimoto M, Narazaki M, Oka H, Kuboki A, Ohira S. Tetrahedron Lett. 2006;47:2287–2290. [Google Scholar]
- 20.Liu XF, Song YL, Zhang HJ, Yang F, Yu HB, Jiao WH, Piao SJ, Chen WS, Lin HW. Org Lett. 2011;13:3154–3157. doi: 10.1021/ol201055w. [DOI] [PubMed] [Google Scholar]
- 21.Compagnone RS, Pina IC, Rangel HR, Dagger F, Suarez AI, Venkata Rami Reddy M, John Faulkner D. Tetrahedron. 1998;54:3057–3068. [Google Scholar]
- 22.Epifanio R, Pinheiro L, Alves N. J Braz Chem Soc. 2005;16:1367–1371. [Google Scholar]
- 23.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 24.Hoye TR, Jeffrey CS, Shao F. Nat Protoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 25.Cai G, Bozhkova N, Odingo J, Berova N, Nakanishi K. J Am Chem Soc. 1993;115:7192–7198. [Google Scholar]
- 26.Huang XH, van Soest R, Roberge M, Andersen R. J Org Lett. 2003;6:75–78. doi: 10.1021/ol0361047. [DOI] [PubMed] [Google Scholar]
- 27.Richard GB, Mary JG, James S. J Chem Soc, Chem Commun. 1980:1165–1167. [Google Scholar]
- 28.Ma G, Khan SI, Jacob MR, Tekwani BL, Li Z, Pasco DS, Walker LA, Khan IA. Antimicrob Agents Chemother. 2004;48:4450–4452. doi: 10.1128/AAC.48.11.4450-4452.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carmichael J, De Graff WG, Gazdar AF, Minna JD, Mitchell JB. Cancer Res. 1987;47:936–942. [PubMed] [Google Scholar]
- 30.Muhammad I, Bedir E, Khan SI, Tekwani BL, Khan IA, Takamatsu S, Pelletier J, Walker LA. J Nat Prod. 2004;67:772–777. doi: 10.1021/np030524n. [DOI] [PubMed] [Google Scholar]
- 31.Mikus J, Steverding D. Parasitol Int. 2000;48:265–269. doi: 10.1016/s1383-5769(99)00020-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.