

Abstract
Dynamic alterations of the extracellular matrix in response to injury directly modulate inflammation and consequently the promotion and resolution of disease. During inflammation, hyaluronan (HA) is increased at sites of inflammation where it may be covalently modified with the heavy chains (HC) of inter-α-trypsin inhibitor. Deposition of this unique, pathological form of HA (HC-HA) leads to the formation of cable-like structures that promote adhesion of leukocytes. Naive mononuclear leukocytes bind specifically to inflammation-associated HA matrices but do not adhere to HA constitutively expressed under homeostatic conditions. In this study, we have directly investigated a role for the blood-coagulation protease thrombin in regulating the adhesion of monocytic cells to smooth muscle cells producing an inflammatory matrix. Our data demonstrate that the proteolytic activity of thrombin negatively regulates the adhesion of monocytes to an inflammatory HC-HA complex. This effect is independent of protease-activated receptor activation but requires proteolytic activity toward a novel substrate. Components of HC-HA complexes were predicted to contain conserved thrombin-susceptible cleavage sites based on sequence analysis, and heavy chain 1 (HC1) was confirmed to be a substrate of thrombin. Thrombin treatment is sufficient to cleave HC1 associated with either cell-surface HA or serum inter-α-trypsin inhibitor. Furthermore, thrombin treatment of the inflammatory matrix leads to dissolution of HC-HA cable structures and abolishes leukocyte adhesion. These data establish a novel mechanism whereby thrombin cleavage of HC1 regulates the adhesive properties of an inflammatory HA matrix.
Keywords: extracellular matrix, glycobiology, glycosaminoglycan, hyaluronan, inflammation, thrombin, heavy-chain hyaluronan complex, inflammation-associated matrix, inter-alpha-inhibitor
Introduction
The extracellular matrix (ECM)2 is a complex environment that functions as a physical scaffold for cells but also provides molecular signals affecting cell shape, proliferation, differentiation, apoptosis, and adhesion. Composed of proteins, proteoglycans, and glycosaminoglycans, the ECM influences cellular function through interactions with cell surface receptors. In response to inflammatory cytokines, the composition of the ECM can be modified by deposition of inflammation-induced matrix components and secretion of matrix proteases (1). The remodeled ECM of inflamed tissues propagates the inflammatory response by altering the behavior of both tissue-resident and infiltrating cells, which amplify immune cell recruitment and activation (2, 3).
Hyaluronan (HA) is a glycosaminoglycan consisting of repeating disaccharide units of glucuronic acid and N-acetylglucosamine joined by alternating (β-1,3 and β-1,4) glycosidic linkages. As an integral component of the ECM, HA exists as a large, hydrophilic polymer often with an average molecular mass of ∼107 Da. Increased accumulation of HA has been reported in numerous inflammatory pathologies, including rheumatoid arthritis (4, 5), atherosclerosis (6), lung disease (7, 8), diabetes (9, 10), and inflammatory bowel disease (IBD) (11). Initially, HA was considered merely an inert space-filling substance, but it has become increasingly clear that HA has multiple roles in inflammation (12).
During inflammation and development (13, 14), HA can be covalently modified with the heavy chains (HCs) of inter-α-inhibitor (IαI) to form a biologically distinct HC-HA complex. Biosynthesis of the IαI family proteins primarily occurs in the liver and results from stepwise assembly in the Golgi and involves several proteolytic processing events (15–17). IαI is unique among proteoglycans in that it is composed of at least two different proteins (bikunin and HCs) covalently attached to the same chondroitin sulfate (CS) chain. HC1, HC2, and HC3 contain a conserved Golgi-processing site in their C terminus and are joined to the CS of bikunin by an ester linkage (18–20). Following assembly, IαI (HC1, HC2, and bikunin) and pre-αI (PαI) (HC3 and bikunin) are secreted into circulation and may reach concentrations of 0.15–0.5 mg/ml (21). Transfer of HCs (from IαI to HA) is catalyzed by the enzyme tumor necrosis factor-stimulated gene 6 (TSG-6) (22, 23), which in most tissues is only expressed during inflammation (24). This evolutionarily conserved reaction (25) results in a unique, pathological form of HA. Inflammation-associated HC-HA has been observed as large “cable”-like structures that promote leukocyte adhesion to the ECM and may contribute to tissue destruction (26–29). Although the HCs are homologous, their distinct biological functions and how they are regulated remain unclear.
The serum protease thrombin is arguably the most extensively studied of all human proteases, functioning both in blood coagulation and as the most potent activator of circulating platelets. In addition to hemostasis, thrombin also has roles in tissue repair (30), development (31), and in inflammation (32). Many of the effects of thrombin on platelets and endothelial cells are elicited through interaction with a family of transmembrane G-protein-coupled receptors, termed protease-activated receptors (PARs) (33). Thrombin binding results in cleavage of PAR-1, the predominant thrombin receptor on endothelial cells. Cleavage generates a new N terminus, also known as the thrombin receptor-activating peptide (TRAP), which acts as a tethered ligand capable of inducing PAR activation (34). Thrombin-mediated activation of PARs on platelets and endothelial cells has been shown to be a central regulator of leukocyte recruitment and adhesion (35–37).
A growing body of evidence suggests that inflammation and hemostasis are closely linked processes capable of amplifying each other (38). In IBD, several elements of the coagulation cascade are altered, and there is evidence for accelerated thrombin generation (39). We hypothesized that thrombin-mediated activation of extravascular tissues such as intestinal mucosa smooth muscle cells (M-SMCs) may be involved in the enhanced leukocyte adhesion seen in IBD. Surprisingly, our data demonstrate that thrombin treatment of poly(I·C)-stimulated M-SMCs negatively regulates leukocyte binding. Our data show that the proteolytic activity of thrombin is required for this effect and that proteolytic cleavage of HC1 results in dissolution of the leukocyte adhesive HC-HA matrix.
Results
Thrombin Abolishes Poly(I:C)-induced M-SMC Leukocyte Adhesion
We previously reported that naive mononuclear leukocytes (e.g. U937 cells, monocytes, and lymphocytes) bind to M-SMCs treated with virus or poly(I·C) specifically via HA cable structures (26). These ECM elements are distinct from native HA because of their modification with hyaluronan-binding proteins, including HC proteins donated by IαI (5, 11, 29, 40, 41) as well as versican (10, 28, 42). The pathological inflammation-associated form of HA is significantly increased in the submucosa of colon tissue in patients with IBD (11) as well as in mice undergoing experimental colitis (43), and deposition of this unique matrix requires vascular leakage of serum-derived IαI into inflamed tissues (11, 23). Thrombin is a serum protease that is generated at an accelerated rate in IBD (39), and we and others have shown that it induces increased leukocyte adhesion to the endothelium, a key role in mediating inflammation (35–37). We therefore hypothesized that thrombin may contribute to the increased HA-mediated leukocyte adhesion/recruitment mediated by intestinal M-SMCs, important contributors to the inflammation associated with IBD.
To test the effect of thrombin on leukocyte adhesion, we first measured U937 monocytic cell adhesion to M-SMCs derived from patients with IBD. Multiple patient isolates of human primary M-SMC cultures were treated for 18 h with poly(I·C) (100 μg/ml) as a positive response stimulus or TNF-α (10 ng/ml) as a negative control. Following treatment, U937 cell adhesion was measured. Consistent with our previous reports (11, 26), U937 cell adhesion to M-SMCs was significantly increased in response to poly(I·C) but not in response to TNF-α, when compared with control media treated with M-SMCs (Fig. 1A). Treatment of M-SMCs with thrombin alone (25 units/ml) had little effect on U937 cell adhesion; surprisingly, however, thrombin addition to poly(I·C)-stimulated M-SMCs abolished U937 binding. To confirm that the effects observed with U937 cells represented a process important to normal leukocytes, we tested the ability of thrombin to abrogate adhesion of normal human peripheral blood leukocytes to poly(I·C)-treated M-SMCs. Fig. 1B demonstrates that treatment of M-SMCs with poly(I·C) results in significantly enhanced binding of total peripheral blood mononuclear cells, purified monocytes and neutrophils, as well as U937 cells, compared with controls. Strikingly, thrombin addition after poly(I·C) stimulation of M-SMCs completely reverses the increased leukocyte attachment for all cell types examined. Together, these data suggest that the HA adhesion mechanism is common to leukocytes and is regulated by thrombin. From these results it seemed plausible that thrombin has the capability to alter the hyaluronan leukocyte adhesive matrix; therefore, we investigated potential mechanisms.
FIGURE 1.

Thrombin abrogates leukocyte adhesion to poly(I·C)-stimulated M-SMCs. Confluent M-SMCs derived from patients with IBD were grown in DME/F-12 medium containing 10% FBS and treated without or with poly(I·C) (100 μg/ml) or TNF-α (10 ng/ml) for 18 h or thrombin (25 units/ml) for 6 h at 37 °C prior to cell adhesion assays. Adhesion of U937 cells to M-SMC cultures isolated from individual patients with IBD (P1, P2, and P3) (A) or adhesion of specific types of leukocytes (B) was performed as described under “Experimental Procedures.” (Mean data are shown with error bars representing ± S.E. between replicates (n = 3); **, p < 0.01; ***, p < 0.001.)
Proteolytic Activity of Thrombin Regulates Leukocyte Adhesion to Poly(I:C)-stimulated M-SMCs
Previously, reports have shown that thrombin increases adhesion of leukocytes to endothelial cells through activation of PARs, which require proteolytic cleavage for receptor activation (33, 34). Therefore, we asked whether the ability of thrombin to attenuate monocyte adhesion was dependent upon proteolytic activity toward PARs or other additional substrates. M-SMCs were treated with poly(I·C) overnight, and 3 h before the leukocyte adhesion assay increasing concentrations of thrombin (0–25 units/ml) were added to the cultures. Thrombin treatment of poly(I·C)-stimulated M-SMCs reduced U937 adhesion in a concentration-dependent manner (Fig. 2A). Although low doses of thrombin capable of activating thrombin receptors (1–5 units/ml) had no discernable impact on U937 adhesion to M-SMCs, we observed a 50% decrease at 15 units/ml and almost a total loss of U937 adhesion at 25 units/ml. To determine whether thrombin was acting directly through a PAR to reduce leukocyte adhesion, poly(I·C)-stimulated M-SMCs were treated with the PAR ligand, TRAP, and its activity was compared with thrombin (25 units/ml). Fig. 2B demonstrates that thrombin receptor activation by TRAP (100 μm) could not reproduce the effects of thrombin protein, suggesting that thrombin was acting independently of PARs. We further determined that the ability of thrombin to abrogate poly(I·C)-induced adhesion was time-dependent, observing a 55% decrease in U937 cell adhesion after 30 min post-thrombin addition and reaching a maximal abrogating effect (81% decrease) by 3 h (Fig. 2C). Importantly, co-incubation of thrombin with the serine protease inhibitor PMSF blocked the ability of thrombin to ablate U937 adhesion to poly(I·C)-induced M-SMCs. These data indicate that the proteolytic activity of thrombin negatively regulates hyaluronan-mediated leukocyte adhesion in a PAR-independent manner.
FIGURE 2.

Thrombin-mediated inhibition of U937 adhesion is independent of PAR activation and requires proteolytic activity. Confluent M-SMCs were grown in DME/F-12 medium containing 10% FBS without or with poly(I·C) for 18 h at 37 °C. A, M-SMCs were treated with varying concentrations of bovine thrombin for 6 h at 37 °C prior to the adhesion assay. B, M-SMCs were treated with poly(I·C) for 18 h, prior to treatment with either thrombin (25 units/ml) or TRAP (100 μm) for 3 h prior to U937 cell adhesion. C, cells were treated with thrombin for the indicated duration in the presence or absence of PMSF prior to addition of U937 cells. (Mean data are shown with error bars representing ± S.E. between replicates (n = 3); *, p < 0.05; ***, p < 0.001.)
Heavy Chains of Inter-α-inhibitor Possess Thrombin Cleavage Sites
We have previously demonstrated that naive mononuclear leukocytes bind specifically to HC-HA and that treatment with specific antibodies to IαI-related proteoglycans during cable formation interferes with leukocyte adhesion (11). The observation that thrombin regulation of leukocyte adhesion to poly(I·C)-stimulated M-SMCs was dependent on proteolytic activity led us to evaluate whether components of the inflammatory HC-HA matrix might be sensitive to proteolysis by thrombin. Known thrombin substrates typically align to three-to-four positions in the reported consensus recognition sequence (44–46), and exosite interactions are known to enhance substrate specificity (47). We therefore queried the Swiss Protein and TrEMBL databases (48) to determine whether any of the known HC-HA components might be novel potential thrombin substrates. The IαI family HCs showed high sequence homology, and with the exception of HC2, at least one predicted thrombin site is present in each HC sequence (data not shown). Human HC1 contains a thrombin cleavage site proximal to the glycosaminoglycan attachment residues where HC1 may be covalently linked to either the CS of IαI during biosynthesis or covalently attached to HA following transfer by TSG-6 (Fig. 3A). The HC1 thrombin consensus sequence (LGPR∼RTF) contains preferable residues at each site, and proteolysis at this site would result in dissociation of most of HC1 from either HA or IαI. By contrast, the consensus sites contained within HC3 and HC4 lie within the pro-peptide region and C terminus, both of which are reported to be removed during biosynthesis (15, 49, 50). Based on this observation, we performed cross-species protein sequence comparisons of HC1 to determine whether the thrombin-susceptible cleavage sequence is conserved. As shown in Fig. 3B, the region of HC1 proximal to the thrombin consensus sequence is highly conserved across all placental mammals analyzed. No thrombin consensus sites were found in either orthologues or paralogues of HC1 in birds, fish, or reptiles (data not shown).
FIGURE 3.
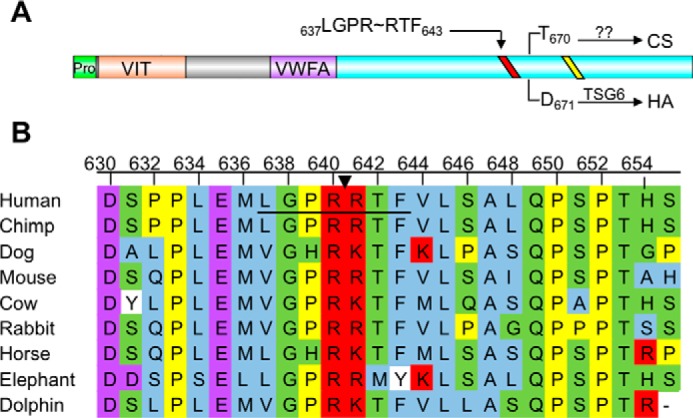
HC1 contains a conserved thrombin cleavage sequence proximal to the glycosaminoglycan attachment site. A, domain structure of inter-α-inhibitor HC1. HC1 has two known domains, the vault protein inter-α-trypsin (VIT) domain and a von Willebrand type A (VWFA) domain. The site of CS linkage or HA attachment (for TSG-6-mediated transfer) is depicted by an arrow from the indicated residue to either CS or HA. The conserved sequence required for processing by an unknown Golgi enzyme and covalent transfer to CS is depicted as a yellow line. The predicted thrombin consensus sequence (LGPRRTF) is shown as a red line. B, partial HC1 sequence alignment of the thrombin consensus sequence. The consensus sequence is underlined, and an arrow indicates the scissile bond. Amino acids are colored corresponding to the following properties. Blue, small and hydrophobic residues; purple, acidic residues; red, basic residues; green, hydroxyl, sulfhydryl, amine, and glycine; yellow, proline; white, aromatic.
Heavy Chain 1 Is Susceptible to Cleavage by Thrombin
Based on these observations, we tested whether thrombin could cleave HC1 directly. We released the cell-associated HC-HA matrix from the surface of poly(I·C)-stimulated M-SMCs with limited Streptomyces hyaluronidase digestion, and we incubated these samples in the presence or absence of 25 units/ml thrombin. Protein samples were then separated on the basis of size, prepared for mass spectrometry, and analyzed for peptides corresponding to HC1 (Fig. 4). This analysis detected 44 unique HC1 peptides with a sequence coverage of 52% (Fig. 4A). In the absence of thrombin, all HC1 peptides detected were present at high molecular mass ranges (within gel regions 1–4) from >250 to 90 kDa, consistent with the molecular masses of IαI, PαI, and free HC1. For samples treated with thrombin, HC1 peptides (31%) were detected at high mass ranges (regions 1–4), but the majority of peptides detected (69%) was found at lower mass ranges (regions 5–9) (Fig. 4, B and C). Next, we selected specific N- and C-terminal HC1 peptides detected in both samples and investigated whether these peptides were observed at different mass ranges upon thrombin treatment (Fig. 4D). The N-terminal peptide was present only at high mass ranges (data shown for region 1) in untreated samples but was present in both high and low mass ranges (data shown for region 7) in thrombin-treated samples, consistent with cleavage by thrombin. By contrast, the majority of the C-terminal peptide was detected only in high mass ranges in both samples. Taken together, these data indicate that HC1 is a genuine substrate of thrombin.
FIGURE 4.

Mass spectrometric analysis of thrombin cleavage of bovine HC1. Confluent M-SMCs were treated with poly(I·C) for 18 h at 37 °C. Cells were rinsed three times with PBS and treated with Streptomyces hyaluronidase (100 milliunits/ml) for 10 min to release HC-HA from the cell surface. The supernatants were collected, split into two equal volumes, and incubated without or with thrombin (25 units/ml) for 1 h. Samples were then prepared for mass spectrometry as described under “Experimental Procedures.” A, coverage of all HC1 peptides detected in untreated (blue) or thrombin-treated (red) samples. The two thrombin cleavage sites in bovine HC1 are underlined in red. The N- and C-terminal peptides analyzed in D are in boxes. Tryptic residues are underlined in black; predicted thrombin sites are underlined in red, and a conserved Golgi processing site is underlined in yellow. B, representative Coomassie stain of cell-surface HC-HA supernatants released from M-SMCs by Streptomyces hyaluronidase digestion. Lanes were divided into nine regions for subsequent MS analysis. C, quantification of tryptic HC1 peptides detected in each sample are represented as a percentage of total HC1 peptides excised from indicated regions of B. D, representative chromatogram of a specific N- or C-terminal peptide of HC1 present in both untreated and treated samples. The N-terminal peptide (MAVDAAVDGVVIR) observed in both samples was found only at high mass ranges (data shown for Region 1, blue) of untreated samples but was detected at lower mass ranges (data shown for Region 7, red) of thrombin-treated samples. The C-terminal peptide (ALQMSLAYQFVTPTSMTVR) observed in both samples was detected only high mass ranges of both samples and was not observed at lower mass ranges of thrombin-treated samples. Data are representative of two independent experiments.
Thrombin Cleaves HC1 from Inflammatory Matrix HA and from Serum IαI
Our previous studies demonstrate that HC1 and HC2 specifically associate with HA on the surface of M-SMCs in response to poly(I·C) (11). We therefore investigated whether thrombin was capable of cleaving HC1 from the HC-HA matrix on the surface of poly(I·C)-treated or untreated M-SMCs. Western blotting analyses using an affinity-purified polyclonal antibody that recognizes intact IαI family members (Fig. 5A) showed a reduction in the bands corresponding to IαI upon thrombin treatment. As expected, we also observed an increase in PαI in the same lane, consistent with a mass shift upon cleavage of one HC from IαI. We verified thrombin cleavage of HC1 by probing with a monoclonal antibody specific to HC1 (Fig. 5B). Altogether, these data indicate that thrombin is capable of cleaving HC1 from the HC-HA matrix on the surface of poly(I·C)-treated M-SMCs.
FIGURE 5.

Thrombin is capable of cleaving HC1 from either cell-surface HC-HA cables or from serum IαI. HA cell-surface layer extracts from untreated, poly(I·C)-treated, or poly(I·C) and thrombin-treated cells were compared by Western blotting. Confluent M-SMCs were treated without or with poly(I·C) for 18 h at 37 °C to form HC-HA cable complexes. Poly(I:C)-treated cells were incubated without or with thrombin (25 units/ml) for 1 h at 37 °C. Cells were rinsed three times with PBS and incubated with Streptomyces hyaluronidase (100 milliunits/ml) for 5 min to release HA-bound cell-surface material. The supernatant was collected and prepared for Western blotting analysis of IαI (A) and HC1 (B). Serum from healthy donors was diluted and incubated without or with thrombin (25 units/ml) for 3 h at 37 °C prior to Western blotting analysis for either IαI (C) or HC1 (D). The green ovals in the schematic models represent HCs attached to bikunin (blue rectangle) via a single CS chain (black line). The schematic with two HCs represents IαI. The schematic with one HC represents PαI. Blots are representative of experiments from three independent patient cell lines and three technical replicates.
We also sought to determine whether thrombin was capable of cleaving HC1 on serum IαI, independent of when it is associated with HA. Western blotting analysis of increasing dilutions of serum incubated without or with thrombin indicates that thrombin treatment of IαI causes a mass shift from IαI to PαI (Fig. 5C), consistent with our findings using the M-SMC-generated HC-HA matrix (Fig. 5A). Analysis with an antibody specific to HC1 (Fig. 5D) indicates a broadening of the band corresponding to HC1 (indicative of HC1 released from IαI), a proteolytic fragment consistent with Fig. 5B, and the presence of multiple proteolytic fragments upon thrombin treatment.
Thrombin Cleavage of HC1 Regulates Leukocyte Binding by Dissolution of HA Cables
Previous studies have demonstrated that the HCs of IαI, but not CD44, are required to form HC-HA cable complexes (26, 42). However, blocking of CD44 on leukocytes has been shown to abolish adhesion to HA, indicating that leukocytes recognize the HA component of HC-HA complexes (26). Because antibodies to IαI have been observed to inhibit the formation of HA cables (11), we investigated whether thrombin-mediated cleavage of HC1 regulates leukocyte adhesion by altering the organization of cell-surface HA. Replicate cultures of M-SMCs were treated either with medium alone or containing poly(I·C) overnight or treated with poly(I·C) overnight followed by treatment with thrombin for 1 h prior to adding peripheral blood leukocytes. Immunofluorescent histochemistry with the HA-binding probe (green) was used to determine the organization of HA on the surface of M-SMCs (Fig. 6). The cells were stained with a monoclonal antibody to CD44 (Fig. 6, red) to detect the cell bodies of both leukocytes (small round cells) and M-SMCs (larger flat cells). Control medium-treated cells produce a relatively small amount of HA, which can be observed in randomly distributed patches along the cell surface. In contrast, poly(I·C) stimulation induces the formation of thicker pericellular coats of HA as well as large HA cables (Fig. 6, arrows) that can span multiple cell lengths. The addition of thrombin to poly(I·C)-stimulated cells abolishes the presence of HA cables, while leaving regions of pericellular HA coats intact.
FIGURE 6.

Thrombin-mediated cleavage of HC1 leads to dissociation of HA cables formed by M-SMCs in response to poly(I·C). Confluent M-SMCs grown on coverslips were treated without or with poly(I·C) for 18 h at 37 °C. Thrombin (25 units/ml) was added to the media of the indicated cells for 1 h prior to the PBMC adhesion assay. The PBMC adhesion assay and immunostaining were performed as described under “Experimental Procedures.” The organization of HA on the surface of M-SMCs was determined using an HA-binding probe (green). Cells were stained with a monoclonal antibody to CD44 (red) to detect the cell bodies of both leukocytes (small round cells) and M-SMCs (lager flat cells), and nuclei were stained with DAPI (blue). Untreated M-SMCs possess a low level of HA on their surface, and few PBMCs appear bound. In contrast, poly(I·C)-treated cells produce more HA as well as large HA cables (arrows), which contain many PBMCs bound to HA in clusters. The addition of thrombin to poly(I·C)-stimulated cells abolishes the presence of HA cables, while leaving regions of pericellular HA coats intact. An immunostaining specificity control (2° Control), in which only secondary reagents are added, is also shown. Scale bar, 50 μm.
Discussion
Using an in vitro system in which a viral mimetic induces the formation of a leukocyte-adhesive HC-HA matrix on the surface of colon mesenchymal cells, we investigated the ability of thrombin to regulate leukocyte adhesion. Here, we have demonstrated that thrombin unexpectedly ablates leukocyte adhesion to the HC-HA matrix. Thrombin treatment of poly(I·C)-stimulated M-SMCs dramatically reduced adhesion of U937 cells, PBMCs, monocytes, and neutrophils (Fig. 1), and this effect is independent of PAR activation but still requires proteolytic activity (Fig. 2). These observations indicated that thrombin acted on a novel substrate, and we determined that HC1 contains a thrombin consensus site, which is highly conserved across a number of mammalian species (Fig. 3). Using mass spectrometric analysis, we confirmed that HC1 is a substrate of thrombin (Fig. 4). Thrombin was shown to cleave HC1 from HA at the cell surface of poly(I·C)-stimulated M-SMCs and from serum IαI, generating fragments cross-reactive with an anti-HC1 antibody (Fig. 5). Importantly, immunofluorescent staining revealed that thrombin treatment of poly(I·C)-stimulated M-SMCs resulted in a dissolution of the HC-HA matrix (Fig. 6). Together, these data demonstrate that thrombin-mediated cleavage of HC1 negatively regulates leukocyte binding to M-SMCs by altering an inflammation-associated HC-HA matrix.
Increasing evidence suggests that the inflammation and coagulation pathways are intertwined and capable of extensive cross-talk. Inflammation enhances activation of coagulation factors, and coagulation in turn amplifies inflammatory activity. Dysregulation of these responses can modulate one another and contribute to disease. Thrombin, which acts as both a pro-coagulant and pro-inflammatory molecule, can activate cellular receptors on platelets, leukocytes, and endothelial cells to promote cytokine production and adhesion. Thrombin generation is reported to be accelerated in IBD (39), and it may contribute to disease pathology through multiple mechanisms. Thrombin activation of PAR-1 on endothelial cells leads to induction of vascular cell adhesion molecule 1 (VCAM-1), which is constitutively expressed in the inflamed mucosa of patients with IBD (51), and in a rat model of colitis (52). Two major pathological changes in IBD in which thrombin may participate are the increased influx of mononuclear leukocytes into the intestinal submucosa and hyperplasia of the muscularis mucosa of smooth muscle cells.
Our present data suggest that the anti-adhesive effect of thrombin results from cleavage of HC1 to prevent leukocyte binding to inflamed smooth muscle cells (Fig. 1). The observation that thrombin treatment leads to decreased leukocyte adhesion independent of PAR activation (Fig. 2) led us to consider novel substrates of thrombin, which might mediate this effect. Thrombin has previously been shown to act upon several ECM adhesion molecules such as nidogen (53), type V collagen (54), fibronectin (55), laminin (56), and integrin αV(46). We have previously demonstrated that the majority of leukocyte adhesion to respiratory syncytial virus-infected M-SMCs was mediated by HA (26). At least in M-SMCs, VCAM-1 function appears to be masked by HA, but it can be restored by removal of HA with hyaluronidase (26). Under normal conditions, cell-surface HA on M-SMCs is minimally adhesive for leukocytes, but viral infection or treatment with poly(I·C) induces the formation of a highly adhesive hyaluronan matrix that is cross-linked with the HCs of IαI (11).
Our evaluation of thrombin-susceptible sequences in human HC proteins identified HC1, HC3, HC4, and HC5 as novel potential targets of thrombin. The location of the predicted site in human HC1 was of particular interest as cleavage of the thrombin-sensitive sequence (LGPRRTF) would result in dissociation of the majority of HC1 from HA, leaving behind a small ∼3.5-kDa fragment attached to HA. We were able to confirm HC1 as a substrate of thrombin, but we observed HC1 fragments at different molecular masses than expected based on our predictions of human HC1 (Fig. 5). This discrepancy can be explained by the fact that bovine HC1 contains an additional thrombin consensus sequence near the N terminus that is also well conserved in placental mammals, including chimps (data not shown). Cleavage at this site is predicted to result in fragments of 55, 16, and 3.5 kDa consistent with our observations of human serum (Fig. 5). Interestingly, the corresponding sequence in human HC1 (VNPQSKV) contains a sub-optimal glutamine at P1 and was missed in our sequence prediction due to the requirement of arginine at P1. Further studies will be needed to determine whether the proteolytic fragments generated by thrombin have biological properties.
Increased matrix deposition of HC-HA has been described in several inflammatory pathologies and is believed to contribute to disease (57). However, in some contexts, such as the amniotic membrane, HC-HA possesses anti-inflammatory effects (41). Some of these divergent functions of HC-HA may be explained by the particular composition of proteins associated with HA. The amniotic membrane endogenously synthesizes both TSG-6 and IαI, producing an HC-HA that primarily contains HC1 (13). This suggests that the in vivo biosynthetic environment of the amniotic membrane is dissimilar from other tissues, in which TSG-6 is produced only in response to inflammation, and HCs are derived from serum IαI.
Increased HA deposition is a consistent feature of tissue injury and inflammation (58, 59). The observation that leukocytes do not bind to HA under normal conditions, but bind strongly to HC-HA, suggests that this inflammation-associated ECM element promotes disease progression. Using an experimental model of colitis, we have demonstrated that HA cable formation precedes inflammatory cell infiltration and that HC-HA cables produced by the endothelium recruit leukocytes (43). Importantly, disruption of endothelial HA synthesis by knock-out of hyaluronan synthase 3 (HAS3) protects HAS3-null mice from colitis (60). Increased HA deposition observed in mouse models during development of colitis strongly parallels human IBD (43). In colon tissue from patients with IBD, inflammation-associated HC-HA is in close contact with CD44-positive leukocytes, suggesting that HCs confer pro-inflammatory properties on native HA (11). Degradation of HA or antibody-mediated blockade of leukocyte CD44 abolishes binding, but antibody blockade of IαI does not, indicating CD44 likely mediates leukocyte adhesion to HC-HA (11, 26). Although a number of groups have established the importance of CD44-HA interactions in inflammation (61–63), the specific contribution of HCs remains unclear. We recently demonstrated that platelets can modify an inflammatory HA matrix through the enzyme hyaluronidase-2 (64). Platelet-mediated degradation of HA-rich inflammatory matrices may modulate inflammatory responses by clearance of HA produced in response to inflammatory stimuli. Dissolution of the HC-HA matrix can abrogate leukocyte binding, as presented in this work. However, cleavage of HA by platelets and other cell types can result in small molecular weight HA fragments, which themselves may contribute to inflammation by promoting chemokine expression, chemotaxis, and angiogenesis (65–67). The direct mechanism through which HC-HA contributes to inflammation is not fully known, but it likely involves organization of additional matrix proteins and increased adhesion/recruitment of leukocytes.
At present, our understanding of how HC-HA matrices are regulated, both in promotion and resolution of inflammation, is incomplete. Our new data suggest that proteolytic cleavage of HC1 leads to loss of the inflammatory HC-HA matrix, and it is therefore likely HCs contribute to the stability of this matrix. Although HC3 and HC4 also contain sites potentially susceptible to thrombin, HC4 has been demonstrated to be a substrate of kallikrein (68). During acute inflammation, hepatic transcription of HC3 and HC4 is reported to be increased, whereas HC1 expression is unchanged and HC2 is decreased (69). Serum-derived serine proteases, such as thrombin and kallikrein, may therefore function as novel ECM remodeling enzymes and act via this additional mechanism to modulate inflammation.
Experimental Procedures
Cell Isolation and Culture
All M-SMC cultures were derived from human colon specimens obtained within 2 h after resection from patients undergoing surgery for conditions that did not involve inflammatory diseases (Department of Anatomical Pathology, Cleveland Clinic) as described previously (26). In brief, the lamina propria (mucosal layer) of each specimen was removed, blotted, cut into strips, washed in 50 ml of Hanks' BSS containing 0.15% DTT (w/v) for 30 min, washed three times in 100 ml of Hanks' BSS containing 1 mm EDTA for 1 h each, and washed four times in Hanks' BSS alone for 30 min each. The washed lamina propria strips were finely minced prior to digestion overnight in 100 ml of Hanks' BSS with collagenase, DNase (0.1 mg/ml each), penicillin (250 units/ml), streptomycin (250 μg/ml), and fungizone (0.625 μg/ml). Cells were then filtered from the undigested tissue by passage through a 10-μm cell strainer and cultured at 37 °C with 5% CO2 in DME/F-12 medium supplemented with 10% fetal bovine serum (Bio-Whittaker, Walkersville, MD) and antibiotics (penicillin, 100 units/ml; streptomycin, 100 μg/ml; fungizone, 0.25 μg/ml). All cultures were used within the first four passages. U937 cells were purchased (ATCC) and cultured according to their specifications.
Separation of Human Leukocytes
Total mononuclear cells were separated from heparinized peripheral blood (100 units heparin/ml) by centrifugation on Ficoll-Hypaque density gradients (70). The isolated peripheral blood mononuclear leukocytes were resuspended in RPMI 1640 medium supplemented with 5% FBS (25–50 × 106 cells/ml) in a Teflon beaker to prevent attachment during labeling. Viability of the peripheral blood mononuclear leukocytes was always greater than 95%, as determined by trypan blue dye exclusion. Neutrophils in the pellet of the Ficoll-Hypaque gradient were further purified according to the method of Stossel et al. (71) using sedimentation in a dextran gradient and hypotonic lysis of residual erythrocytes. Cells isolated by this procedure were routinely greater than 95% neutrophils, as estimated by differential counting.
Monocytes were separated from peripheral blood (100 units of heparin/ml) by a modification of the method of Recalde (72). Briefly, total mononuclear cells were collected by Ficoll-Hypaque density gradient centrifugation, washed, and resuspended in FBS. The cells were then maintained at 37 °C, and 9% NaCl was added at three 10-min intervals (5 μl/ml, then 10 μl/ml, and then another 10 μl/ml). After the last 10-min incubation, the cell suspension was mixed with 2 volumes of PBS (with 27 μl of 9% NaCl/ml added) and underlaid with 1 volume of Ficoll-Hypaque (with 2.8 mg of NaCl/ml added). The gradient was spun at 600 × g for 20 min, and the monocytes were separated from the Ficoll interface, washed twice with cold PBS, and finally resuspended in DME/F-12 + 5% FBS in a Teflon beaker to prevent attachment. Isolated populations were routinely 78–90% monocytes by differential count with buffered Wright-Giemsa stain, and viability was always greater than 90% by trypan blue dye exclusion.
Assay for Leukocyte Adhesion to M-SMCs
Adhesion of leukocytes to M-SMCs was measured as described previously (11, 26). Briefly, M-SMCs were plated into 24-well plates in their appropriate medium (2 to 3 × 104cells/well in 0.5 ml) 3–5 days before the assay and grown to confluence. Treatment of M-SMCs with poly(I·C) (100 μg/ml) was performed for 18 h before assay. On the day of the adhesion assay, U937 cells (up to 70 × 106 cells/ml) were labeled for 90 min at 37 °C with 100 μCi of 51Cr as sodium chromate (PerkinElmer Life Sciences) in 1 ml of culture medium. The labeled cells were washed three times with culture medium, counted on a hemocytometer, and resuspended to 106 viable cells (as determined by trypan blue dye exclusion) per 0.5 ml of culture medium. Incubation medium was aspirated from M-SMCs, and 106 labeled leukocytes were added to each well.
Prior to leukocyte adhesion, monolayers of M-SMCs were incubated in the presence or absence of thrombin (25 units/ml, 5 ml/75-cm2 culture; United States Biochemical Corp.), thrombin receptor peptide (SFLLRN, 100uM, Immuno-Dynamics, Inc.), or TNF-α (10 ng/ml, Shenandoah Biotechnology) for 3 h (unless otherwise noted) in complete media and washed three times with Hanks' BSS. The binding phase of the assay was then performed at 4 °C for 1 h. Subsequently, the wells were gently washed three times with cold medium to removed unbound leukocytes. The cells were lysed with 1% Triton X-100, and an aliquot was removed for quantitation of radiolabel. The number of leukocytes bound per well was calculated from the initial specific activity (cpm/cell). Spontaneous release of chromium from the monocytes in control incubations without M-SMCs was typically less than 5%.
Bioinformatics Analysis of HC Sequences
The Swiss Protein and TrEMBL databases were searched using PeptideCutter (73) and PROSITE Pattern Scan (74) for human HC protein family members with the thrombin consensus motif (A/F/G/I/L/T/V/M)(A/F/G/I/L/T/V/W/A)(P)R(not D/E)(not D/E). No description filter was chosen, and at most one character was allowed to match a conserved position in the pattern, and the match mode was set to “greedy, overlap, and no includes.” Protein hits were assessed individually for the consensus sequence and confirmed in a secondary database search. Illustration of HC1 domains and post-translational modifications was produced using Illustrator for Biological Sciences tool (75). HC1 sequences were aligned using Clustal Omega (76).
Proteomic Analysis of Thrombin-treated HC-HA
Protein samples were fractionated on a TGX 4–25% SDS-polyacrylamide gel (Bio-Rad) for GeLC analysis. A dual-color pre-stained Precision Plus Protein standard (Bio-Rad) was run as the molecular weight marker. The gel was fixed for 30 min in 50% ethanol, 10% acetic acid, transferred to a plastic dish, and stained in the dark with Gel-Code blue.
Each sample was divided into nine bands, and each band was digested according to an in-gel digestion procedure. The gel pieces were washed with water and dehydrated in acetonitrile. The bands then were reduced with dithiothreitol and alkylated with iodoacetamide before the in-gel digestion. All bands were treated with trypsin by adding 5 μl of 10 ng/μl trypsin in 50 mmol/liter ammonium bicarbonate and incubating overnight at room temperature to achieve complete digestion. The peptides that were formed were extracted from the polyacrylamide in 2 aliquots of 30 μl of 50% acetonitrile with 5% formic acid. These extracts were combined and evaporated to less than 10 μl in a Speedvac (Thermo Fisher Scientific) and then resuspended in 1% acetic acid to make a final volume of ∼30 μl for liquid chromatography-mass spectrometry (LC-MS) analysis.
The LC-MS system was a Finnigan LTQ-Orbitrap Elite hybrid mass spectrometer system interphased with a Dionex Ultimate 3000 high performance liquid chromatography system (Thermo Fisher Scientific). The high performance liquid chromatography column was a Dionex 15-cm × 75-μm internal diameter Acclaim Pepmap C18, 2 μm, 100 Å, reversed-phase capillary chromatography column. Five microliters of the extract was injected, and the peptides were eluted from the column by an acetonitrile, 0.1% formic acid gradient at a flow rate of 0.25 μl/min and were introduced into the source of the mass spectrometer online. The digest was analyzed using the data-dependent multitask capability of the instrument acquiring full scan mass spectra to determine peptide molecular weights and tandem mass spectra (MS/MS) to determine amino acid sequence in successive instrument scans.
Tandem mass spectra were extracted by Proteome Discoverer version 1.4.1.288. All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.3.02), Sequest (Thermo Fisher Scientific; version 1.4.0.288), and X! Tandem (The GPM, thegpm.org; version CYCLONE (2010.12.01.1)). Mascot, Sequest, and X! Tandem were set up to search the Bovine and Human Reference Sequence database assuming the digestion enzyme trypsin, fragment ion mass tolerance of 0.6 Da, and a parent ion tolerance of 10 ppm. Carbamidomethyl of cysteine was specified as a fixed modification, and oxidation of methionine was specified as a variable modification.
Scaffold (version Scaffold_4.4.6, Proteome Software Inc.) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability by the PeptideProphet algorithm (77). Protein identifications were accepted if they could be established at greater than 99.0% probability to achieve a false discovery rate of less than 1.0% and contained at least two identified peptides. Protein probabilities were assigned by the ProteinProphet algorithm. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Western Blotting Analysis
Confluent cultures of M-SMCs were treated with DME/F-12 medium containing 10% FBS with or without poly(I·C) (100 μg/ml) for 18 h. Cultures were rinsed three times with 5 ml of Hanks' BSS and treated with or without thrombin (25 units/ml, 5 ml for a T75-cm2 culture flask; United States Biochemical Corp.) for 3 h, then rinsed three additional times with Hanks' BSS, and treated with Streptomyces hyaluronidase (100 milliunits/ml, 1 ml/75-cm2 culture; Seikagaku) for 5 min at 37 °C to release HA-associated material from the cell surface as described previously (11). The HA digest supernatant was collected, and protein concentration was determined by using the Bradford assay (Bio-Rad). Protein samples were mixed with Laemmli sample buffer (Bio-Rad,) and 2-mercaptoethanol was added as a reducing agent to a final concentration of 5%. Equivalent volumes of either HA-released supernatant or diluted human serum (Equitech) (25 μl) were loaded onto 4–20% Mini-PROTEAN TGX gels (Bio-Rad) and blotted using the Trans-Blot Turbo System (Bio-Rad). Blocking was performed for 1 h in phosphate-buffered saline (PBS) containing 5% nonfat dry milk at room temperature. After blocking, membranes were incubated with a rabbit polyclonal antibody to IαI (at a 1:1000 dilution, A0301, DAKO) or with a mouse anti-human ITIH1 antibody against full-length ITIH1 (1:1000, Abcam) in PBS containing 0.1% Tween 20 (PBST) with 5% nonfat dry milk overnight at 4 °C. Membranes were washed three times in PBST (10 min/each), incubated with a horseradish peroxidase-conjugated secondary antibody (1:15,000 dilution in PBST with 5% milk) for 1 h at room temperature, and washed three times with PBST and twice with PBS (for 10 min each). Signal was detected using an enhanced chemiluminescence solution (GE Healthcare).
Immunofluorescent Analysis
M-SMCs were grown using glass chamber slides (ibidi) and treated as described previously (11, 26). Immediately after the PBMC adhesion assay, slides were gently rinsed with Hanks' BSS, fixed with ice-cold methanol for 15 min at 4 °C, and then air-dried. Chamber slides were preincubated with Hanks' BSS containing 2% FBS for 30 min at room temperature to block nonspecific interactions. Slides were incubated with primary reagents consisting of biotinylated HA-binding protein (Calbiochem-EMD Millipore) (5 μg/ml) and a monoclonal antibody to CD44 (clone A3D8, Sigma) in Hanks' BSS containing 2% FBS overnight at 4 °C. After primary incubation, slides were washed three times with Hanks' BSS and then incubated with secondary detection reagents: AlexaFluor 488-tagged streptavidin (at a 1:500 dilution) and anti-mouse AlexaFluor 568 (at a 1:500) in Hanks' BSS containing 2% FBS for 1 h at room temperature. Slides were then washed and mounted using Vectashield mounting medium containing DAPI (Vector Laboratories). All images were obtained using a Leica upright microscope DM5500 B (Leica), HCX PLAN APO ×20/1.32NA oil immersion objective, QImaging Retiga cooled CCD camera, and QCapture Suite software (QImaging).
Statistical Analysis
All data were analyzed with GraphPad Prism 5 software. The data collected from each experiment is represented as the mean, with error bars signifying the standard error of the mean (S.E.). All data were analyzed using a two-tailed paired Student's t test for parametric data, and a p value less than or equal to 0.05 was considered as statistically significant.
Author Contributions
A. C. P. designed the research, performed experiments, and wrote the paper; C. A. d. l. M. designed the research, performed experiments, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Melissa Michaud for assistance with cell culture and Western blotting. We also thank Dr. Vincent Hascall and Dr. Jarrod Barnes for critical reading of the manuscript.
This work was supported by National Institutes of Health Programs of Excellence in Glycosciences Grant HL107147 from the NHLBI (to C. A. d. l. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.
- ECM
- extracellular matrix
- HA
- hyaluronan
- IBD
- inflammatory bowel disease
- HC
- heavy chain
- IαI
- inter-α-inhibitor
- PαI
- pre-α-inhibitor
- CS
- chondroitin sulfate
- PAR
- protease-activated receptor
- TRAP
- thrombin-receptor activating peptide
- M-SMC
- mucosa smooth muscle cell
- BSS
- balanced saline solution
- PBMC
- peripheral blood mononuclear cell.
References
- 1. Sorokin L. (2010) The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 10, 712–723 [DOI] [PubMed] [Google Scholar]
- 2. Davis G. E., Bayless K. J., Davis M. J., and Meininger G. A. (2000) Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am. J. Pathol. 156, 1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pakianathan D. R. (1995) Extracellular matrix proteins and leukocyte function. J. Leukocyte Biol. 57, 699–702 [DOI] [PubMed] [Google Scholar]
- 4. Dahl L. B., Dahl I. M., Engström-Laurent A., and Granath K. (1985) Concentration and molecular weight of sodium hyaluronate in synovial fluid from patients with rheumatoid arthritis and other arthropathies. Ann. Rheum. Dis. 44, 817–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yingsung W., Zhuo L., Morgelin M., Yoneda M., Kida D., Watanabe H., Ishiguro N., Iwata H., and Kimata K. (2003) Molecular heterogeneity of the SHAP-hyaluronan complex. Isolation and characterization of the complex in synovial fluid from patients with rheumatoid arthritis. J. Biol. Chem. 278, 32710–32718 [DOI] [PubMed] [Google Scholar]
- 6. Evanko S. P., Raines E. W., Ross R., Gold L. I., and Wight T. N. (1998) Proteoglycan distribution in lesions of atherosclerosis depends on lesion severity, structural characteristics, and the proximity of platelet-derived growth factor and transforming growth factor-β. Am. J. Pathol. 152, 533–546 [PMC free article] [PubMed] [Google Scholar]
- 7. Hällgren R., Samuelsson T., Laurent T. C., and Modig J. (1989) Accumulation of hyaluronan (hyaluronic acid) in the lung in adult respiratory distress syndrome. Am. Rev. Respir. Dis. 139, 682–687 [DOI] [PubMed] [Google Scholar]
- 8. Lauer M. E., Majors A. K., Comhair S., Ruple L. M., Matuska B., Subramanian A., Farver C., Dworski R., Grandon D., Laskowski D., Dweik R. A., Erzurum S. C., Hascall V. C., and Aronica M. A. (2015) Hyaluronan and its heavy chain modification in asthma severity and experimental asthma exacerbation. J. Biol. Chem. 290, 23124–23134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lewis A., Steadman R., Manley P., Craig K., de la Motte C., Hascall V., and Phillips A. O. (2008) Diabetic nephropathy, inflammation, hyaluronan and interstitial fibrosis. Histol. Histopathol. 23, 731–739 [DOI] [PubMed] [Google Scholar]
- 10. Bogdani M., Johnson P. Y., Potter-Perigo S., Nagy N., Day A. J., Bollyky P. L., and Wight T. N. (2014) Hyaluronan and hyaluronan-binding proteins accumulate in both human type 1 diabetic islets and lymphoid tissues and associate with inflammatory cells in insulitis. Diabetes 63, 2727–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de la Motte C. A., Hascall V. C., Drazba J., Bandyopadhyay S. K., and Strong S. A. (2003) Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyinosinic acid:polycytidylic acid: inter-α-trypsin inhibitor is crucial to structure and function. Am. J. Pathol. 163, 121–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Petrey A. C., and de la Motte C. A. (2014) Hyaluronan, a crucial regulator of inflammation. Front. Immunol. 5, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang S., He H., Day A. J., and Tseng S. C. (2012) Constitutive expression of inter-α-inhibitor (IαI) family proteins and tumor necrosis factor-stimulated gene-6 (TSG-6) by human amniotic membrane epithelial and stromal cells supporting formation of the heavy chain-hyaluronan (HC-HA) complex. J. Biol. Chem. 287, 12433–12444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salier J. P., Chan P., Raguenez G., Zwingman T., and Erickson R. P. (1993) Developmentally regulated transcription of the four liver-specific genes for inter-α-inhibitor family in mouse. Biochem. J. 296, 85–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blom A. M., Thuveson M., and Fries E. (1997) Intracellular coupling of bikunin and the heavy chain of rat pre-α-inhibitor in COS-1 cells. Biochem. J. 328, 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lindqvist A., Bratt T., Altieri M., Kastern W., and Akerström B. (1992) Rat α 1-microglobulin: co-expression in liver with the light chain of inter-α-trypsin inhibitor. Biochim. Biophys. Acta 1130, 63–67 [DOI] [PubMed] [Google Scholar]
- 17. Bratt T., Olsson H., Sjöberg E. M., Jergil B., and Akerström B. (1993) Cleavage of the α1-microglobulin-bikunin precursor is localized to the Golgi apparatus of rat liver cells. Biochim. Biophys. Acta 1157, 147–154 [DOI] [PubMed] [Google Scholar]
- 18. Enghild J. J., Salvesen G., Hefta S. A., Thøgersen I. B., Rutherfurd S., and Pizzo S. V. (1991) Chondroitin 4-sulfate covalently cross-links the chains of the human blood protein pre-α-inhibitor. J. Biol. Chem. 266, 747–751 [PubMed] [Google Scholar]
- 19. Enghild J. J., Salvesen G., Thøgersen I. B., Valnickova Z., Pizzo S. V., and Hefta S. A. (1993) Presence of the protein-glycosaminoglycan-protein covalent cross-link in the inter-α-inhibitor-related proteinase inhibitor heavy chain 2/bikunin. J. Biol. Chem. 268, 8711–8716 [PubMed] [Google Scholar]
- 20. Enghild J. J., Thøgersen I. B., Cheng F., Fransson L. A., Roepstorff P., and Rahbek-Nielsen H. (1999) Organization of the inter-α-inhibitor heavy chains on the chondroitin sulfate originating from Ser(10) of bikunin: posttranslational modification of IαI-derived bikunin. Biochemistry 38, 11804–11813 [DOI] [PubMed] [Google Scholar]
- 21. Mizon C., Balduyck M., Albani D., Michalski C., Burnouf T., and Mizon J. (1996) Development of an enzyme-linked immunosorbent assay for human plasma inter-α-trypsin inhibitor (ITI) using specific antibodies against each of the H1 and H2 heavy chains. J. Immunol. Methods 190, 61–70 [DOI] [PubMed] [Google Scholar]
- 22. Mukhopadhyay D., Asari A., Rugg M. S., Day A. J., and Fülöp C. (2004) Specificity of the tumor necrosis factor-induced protein 6-mediated heavy chain transfer from inter-α-trypsin inhibitor to hyaluronan: implications for the assembly of the cumulus extracellular matrix. J. Biol. Chem. 279, 11119–11128 [DOI] [PubMed] [Google Scholar]
- 23. Rugg M. S., Willis A. C., Mukhopadhyay D., Hascall V. C., Fries E., Fülöp C., Milner C. M., and Day A. J. (2005) Characterization of complexes formed between TSG-6 and inter-α-inhibitor that act as intermediates in the covalent transfer of heavy chains onto hyaluronan. J. Biol. Chem. 280, 25674–25686 [DOI] [PubMed] [Google Scholar]
- 24. Milner C. M., Higman V. A., and Day A. J. (2006) TSG-6: a pluripotent inflammatory mediator? Biochem. Soc. Trans. 34, 446–450 [DOI] [PubMed] [Google Scholar]
- 25. Sanggaard K. W., Hansen L., Scavenius C., Wisniewski H. G., Kristensen T., Thøgersen I. B., and Enghild J. J. (2010) Evolutionary conservation of heavy chain protein transfer between glycosaminoglycans. Biochim. Biophys. Acta 1804, 1011–1019 [DOI] [PubMed] [Google Scholar]
- 26. de La Motte C. A., Hascall V. C., Calabro A., Yen-Lieberman B., and Strong S. A. (1999) Mononuclear leukocytes preferentially bind via CD44 to hyaluronan on human intestinal mucosal smooth muscle cells after virus infection or treatment with poly(I·C). J. Biol. Chem. 274, 30747–30755 [DOI] [PubMed] [Google Scholar]
- 27. Mahadevan P., Larkins R. G., Fraser J. R., Fosang A. J., and Dunlop M. E. (1995) Increased hyaluronan production in the glomeruli from diabetic rats: a link between glucose-induced prostaglandin production and reduced sulphated proteoglycan. Diabetologia 38, 298–305 [DOI] [PubMed] [Google Scholar]
- 28. Evanko S. P., Potter-Perigo S., Johnson P. Y., and Wight T. N. (2009) Organization of hyaluronan and versican in the extracellular matrix of human fibroblasts treated with the viral mimetic poly I:C. J. Histochem. Cytochem. 57, 1041–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lauer M. E., Fulop C., Mukhopadhyay D., Comhair S., Erzurum S. C., and Hascall V. C. (2009) Airway smooth muscle cells synthesize hyaluronan cable structures independent of inter-α-inhibitor heavy chain attachment. J. Biol. Chem. 284, 5313–5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olszewska-Pazdrak B., Bergmann J. S., Fuller G. M., and Carney D. H. (2009) in Thrombin: Physiology and Disease (Maragoudakis E. M., and Tsopanoglou E. N., eds) pp. 115–132, Springer, New York [Google Scholar]
- 31. Rohatgi T., Sedehizade F., Reymann K. G., and Reiser G. (2004) Protease-activated receptors in neuronal development, neurodegeneration, and neuroprotection: thrombin as signaling molecule in the brain. Neuroscientist 10, 501–512 [DOI] [PubMed] [Google Scholar]
- 32. Popović M., Smiljanić K., Dobutović B., Syrovets T., Simmet T., and Isenovic E. R. (2012) Thrombin and vascular inflammation. Mol. Cell. Biochem. 359, 301–313 [DOI] [PubMed] [Google Scholar]
- 33. Coughlin S. R. (2000) Thrombin signalling and protease-activated receptors. Nature 407, 258–264 [DOI] [PubMed] [Google Scholar]
- 34. Vu T. K., Hung D. T., Wheaton V. I., and Coughlin S. R. (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068 [DOI] [PubMed] [Google Scholar]
- 35. DiCorleto P. E., and de la Motte C. A. (1989) Thrombin causes increased monocytic-cell adhesion to endothelial cells through a protein kinase C-dependent pathway. Biochem. J. 264, 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaplanski G., Marin V., Fabrigoule M., Boulay V., Benoliel A. M., Bongrand P., Kaplanski S., and Farnarier C. (1998) Thrombin-activated human endothelial cells support monocyte adhesion in vitro following expression of intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106). Blood 92, 1259–1267 [PubMed] [Google Scholar]
- 37. Kaplan Z. S., Zarpellon A., Alwis I., Yuan Y., McFadyen J., Ghasemzadeh M., Schoenwaelder S. M., Ruggeri Z. M., and Jackson S. P. (2015) Thrombin-dependent intravascular leukocyte trafficking regulated by fibrin and the platelet receptors GPIb and PAR4. Nat. Commun. 6, 7835. [DOI] [PubMed] [Google Scholar]
- 38. Levi M., and van der Poll T. (2010) Inflammation and coagulation. Crit. Care Med. 38, S26–S34 [DOI] [PubMed] [Google Scholar]
- 39. Chamouard P., Grunebaum L., Wiesel M. L., Frey P. L., Wittersheim C., Sapin R., Baumann R., and Cazenave J. P. (1995) Prothrombin fragment 1 + 2 and thrombin-antithrombin III complex as markers of activation of blood coagulation in inflammatory bowel diseases. Eur. J. Gastroenterol. Hepatol. 7, 1183–1188 [DOI] [PubMed] [Google Scholar]
- 40. Garantziotis S., Zudaire E., Trempus C. S., Hollingsworth J. W., Jiang D., Lancaster L. H., Richardson E., Zhuo L., Cuttitta F., Brown K. K., Noble P. W., Kimata K., and Schwartz D. A. (2008) Serum inter-α-trypsin inhibitor and matrix hyaluronan promote angiogenesis in fibrotic lung injury. Am. J. Respir. Crit. Care Med. 178, 939–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He H., Li W., Tseng D. Y., Zhang S., Chen S. Y., Day A. J., and Tseng S. C. (2009) Biochemical characterization and function of complexes formed by hyaluronan and the heavy chains of inter-α-inhibitor (HC·HA) purified from extracts of human amniotic membrane. J. Biol. Chem. 284, 20136–20146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Selbi W., de la Motte C. A., Hascall V. C., Day A. J., Bowen T., and Phillips A. O. (2006) Characterization of hyaluronan cable structure and function in renal proximal tubular epithelial cells. Kidney Int. 70, 1287–1295 [DOI] [PubMed] [Google Scholar]
- 43. Kessler S., Rho H., West G., Fiocchi C., Drazba J., and de la Motte C. (2008) Hyaluronan (HA) deposition precedes and promotes leukocyte recruitment in intestinal inflammation. Clin. Transl. Sci. 1, 57–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pozsgay M., Szabó G., Bajusz S., Simonsson R., Gáspár R., and Elödi P. (1981) Study of the specificity of thrombin with tripeptidyl-p-nitroanilide substrates. Eur. J. Biochem. 115, 491–495 [DOI] [PubMed] [Google Scholar]
- 45. Lottenberg R., Hall J. A., Blinder M., Binder E. P., and Jackson C. M. (1983) The action of thrombin on peptide p-nitroanilide substrates. Substrate selectivity and examination of hydrolysis under different reaction conditions. Biochim. Biophys. Acta 742, 539–557 [DOI] [PubMed] [Google Scholar]
- 46. Gallwitz M., Enoksson M., Thorpe M., and Hellman L. (2012) The extended cleavage specificity of human thrombin. PloS one 7, e31756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bock P. E., Panizzi P., and Verhamme I. M. (2007) Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost. 5 suppl. 1, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. UniProt Consortium (2015) UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chan P., Risler J. L., Raguenez G., and Salier J. P. (1995) The three heavy-chain precursors for the inter-α-inhibitor family in mouse: new members of the multicopper oxidase protein group with differential transcription in liver and brain. Biochem. J. 306, 505–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thøgersen I. B., and Enghild J. J. (1995) Biosynthesis of bikunin proteins in the human carcinoma cell line HepG2 and in primary human hepatocytes. Polypeptide assembly by glycosaminoglycan. J. Biol. Chem. 270, 18700–18709 [DOI] [PubMed] [Google Scholar]
- 51. Koizumi M., King N., Lobb R., Benjamin C., and Podolsky D. K. (1992) Expression of vascular adhesion molecules in inflammatory bowel disease. Gastroenterology 103, 840–847 [DOI] [PubMed] [Google Scholar]
- 52. Sans M., Panés J., Ardite E., Elizalde J. I., Arce Y., Elena M., Palacín A., Fernández-Checa J. C., Anderson D. C., Lobb R., and Piqué J. M. (1999) VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology 116, 874–883 [DOI] [PubMed] [Google Scholar]
- 53. Mayer U., Mann K., Timpl R., and Murphy G. (1993) Sites of nidogen cleavage by proteases involved in tissue homeostasis and remodelling. Eur. J. Biochem. 217, 877–884 [DOI] [PubMed] [Google Scholar]
- 54. Sage H., Pritzl P., and Bornstein P. (1981) Susceptibility of type V collagen to neutral proteases: evidence that the major molecular species is a thrombin-sensitive heteropolymer, [α1(V)]2 α2(V). Biochemistry 20, 3778–3784 [DOI] [PubMed] [Google Scholar]
- 55. Liotta L. A., Goldfarb R. H., Brundage R., Siegal G. P., Terranova V., and Garbisa S. (1981) Effect of plasminogen activator (urokinase), plasmin, and thrombin on glycoprotein and collagenous components of basement membrane. Cancer Res. 41, 4629–4636 [PubMed] [Google Scholar]
- 56. Sigle R. O., Gil S. G., Bhattacharya M., Ryan M. C., Yang T. M., Brown T. A., Boutaud A., Miyashita Y., Olerud J., and Carter W. G. (2004) Globular domains 4/5 of the laminin α3 chain mediate deposition of precursor laminin 5. J. Cell Sci. 117, 4481–4494 [DOI] [PubMed] [Google Scholar]
- 57. Day A. J., and de la Motte C. A. (2005) Hyaluronan cross-linking: a protective mechanism in inflammation? Trends Immunol. 26, 637–643 [DOI] [PubMed] [Google Scholar]
- 58. Taylor K. R., Yamasaki K., Radek K. A., Di Nardo A., Goodarzi H., Golenbock D., Beutler B., and Gallo R. L. (2007) Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. J. Biol. Chem. 282, 18265–18275 [DOI] [PubMed] [Google Scholar]
- 59. Zaman A., Cui Z., Foley J. P., Zhao H., Grimm P. C., Delisser H. M., and Savani R. C. (2005) Expression and role of the hyaluronan receptor RHAMM in inflammation after bleomycin injury. Am. J. Respir. Cell Mol. Biol. 33, 447–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kessler S. P., Obery D. R., and de la Motte C. (2015) Hyaluronan synthase 3 null mice exhibit decreased intestinal inflammation and tissue damage in the DSS-induced colitis model. Int. J. Cell Biol. 2015, 745237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. DeGrendele H. C., Estess P., Picker L. J., and Siegelman M. H. (1996) CD44 and its ligand hyaluronate mediate rolling under physiologic flow: a novel lymphocyte-endothelial cell primary adhesion pathway. J. Exp. Med. 183, 1119–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. DeGrendele H. C., Estess P., and Siegelman M. H. (1997) Requirement for CD44 in activated T cell extravasation into an inflammatory site. Science 278, 672–675 [DOI] [PubMed] [Google Scholar]
- 63. Teder P., Vandivier R. W., Jiang D., Liang J., Cohn L., Puré E., Henson P. M., and Noble P. W. (2002) Resolution of lung inflammation by CD44. Science 296, 155–158 [DOI] [PubMed] [Google Scholar]
- 64. Albeiroti S., Ayasoufi K., Hill D. R., Shen B., and de la Motte C. A. (2015) Platelet hyaluronidase-2: an enzyme that translocates to the surface upon activation to function in extracellular matrix degradation. Blood 125, 1460–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aruffo A., Stamenkovic I., Melnick M., Underhill C. B., and Seed B. (1990) CD44 is the principal cell surface receptor for hyaluronate. Cell 61, 1303–1313 [DOI] [PubMed] [Google Scholar]
- 66. Black K. E., Collins S. L., Hagan R. S., Hamblin M. J., Chan-Li Y., Hallowell R. W., Powell J. D., and Horton M. R. (2013) Hyaluronan fragments induce IFNβ via a novel TLR4-TRIF-TBK1-IRF3-dependent pathway. Journal of Inflammation 10, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. de la Motte C., Nigro J., Vasanji A., Rho H., Kessler S., Bandyopadhyay S., Danese S., Fiocchi C., and Stern R. (2009) Platelet-derived hyaluronidase 2 cleaves hyaluronan into fragments that trigger monocyte-mediated production of proinflammatory cytokines. Am. J. Pathol. 174, 2254–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nishimura H., Kakizaki I., Muta T., Sasaki N., Pu P. X., Yamashita T., and Nagasawa S. (1995) cDNA and deduced amino acid sequence of human PK-120, a plasma kallikrein-sensitive glycoprotein. FEBS Lett. 357, 207–211 [DOI] [PubMed] [Google Scholar]
- 69. Daveau M., Jean L., Soury E., Olivier E., Masson S., Lyoumi S., Chan P., Hiron M., Lebreton J. P., Husson A., Jegou S., Vaudry H., and Salier J. P. (1998) Hepatic and extra-hepatic transcription of inter-α-inhibitor family genes under normal or acute inflammatory conditions in rat. Arch. Biochem. Biophys. 350, 315–323 [DOI] [PubMed] [Google Scholar]
- 70. Boyum A. (1968) Isolation of mononuclear cells and granulocytes from human peripheral blood. Scand. J. Clin. Lab. Invest. 21, 77–89 [PubMed] [Google Scholar]
- 71. Stossel T. P., Pollard T. D., Mason R. J., and Vaughan M. (1971) Isolation and properties of phagocytic vesicles from polymorphonuclear leukocytes. J. Clin. Invest. 50, 1745–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fogelman A. M., Elahi F., Sykes K., Van Lenten B. J., Territo M. C., and Berliner J. A. (1988) Modification of the Recalde method for the isolation of human monocytes. J. Lipid Res. 29, 1243–1247 [PubMed] [Google Scholar]
- 73. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., and Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 74. de Castro E., Sigrist C. J., Gattiker A., Bulliard V., Langendijk-Genevaux P. S., Gasteiger E., Bairoch A., and Hulo N. (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu W., Xie Y., Ma J., Luo X., Nie P., Zuo Z., Lahrmann U., Zhao Q., Zheng Y., Zhao Y., Xue Y., and Ren J. (2015) IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics 31, 3359–3361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]