

Abstract
Purpose
To report a case of severe, bilateral, rapidly progressing peripheral retinal non-perfusion caused by underlying aplastic anemia.
Methods
An interventional case report.
Results
A 4-year-old female presented with decreased visual acuity. On clinical examination she was found to have RAPD, elevated intraocular pressure, 360 degree rubeosis, vitreous hemorrhage, severe exudative retinal detachment and telangiectasia with severe peripheral retinal non-perfusion. Laboratory workup was significant for pancytopenia and a bone marrow biopsy showed extreme hypocellularity with no malignant cells. The patient was diagnosed with primary aplastic anemia. She developed dramatic progression of retinal non-perfusion in the left eye, as well as in the fellow right eye. This bilateral retinopathy was poorly responsive to aggressive management, which included laser photocoagulation and intravitreal injections of anti-VEGF medications.
Conclusion
Asymmetric, bilateral quickly progressing peripheral retinal ischemia, in conjunction with pancytopenia and otherwise negative workup may be related to underlying bone marrow failure and aplastic anemia.
Keywords: aplastic anemia, Coat’s, peripheral retinal non-perfusion
Case report
A four year-old girl presented to the emergency room with a dilated left pupil. Visual acuity was 20/40 OD and counting fingers OS. Pupillary exam revealed a fixed and dilated left pupil with a relative afferent pupillary defect. Intraocular pressure was 11 mmHg OD and 29 mmHg OS. Anterior segment examination of the right eye was completely normal. In the left eye, band keratopathy, trace anterior chamber flare, and 360-degree iris rubeosis with ectropion uveae was noted. Dilated fundus examination in the left eye (Figure 1) revealed a slight vitreous hemorrhage; the optic disc was edematous and had overlaying neovascularization; the macula had a diffuse exudative detachment extending through the fovea; and the vasculature was engorged. The peripheral retina was detached due to an extensive exudative process associated with telangiectatic blood vessels and hemorrhages.
Figure 1.
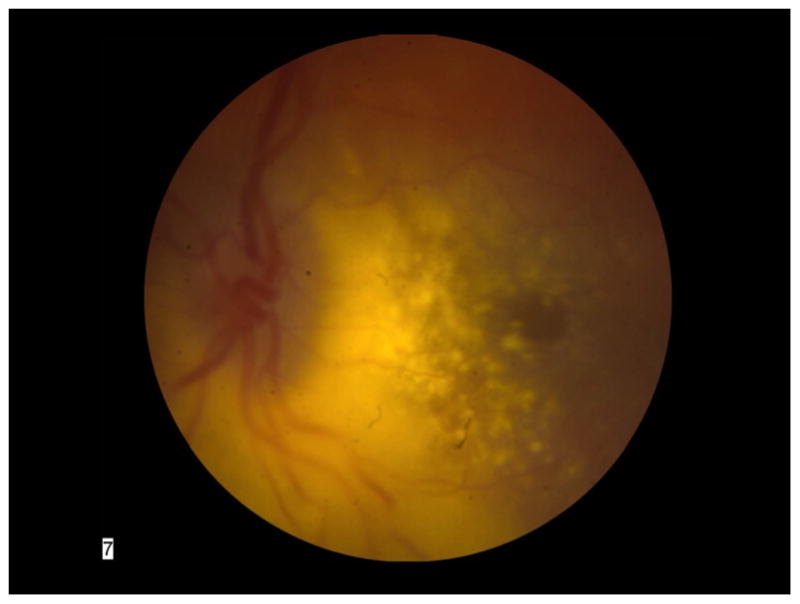
Color fundus photograph of the patient’s left eye showing severe exudative retinal detachment involving the macula; an edematous optic nerve; and dilated, engorged blood vessels.
Examination under anesthesia (EUA) revealed subtle vascular changes and retinal hemorrhages in the temporal periphery of the fellow right eye. On fluorescein angiography, this area of the right fundus demonstrated a telangiectatic microvasculature with peripheral retinal non-perfusion (Figure 2).
Figure 2.
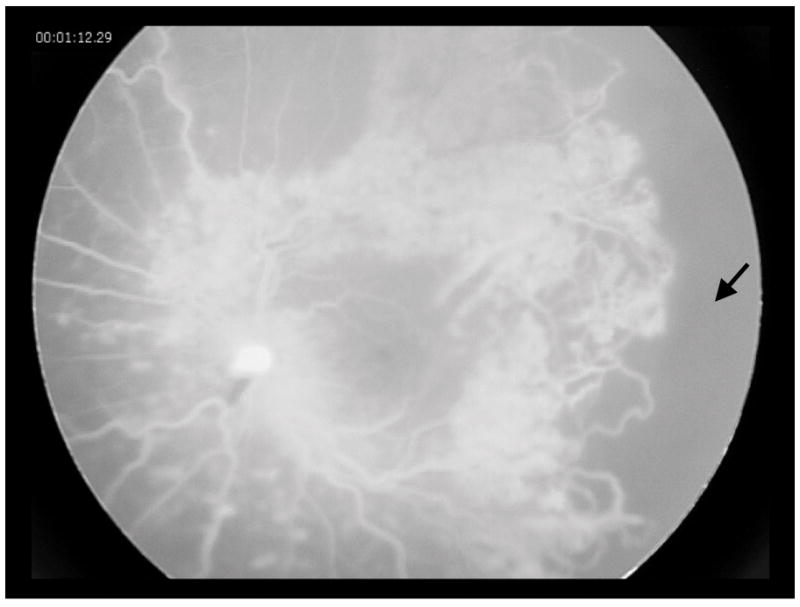
Fluorescein angiography of the right eye, showing peripheral vascular telangiectasia and capillary drop out (arrow) in the temporal retina.
In the left eye, fluorescein angiography showed normal transit times, blockage associated with the exudates, and hyperfluorescence of the disc with leakage in the mid and late phases. The vasculature was teleangiectatic with multiple areas of capillary drop out and aneurysmal dilatation (Figure 3). There was diffuse leakage evident from retinal neovascularization and microaneurysms in the late phases.
Figure 3.
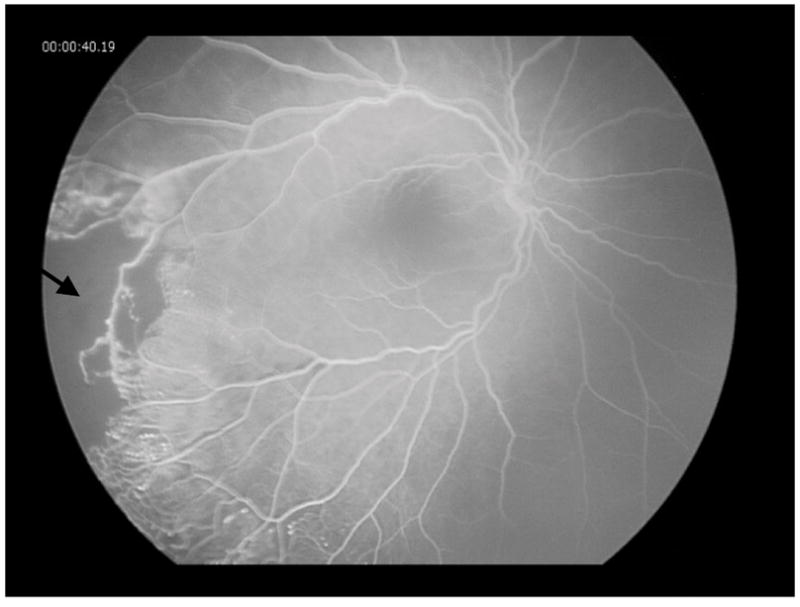
Fluorescein angiography of the patient’s left eye showing vascular telangiectasia, microaneurysms, collateral vessels, and severe retinal non-perfusion (arrow) with fluorescein leakage caused by neovascularization of the disc and retina.
Ultrasonography of the left eye confirmed an exudative retinal detachment from twelve to ten o’clock associated with diffuse chorioretinal thickening. There was no evidence of a discrete mass or calcifications.
Laboratory workup was significant for pancytopenia (white blood cell count 3.3 thous/cu.mm [5.5–15.5]; hemoglobin 10.6 g/dl [11.5–14.5]; platelets 94 thous/cu.mm [150–440]); no eosinophils or blasts were noted in the smear. A bone marrow biopsy showed extreme hypocellularity but no malignant cells.
The patient’s mother and maternal grandmother had normal eye examinations.
Karyotype and DNA copy number variation analysis of the patient were normal. Genetic testing for dyskeratosis congenitii, Norrie’s disease and familial exudative vitreoretinopathy (FEVR) did not identify any disease causing variants.
A full dermatologic evaluation was normal. A bone scan and audiogram were normal.
The patient remained pancytopenic throughout her course due to a presumed primary aplastic anemia. Her retinopathy progressed dramatically with a rapid increase in areas of retinal non-perfusion and capillary drop out in the left eye as, well as in the fellow right eye (Figure 4), despite extensive laser photocoagulation and intravitreal injections of anti-VEGF medications. Increasingly severe proliferative disease with associated vitreous hemorrhage and traction necessitated scleral buckle, vitrectomy, membranectomy, silicone oil, and lensectomy in the right eye. Cyclophotocoagulation was ultimately required to control the pressure in the left eye. The patient was given multiple blood transfusions and eventually was treated with hematopoietic stem cell transplantation for the refractory aplastic anemia. Her most recent vision was light perception in the right eye and no light perception in the left eye.
Figure 4.
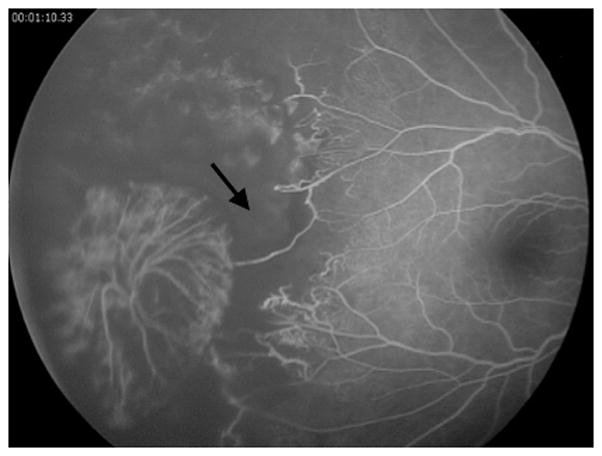
Fluorescein angiography of the right eye, showing rapid progression of retinal non-perfusion (arrow) compared to initial presentation 3 months previously.
Discussion
The initial presentation included anterior segment neovascularization, neovascular glaucoma, vitreous hemorrhage, optic disc edema with neovascularization of the disc, a large exudative retinal detachment, teleangiectatic blood vessels and retinal hemorrhages. These findings initially led to a broad differential diagnosis that included diffuse infiltrating retinoblastoma, Coat’s disease, incontinentia pigmenti and FEVR. The evidence of asymmetric bilateral involvement, in conjunction with pancytopenia, in the setting of a very rapid progression with an otherwise negative work-up suggested that the retinal vasculopathy may be due to the underlying bone marrow failure and aplastic anemia.
Retinal vasculopathies associated with bone marrow failure and aplastic anemia have been described in the past. Mansour and Lee et al. described retinal findings in 37 patients, including 30 patients with bilateral retinal hemorrhages in a cohort of 156 patients with aplastic anemia (1). A literature review (1958–2010) of 200 aplastic anemia cases revealed retinal hemorrhages in 56%, subhyaloid or vitreous hemorrhage in 9%, and peripheral retinal vasculopathy in 5.5%.
Other rare, genetic syndromes with bone marrow failure have been reported to have an associated peripheral retinal vasculopathy. For instance, peripheral retinal neovascularization, areas of retinal capillary nonperfusion and peripheral ischemic retinopathy were described in a patient with Fanconi anemia, a type of aplastic anemia (2). Patients with the telomere biology syndrome of dyskeratosis congenita have been reported to have retinal neovascularization, retinal detachments, vascular sheathing and exudative retinopathy (3). These individuals are at increased risk for progressive bone marrow failure. Revesz and al-Gazali et al., described a six-month-old male infant with bilateral exudative retinopathy, aplastic anemia, intrauterine growth retardation, reticulate skin pigmentation, and central nervous system abnormalities (4). Exudative retinopathy appears to be specific to Revesz syndrome which is now recognized as a severe variant of dyskeratosis congenita. Kajtar and Mehes reported a case of a two-year-old girl with bilateral exudative retinopathy, aplastic anemia, a variety of integumentary findings, cerebellar hypoplasia, mental retardation, and intrauterine growth retardation (5). Our patient did not have any dermatologic findings.
A mechanistic link between aplastic anemia and bilateral retinopathy is biologically plausible. In patients with a dysplastic bone marrow, a paucity or absence of progenitor cell populations could result in the vascular pathology seen in our patient and those described in previous papers. If this is the case, the retina of these patients may benefit by not only addressing the local ischemia through laser photocoagulation and anti-VEGF drugs but also through a more systemic intervention like bone marrow transplant.
Summary.
We report a case of a four-year-old girl with severe, bilateral, rapidly progressing peripheral retinal non-perfusion causing secondary exudation, severe neovascularization of the anterior and posterior segment, vitreous hemorrhage and retinal detachment presumed due to underlying aplastic anemia.
Acknowledgments
The authors would like to thank the patient and her family. This study was supported by funding from Research to Prevent Blindness and NEI core grant EY001792.
References
- 1.Mansour AM, Lee JW, Yahng SA, Kim KS, Shahin M, Hamerschlak N, et al. Ocular manifestations of idiopathic aplastic anemia: retrospective study and literature review. Clinical ophthalmology. 2014;8:777–87. doi: 10.2147/OPTH.S62163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yahia SB, Touffahi SA, Zeghidi H, Zaouali S, Khairallah M. Ocular neovascularization in a patient with Fanconi anemia. Can J Ophthalmol. 2006 Dec;41(6):778–9. doi: 10.3129/i06-078. [DOI] [PubMed] [Google Scholar]
- 3.Tsilou ET, Giri N, Weinstein S, Mueller C, Savage SA, Alter BP. Ocular and orbital manifestations of the inherited bone marrow failure syndromes: Fanconi anemia and dyskeratosis congenita. Ophthalmology. 2010 Mar;117(3):615–22. doi: 10.1016/j.ophtha.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Revesz T, Fletcher S, al-Gazali LI, DeBuse P. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: a new syndrome? Journal of medical genetics. 1992 Sep;29(9):673–5. doi: 10.1136/jmg.29.9.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kajtar P, Mehes K. Bilateral coats retinopathy associated with aplastic anaemia and mild dyskeratotic signs. American journal of medical genetics. 1994 Feb 15;49(4):374–7. doi: 10.1002/ajmg.1320490404. [DOI] [PubMed] [Google Scholar]