

Abstract
In the retina, aberrant opsin transport from cell bodies to outer segments leads to retinal degenerative diseases such as retinitis pigmentosa. Opsin transport is facilitated by the intraflagellar transport (IFT) system that mediates the bidirectional movement of proteins within cilia. In contrast to functions of the anterograde transport executed by IFT complex B (IFT-B), the precise functions of the retrograde transport mediated by IFT complex A (IFT-A) have not been well studied in photoreceptor cilia. Here, we analyzed developing zebrafish larvae carrying a null mutation in ift122 encoding a component of IFT-A. ift122 mutant larvae show unexpectedly mild phenotypes, compared with those of mutants defective in IFT-B. ift122 mutants exhibit a slow onset of progressive photoreceptor degeneration mainly after 7 days post-fertilization. ift122 mutant larvae also develop cystic kidney but not curly body, both of which are typically observed in various ciliary mutants. ift122 mutants display a loss of cilia in the inner ear hair cells and nasal pit epithelia. Loss of ift122 causes disorganization of outer segment discs. Ectopic accumulation of an IFT-B component, ift88, is observed in the ift122 mutant photoreceptor cilia. In addition, pulse-chase experiments using GFP-opsin fusion proteins revealed that ift122 is required for the efficient transport of opsin and the distal elongation of outer segments. These results show that IFT-A is essential for the efficient transport of outer segment proteins, including opsin, and for the survival of retinal photoreceptor cells, rendering the ift122 mutant a unique model for human retinal degenerative diseases.
Keywords: cilia, hair cell, photoreceptor, retinal degeneration, rhodopsin
Introduction
In vertebrates, cilia are essential for the function of sensory cells, including retinal photoreceptor cells, inner ear hair cells, and nasal olfactory cells (1–4). Retinal photoreceptor cells develop photosensitive organelles called outer segments, in which photosensitive G protein-coupled receptors (GPCRs),2 opsins, are densely clustered. Outer segments are highly modified cilia that develop from the primary cilia of photoreceptor precursor cells (5, 6). In the human eye, impairment of opsin transport from cell bodies to outer segments leads to retinal degenerative diseases such as retinitis pigmentosa (RP) and Leber congenital amaurosis (2, 7). Consistent with this, defects of ciliary function induced by mutations in genes encoding ciliary proteins cause RP and Leber congenital amaurosis (1, 2, 7).
The assembly and maintenance of cilia require bidirectional movement of protein complexes along the microtubule-based axoneme (5). This motor system is termed intraflagellar transport (IFT). Transport from the ciliary base toward the ciliary tip (anterograde) is mediated by the IFT complex B (IFT-B) and kinesin-2 motors, whereas transport from the ciliary tip back to the base (retrograde) is executed by the IFT complex A (IFT-A) and dynein motors (8). Mutations in genes encoding IFT-A components cause ciliopathies characterized by skeletal abnormalities, renal malformations, and retinal degeneration. In humans, mutations in IFT122 (associated with Sensenbrenner syndrome) (9), WDR35/IFT121 (associated with Sensenbrenner and short-rib polydactyly syndromes) (10, 11), TTC21B/IFT139 (associated with Jeune syndrome and nephronophthisis) (12), IFT43 (associated with Sensenbrenner syndrome) (13), and WDR19/IFT144 (associated with Sensenbrenner syndrome) (14) produce ciliopathies. Several studies have reported retinal abnormalities in these patients (12, 14). Patients with mutations in IFT144 exhibited extinguished rod signals in electroretinograms and attenuated blood vessels in ophthalmoscopic analysis (14). However, the pathological basis underlying the retinal abnormalities caused by IFT-A dysfunction has been poorly explored.
Several IFT-A deficiencies have been reported in mice. Sister of open brain (sopb), a mouse null mutant of Ift122, and the Ift122 knock-out mouse display exencephaly, caudal neural tube defects, preaxial polydactyly, and left-right asymmetry, and are embryonic lethal with the impairment of the Shh pathway (15, 16). In fact, Ift122 was found to control the distribution of the Shh pathway activators and repressors along the cilium. In addition to the patterning defects during development, shortened cilia that display IFT-B mislocalization have been described to appear as a consequence of IFT-A defect (16). Another mouse mutant, Alien (aln) (Ift139/Thm1/Ttc21b) shows polydactyly, microphthalmia, irregular shape of long bones, rib fusion and truncation, neural tube defects, and abnormal brain structure (17). The embryonic lethality of these animals conceals the IFT-A functions at later developmental stages. Ift144, Ift140, and Ift122 encode components of a stable IFT-A core subcomplex that may explain why mutations in these genes result in the similar disease states (18). The structural details of each component of the IFT core machinery and further functional analysis are required to elucidate the molecular mechanism in which they operate and shed light on phenotypic similarities observed in mutants of IFT-A components.
In zebrafish, IFT-B mutants, including fleer/IFT70, elipsa/Traf3ip1, oval/IFT88, and qilin/Cluap1, display specific phenotypes such as cystic kidney, early photoreceptor degeneration, and curly body axis (19–24). However, no IFT-A mutants have been reported so far. Although zebrafish ift122 knockdown was reported to lead to pronephric cyst with shortened cilia (9), the loss of ift122 function phenotype in sensory organs has not yet been analyzed. To our knowledge, this is the first report on the phenotypic analysis of a zebrafish IFT-A mutant in sensory organs. Our results show essential roles for IFT-A in the formation and maintenance of sensory cells.
Results
jj263 Mutant Larvae Exhibit Photoreceptor Cell Degeneration in the Retina
To identify mutations affecting photoreceptor survival, we performed an N-ethyl-N-nitrosourea mutagenesis screen in zebrafish. To analyze the morphological integrity of photoreceptor cells, we used a transgenic zebrafish strain that expresses GFP in retinal rod photoreceptor cells under the control of the rhodopsin promoter (25). During this screen, we identified a recessive mutant line, jj263, which exhibited progressive degeneration of retinal photoreceptor cells (Fig. 1, A–D). By 10 dpf, wild-type larvae develop three distinct nuclear layers in the retina as follows: the ganglion cell layer, the inner nuclear layer (INL), and the outer nuclear layer (ONL, photoreceptor cell layer) (Fig. 1, A and B). We found that jj263 larvae exhibited ONL degeneration at 10 dpf (Fig. 1, C and D), whereas the INL and ganglion cell layer appeared to be normal. This result suggests that the jj263 locus is critical for the development and/or survival of photoreceptor cells. Several mutants with photoreceptor degeneration were previously reported to carry mutations in IFT-B, including fleer/IFT70, elipsa/Traf3ip1, oval/IFT88, and qilin/Cluap1 (19–24). Although all of these IFT-B mutants display a curly body, the jj263 mutant larvae showed no significant curly phenotype (Fig. 1, E and F). Ciliary mutants with photoreceptor degeneration often exhibit functional defects of the kidney, leading to a cystic kidney phenotype (26). We observed pronephric cyst formation in jj263 mutant larvae at 6 dpf (Fig. 1, E–H). To investigate whether jj263 phenotypes were caused by ciliary defects, we visualized cilia in the pronephric tubules at 7 dpf by antibody staining. Bundles of cilia extending into the lumen of pronephric tubules were observed in wild-type larvae (Fig. 1, I and I′). In contrast, ciliary bundles in mutant pronephric ducts were disorganized and shorter compared with those of the control animals (Fig. 1, J and J′). These results suggest that the pronephric cyst in jj263 mutant larvae is attributable to ciliary defects in the kidney.
FIGURE 1.

jj263 mutant larvae exhibited photoreceptor degeneration and cystic kidney without curly body axis phenotype. A–D, jj263 mutant larvae showed photoreceptor degeneration in the retina. Retinal sections at 10 dpf of wild-type (A and B) and jj263 mutant (C and D) larvae were immunostained with anti-Zpr1 (an-arrestin, double cone photoreceptors, green) and anti-M-opsin (a cone outer segment marker, red) antibodies. Nuclei were stained with DAPI (blue). In the wild-type retina, cone photoreceptors are localized in the ONL and form the outer segments on apical sides of the ONL. In the jj263 mutant larvae, photoreceptor cells in the ONL were severely degenerated. In contrast, the INL in the jj263 mutant larvae was almost normal. Dotted lines indicate the boundary of retinal pigment epithelia. E–H, lateral (E and F) and dorsal (G and H) views of wild-type (E and G) and jj263 mutant (F and H) larvae at 6 dpf. A cystic kidney phenotype was observed in the jj263 mutant larvae (F and H, arrow). The curly body axis phenotype, a common feature of ciliary mutants, was not observed in the jj263 mutant larvae. I–J′, pronephric cilia are disorganized and shortened in the jj263 mutant larvae. Pronephric cilia in the wild-type (I and I′) and mutant (J and J′) larvae were immunostained with an acetylated α-tubulin antibody (red). Pronephric epithelia in pronephric tubules were stained with phalloidin (green). Nuclei were stained with DAPI (blue). Dotted lines indicate boundaries of pronephric tubules. ONL, outer nuclear layer; INL, inner nuclear layer; LE, lens.
Zebrafish jj263 Locus Encodes ift122, an IFT-A Component
To identify the gene responsible for the jj263 mutant phenotypes, we meiotically mapped the mutation to a region in chromosome 8 defined by a microsatellite Z8703 (0 recombination in 177 meiotic events; Fig. 2A). ift122, a component of IFT-A, is located in this region of zebrafish chromosome 8. To assess whether jj263 harbors a mutation in the ift122 gene, we sequenced the ift122 coding region. We found a nonsense mutation at position 63 of the ift122 gene in the mutant larvae, resulting in a severely truncated ift122 protein (Fig. 2, B and C). We confirmed that there was no recombination between the ift122 gene and the polymorphic marker ift122p17p18 in 177 meioses. To provide further evidence that ift122 is responsible for the jj263 mutant phenotype, we performed an mRNA rescue experiment. We prepared full-length human IFT122 mRNA and injected it into embryos obtained from crosses between jj263 mutant heterozygotes. We found that 25.0% of embryos formed kidney cysts when control GFP mRNA was injected, whereas only 6.8% of embryos injected with IFT122 mRNA formed kidney cysts at 5 dpf (212 and 88 embryos were injected in these two groups respectively, p < 0.03, Fig. 2, D and E). Taken together, these results provide convincing evidence that jj263 encodes ift122. In zebrafish, zygotic transcription starts at around the 512-cell stage (27), and the early period of embryonic development is controlled by maternal gene products. Several components of IFT-B have been shown to be maternally deposited in zebrafish (28, 29). To evaluate the maternal contribution of the ift122 transcript, RT-PCR was carried out on RNA extracted from wild-type embryos at four different developmental stages (Fig. 2F). At the 4–16-cell stage as well as at later stages, we detected the ift122 transcript, confirming that maternal contribution of ift122 mRNA is present in early embryos. This result suggests a possibility that the lack of an early cilia phenotype, including curved body axis and hydrocephalus in the ift122 mutant, is due to the presence of maternal ift122 mRNA in early embryos.
FIGURE 2.

jj263 locus encodes the zebrafish ift122. A, genetic map of the jj263 locus and the exon/intron structure of the ift122 gene. The ift122 locus was mapped on zebrafish chromosome 8 in the vicinity of a genetic marker Z8703. B, domain structure of the zebrafish ift122 protein and the mutation of the jj263 allele. The N-terminal WD40 repeats and the C-terminal TPR domain were indicated. The position of the ift122jj263 mutation is indicated with an arrow. This mutation produces a premature stop codon resulting in a 62-amino acid truncated product of ift122. C, sequencing trace data for the wild-type (left) and jj263 allele (right). The tyrosine residue at position 63 was changed to stop codon in the jj263 mutant. D and E, rescue by injection of IFT122 mRNA into embryos from crosses between jj263 heterozygotes. D, percentages of larvae that display pronephric cysts at 5 dpf following injection of GFP mRNA or IFT122 mRNA into embryos. E, images of ift122 mutant larvae at 5 dpf following injection of GFP (left panel) or IFT122 mRNA (right panel). Pronephric cyst formation is partially eliminated in homozygous ift122 mutants by injection of IFT122 mRNA (right panel). Arrowheads point to pronephric cysts. F, RT-PCR analysis of ift122 expression in embryos at several developmental stages. Maternal ift122 transcript was detected in embryos at 4–16-cell stage. A housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (gapdh), was used as a loading control. Three different concentrations of cDNA templates (1×, 0.2×, and 0.04×) were used.
Ciliary Defects in Sensory Organs of ift122 Mutants
To address ift122 function in sensory neurons, we first investigated ciliary integrity in the inner ear (Fig. 3, A–D). Hair cells in inner ear cristae develop kinocilia until 3 dpf in wild-type larvae (Fig. 3A). Compared with the wild type, we observed slightly shortened kinocilia in ift122 mutant hair cells at 3 dpf (Fig. 3B). At 5 dpf, kinocilia were often disrupted in ift122 mutants (Fig. 3D), compared with those of wild-type larvae (Fig. 3C). Hair cells of wild-type neuromast have long kinocilia at 4 dpf, whereas kinocilia are severely disrupted in ift122 mutants (Fig. 3, E and F). In contrast to the disorganization of kinocilia in ift122 mutants, stereocilia, which are actin-based structures, appeared to be normal in ift122 mutant cristae and neuromast hair cells (Fig. 3, D and F), showing that the loss of ift122 specifically affects kinocilia in hair cells.
FIGURE 3.
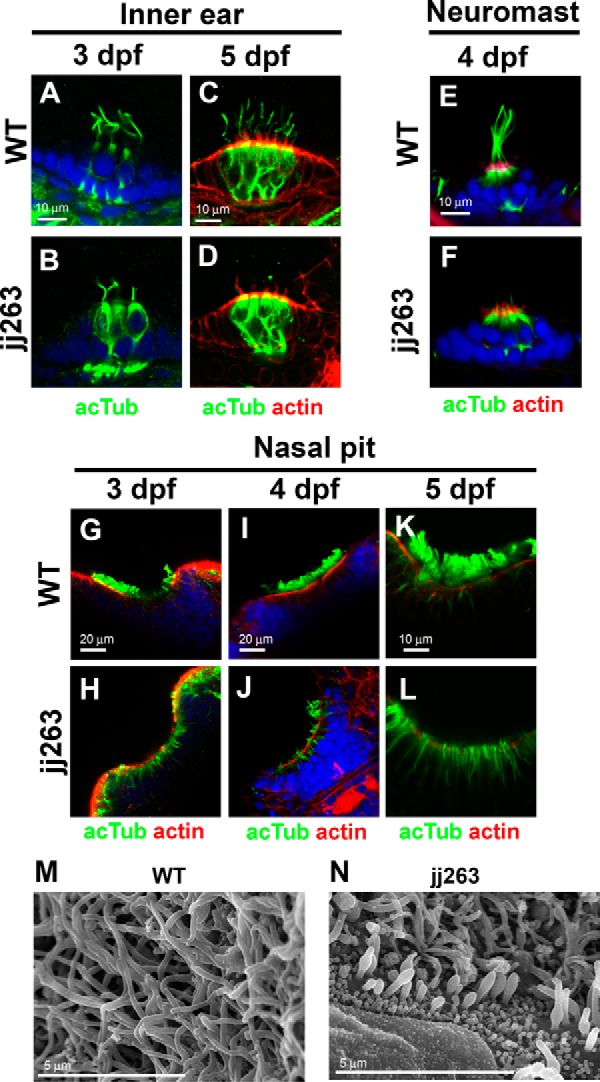
Ciliary defects in sensory organs of the ift122 mutant. A–F, hair cell kinocilium in the inner ear (A–D) and neuromasts (E and F) of wild-type (A, C, and E) and jj263 (B, D, and F) embryos at 3 dpf (A and B), 4 dpf (E and F), and 5 dpf (C and D). Cilia are immunostained with an anti-acetylated α-tubulin antibody (green). Stereocilia of hair cells were stained with phalloidin (actin staining, red). Nuclei were stained with DAPI (blue). Kinocilia of hair cells were disorganized in the ift122 mutant inner ear and neuromast. G–L, cilia in the wild-type (G, I, and K) and ift122 mutant (H, J, and L) nasal pit at 3 dpf (G and H), 4 dpf (I and J), and 5 dpf (K and L). Cilia are immunostained with an anti-acetylated α-tubulin antibody (green). Apical surface of epithelia were labeled with phalloidin (actin staining, red). Nuclei were stained with DAPI (blue). Cilia do not develop in mutant nasal pit at 3 dpf. M and N, ultrastructural analysis of nasal cilia in wild-type and ift122 mutant larvae at 4 dpf. Swollen cilia were observed in the ift122 mutant.
To investigate the formation and maintenance of cilia in nasal pits, we immunostained wild-type and mutant nasal pits at several developmental stages using an acetylated α-tubulin antibody (Fig. 3, G–L). We found cilia in the nasal pit of wild-type larvae at 3 dpf (Fig. 3G), whereas cilia were almost absent in mutant nasal pits at the same stage (Fig. 3H). At 4 dpf, cilia elongate in wild-type nasal pits (Fig. 3I). In ift122 mutant nasal pits, we often found short and disorganized cilia (Fig. 3J). At 5 dpf, cilia are fully formed in nasal pits of wild-type larvae (Fig. 3K), whereas the cilia are largely absent in mutant larvae (Fig. 3L). To further investigate ciliary structure in nasal pits, we analyzed nasal cilia of ift122 mutants using electron microscopy at 4 dpf. In the wild-type larvae, multiple long cilia are observed on the surface of the nasal pit (Fig. 3M). In contrast, consistent with the results of immunostaining, we often found swollen and shortened cilia in ift122 mutant nasal pits (Fig. 3N). These results show that ift122 is essential for proper formation and maintenance of cilia in the nasal pit.
Slow Progressive Photoreceptor Degeneration in the ift122jj263 Mutant Retina
Previously, IFT-B mutants were reported to exhibit severe photoreceptor degeneration around 5 dpf (19–24). To determine precisely the time course of photoreceptor degeneration in the ift122 mutant retina, we analyzed photoreceptor cell body and outer segments at 4, 5, 7, and 10 dpf (Fig. 4, A–L). Photoreceptor degeneration in the mutant retina was not observed at 4 and 5 dpf (Fig. 4, B, C, E, and F). At 7 dpf, only a small portion of the photoreceptor layer had degenerated in the ift122 mutant retina (Fig. 4. H and I, dotted lines). We found, however, that most regions of the photoreceptor layer underwent degeneration in the mutant retinae by 10 dpf, with the exception of the retinal periphery where retinal neurons are generated (Fig. 4, K and L). Our quantitative analysis of the degenerated region confirmed photoreceptor degeneration at 7 and 10 dpf (Fig. 4M, n = 9–18). These results show that photoreceptor degeneration in the ift122 mutant retina is significantly slower, compared with the early onset degeneration in the IFT-B mutant retina.
FIGURE 4.
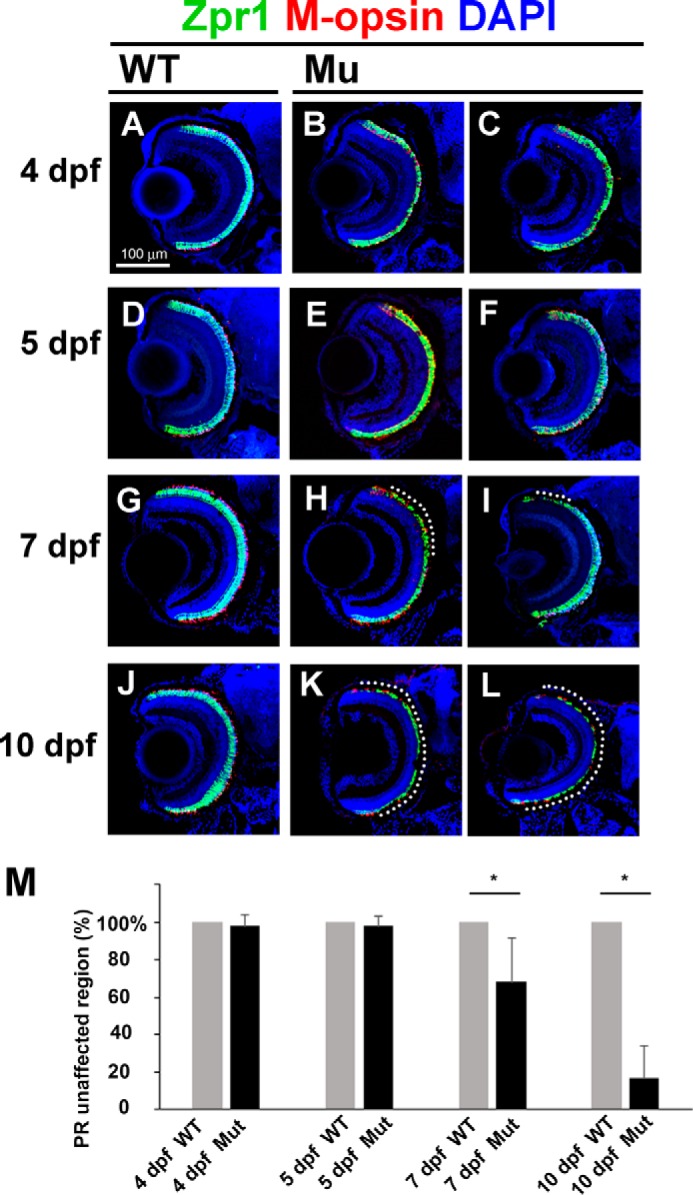
Slow progressive photoreceptor degeneration in the ift122 mutant retina. A–L, ift122 mutant larvae showed slow photoreceptor degeneration in the retina. Retinal sections at 4 dpf (A–C), 5 dpf (D–F), 7 dpf (G–I), and 10 dpf (J–L) of wild-type (A, D, G, and J) and ift122 mutant (B, C, E, F, H, I, K, and L) larvae were stained with anti-Zpr1 (double cone photoreceptors, green), and M-opsin (a cone outer segment marker, red) antibodies. Nuclei were stained with DAPI (blue). No photoreceptor degeneration was observed in the ift122 mutant retina between 4 and 5 dpf. Only small portions of the photoreceptor layer degenerate at 7 dpf (H and I, dotted lines). Most of the photoreceptor layer degenerated by 10 dpf in the ift122 mutant retina (K and L). M, we measured the depth of the degenerated photoreceptor region and calculated the size of the unaffected region. Significant photoreceptor degeneration was observed at 7 and 10 dpf. n = 9–18.
Defects of Opsin Transport Machinery in the ift122jj263 Mutant Retina
ift122 mutant larvae show photoreceptor loss at 10 dpf (Fig. 1). To determine the mechanism of photoreceptor degeneration in the ift122 mutant retina, we first analyzed the distribution of opsin in photoreceptor cells (Fig. 5, A–L). In the wild-type retina, M-opsin was mostly localized to the outer segments in Zpr1-positive double cone photoreceptors (Fig. 5A). In contrast, M-opsin was broadly distributed to the outer segments and cell bodies in ift122 mutant photoreceptors at 4 dpf (Fig. 5, B and C). At 5dpf, M-opsin was enriched in the outer segments of mutant photoreceptors, although ectopic M-opsin accumulation in the cell bodies continued to be observed (Fig. 5, E and F). At 7dpf, although photoreceptor cell bodies in the mutant retina are smaller than those in the wild-type retina, M-opsin signal was detected in mutant outer segments (Fig. 5, G–I). At 10 dpf, only degenerated photoreceptors were observed in most parts of the photoreceptor layer in mutants (Fig. 5, K and L). Opsins are produced in the photoreceptor cell body and transported to the outer segment through the connecting cilium. In connecting cilia, the anterograde transport of opsins is likely mediated by IFT-B machinery with the kinesin-2 motor (30). Previous reports showed that loss of function of IFT-B machinery components causes the accumulation of opsins in photoreceptor cell bodies and induces photoreceptor cell degeneration (20, 23). To investigate the IFT-B contribution to photoreceptor degeneration, we next observed subcellular localization of an IFT-B component, ift88, in the ift122 mutant retina (Fig. 5, M–R). In this assay, cilia were visualized in photoreceptors using anti-acetylated α-tubulin labeling. In the ift122 mutant, cilia were observed, but their length was shorter, compared with that of wild-type cilia (Fig. 5, N, N″, P, P″, and R, 25 cilia from five larvae for each group). This result indicates that ift122 is essential for cilia elongation and/or the maintenance of cilia length. In addition, we found strong accumulation of ift88 in the cilia of photoreceptor cells in the ift122 mutant retina (Fig. 5, N, N′, P, and P′) and the ift88 signal intensity was significantly higher in ift122 mutant photoreceptor cilia (Fig. 5Q, 25 cilia from five larvae for each group), suggesting that the loss of ift122 affects the retrograde transport of IFT-B proteins back into the cell body from the outer segments.
FIGURE 5.

Defect of the opsin transport machinery in the ift122 mutant retina. A–L, photoreceptor cells at 4 dpf (A–C), 5 dpf (D–F), 7 dpf (G–I), and 10 dpf (J–L) in wild-type (A, D, G, and J) and ift122 mutant (B, C, E, F, H, I, K, and L) larvae were immunostained with anti-Zpr1 (green) and anti-M-opsin (red) antibodies. In the 4 dpf wild type, the M-opsin was localized at the outer segments (A). In contrast, the M-opsin signal was widely distributed in photoreceptor cell bodies in ift122 mutant larvae at 4 dpf (B and C). At 5 dpf, ectopic accumulation of M-opsin signal in photoreceptor cell bodies was still observed (E and F), but enrichment of the M-opsin in the outer segment was also observed in mutant photoreceptors. At 7 dpf, the cell bodies of mutant photoreceptors were smaller compared with those of wild-type photoreceptors (H and I). At 10 dpf, only degenerated photoreceptors were observed in most of the photoreceptor layer in the mutant retina (K and L). Arrows indicate outer segments. M–P″, retinal sections were immunostained with an anti-Ift88 antibody (green) in the wild-type (M–N″) and mutant larvae (O–P″) at 4 dpf. Photoreceptor cilia are immunostained with an anti-acetylated α-tubulin antibody (red). Accumulations of the ift88 signals are observed in the photoreceptor cilia of mutant larvae. Q and R, quantification of length and ift88 signal intensity in photoreceptor cilia of ift122 mutant larvae at 4 dpf. Accumulation of ift88 at ciliary tips was significantly increased in ift122 photoreceptor cilia (Q). Photoreceptor cilia stained with acetylated α-tubulin were significantly shorter in ift122 mutant retina (R). Nuclei were stained with DAPI (blue). ONL, outer nuclear layer.
To examine opsin transport efficiency, we performed a pulse-chase experiment using GFP fused with the 44 C-terminal amino acids of opsin, a sequence sufficient for outer segment targeting (GFP-opsin) (31, 32). We generated a pulse of GFP-opsin expression in photoreceptors of the wild-type and ift122 mutant retina by heat-shock treatment (Fig. 6, A–F). In wild-type photoreceptors, GFP-opsin was rapidly transported from the cell body to the outer segment within 4 h after heat-shock induction and was maintained in the outer segment for 24 h (Fig. 6, B, D, and F). In contrast, GFP-opsin initially accumulated in the cell bodies of ift122 mutant photoreceptors (Fig. 6, C and F). GFP-opsin was eventually transported from the cell body to outer segments by 24 h after heat-shock treatment in ift122 mutant retinas (Fig. 6, D–F). These results suggest that ift122 function is required for opsin transport in vertebrate photoreceptor cells.
FIGURE 6.

Pulse-chase experiment using GFP-opsin fusion protein in ift122-deficient photoreceptor cells. A, schematic diagram of the heat-shock promoter-driven GFP-opsin expression construct used in this study. Time course of heat-shock induction of GFP-opsin in the larvae (lower panel). B–E, representative confocal images of transverse cryosections through the central retina of wild-type and ift122 mutant larvae at 4 and 24 h after heat-shock induction. The subcellular distribution of the GFP-opsin (green) was evaluated. Sections are counterstained with phalloidin (red) to visualize the outer limiting membrane and the outer plexiform layer and DAPI (blue) to visualize cell nuclei. Signal intensity in cell bodies was measured between the outer limiting membrane and the outer plexiform layer. Asterisks indicate outer segments; arrowheads indicate the outer limiting membrane, and arrows indicate the outer plexiform layer. F, graph representing signal intensities of GFP-opsin in photoreceptor cell bodies of wild-type and ift122 mutant larvae at 4 and 24 h after heat-shock induction. Percentages of GFP signal intensity in the photoreceptor cell body relative to the entire cell are shown.
Ultrastructural Analysis of Photoreceptor Outer Segments in the ift122 Mutant Retina
Electron microscopy was performed to evaluate photoreceptor outer segment integrity in mutant larvae (Fig. 7). At 4 dpf, wild-type photoreceptor outer segments were elongated and contained well organized stacks of discs (Fig. 7, A and B). In contrast, outer segment discs were severely disorganized in mutant photoreceptor cells (Fig. 7, C–H), showing that ift122 is required for the proper stacking of outer segment discs and continuous elongation of outer segments. Interestingly, most photoreceptor cells in ift122 mutants have outer segments, which were abnormally shortened with disorganized disc stacks. Previous studies showed that mutations in IFT-B components cause nearly complete loss of photoreceptor outer segments in photoreceptor cells (19, 20). Our results suggest that ift122 is essential for their proper elongation and/or maintenance.
FIGURE 7.

Ultrastructural analysis of photoreceptor outer segments in the ift122 mutant retina. A–H, electron microscopic images of sections in the wild-type (A and B) and ift122 mutant (C–H) photoreceptor cells at 4 dpf. EM images of sections perpendicular to outer segments in wild-type and mutant photoreceptors. Severely disorganized outer segment discs were observed in mutant photoreceptor cells. Arrows indicate outer segments and arrowheads in enlarged images point to outer segment discs.
Discussion
In contrast to the fairly good understanding of anterograde ciliary transport machinery mediated by IFT-B and kinesin-2 motors, the biological functions of retrograde transport mediated by IFT-A and dynein motors are poorly understood in vertebrate photoreceptors. Here, we performed phenotypic analysis of a novel zebrafish mutant defective in ift122, a component of IFT-A. We showed that ift122 mutant larvae exhibit a progressive degeneration of retinal photoreceptor cells accompanied by the accumulation of opsin in photoreceptor cell bodies and the disorganization of the outer segment disc array. We also found that loss of ift122 impaired ciliogenesis in the inner ear, neuromasts, and the nasal pit. Accumulation of an IFT-B component is observed in mutant photoreceptor cilia. Thus, our results reveal essential roles for IFT-A in ciliogenesis in the vertebrate sensory organs and in the survival of retinal photoreceptor cells.
In previous studies, several lines of evidence supported the idea that IFT-A may have other functions in addition to its well established role in retrograde IFT. First, in mice, mutations affecting the IFT dynein motor subunit, Dync2h1, show disruption of the Hedgehog activation pathway, whereas mutations in IFT-A subunits, including Ift122 and Thm1, show overactivation of the Shh pathway (16, 17). These phenotypic differences indicate that IFT-A and dynein display some non-overlapping functions in ciliary transport. Second, a modulator of the Shh pathway, Tulp3, localizes to the ciliary tip both in wild-type and Dync2h1 mutants; however, ciliary localization of Tulp3 is disrupted in Ift122 mutants (16). Third, double mutant embryos lacking both Ift122 and Dync2h1 develop cilia resembling those of the Ift122 single mutants (33). Finally, IFT-A and Tulp3 promote trafficking of a subset of GPCRs to cilia (34). In this study, we observed an abnormal accumulation of opsin in photoreceptor cell bodies and progressive photoreceptor degeneration in ift122 mutants. Our findings contradict a previous study that suggested that the lack of the ciliary retrograde transport motor, cytoplasmic dynein-2, by antisense morpholino knockdown causes zebrafish larvae to exhibit small eyes and outer segment malformation without affecting the localization of opsin (35). A possible interpretation for these phenotypic discrepancies is that IFT-A and dynein-2 function differently. IFT-A may have additional specific functions in retrograde transport in cilia. Another possible explanation is the technical limitations of morpholino knockdown experiments. Because photoreceptor outer segment extension occurs after 60 hours post-fertilization in the zebrafish retina, it is technically difficult to investigate phenotypes such as opsin transport, outer segment extension, or degeneration of photoreceptor cells by morpholino knockdown experiments. Analysis of genetic dynein mutants generated by genome-editing methods would help to understand the similarities and differences between IFT-A and dynein-2 functions in the retrograde IFT system.
We observed the accumulation of opsin in photoreceptor cell bodies in ift122 mutant larvae, suggesting that opsin transport through the cilium was impaired in the mutants. Similar ectopic accumulation of opsins in the cell bodies causes photoreceptor cell death in humans and mice (7). Taken together, loss of ift122 causes the accumulation of opsins in photoreceptor cell bodies, and this accumulation probably triggers the progressive cell death of photoreceptor cells in the ift122 mutant retina. What is the basis of this transport defect? There are at least two possible mechanisms. First, loss of IFT-A not only causes defective retrograde opsin transport but also affects anterograde transport. In support of this idea, depletion of IFT-A components decreases ciliary transport of GPCRs in mammalian cells (34). It is thus possible that ift122 directly regulates opsin trafficking into cilia. Second, an IFT-A defect causes deficiency of IFT-B recycling, which, in turn, affects the anterograde transport of opsin through the connecting cilium. A previous study showed that loss of mouse Ift122 caused accumulation of Ift88 in cilia (16). In this case, dynein-2 mutants are expected to show photoreceptor degeneration similar to that observed in IFT-A mutants. Further investigation using retrograde IFT mutants is needed to clarify this issue.
We previously reported that the loss of a ciliary kinase, Mak, affects photoreceptor ciliary length regulation and causes progressive photoreceptor loss in mice (36). Indeed, MAK mutation in humans causes RP (37, 38). Interestingly, a mutation of the nematode homolog of Mak, Dyf-5, affects the anterograde transport of IFT-A and -B proteins in sensory cilia (39). Recently, we showed that a paralog of Mak, ICK, regulates IFT turnaround at ciliary tips (40). It may be possible that ciliary kinases such as Mak and ICK are also involved in the regulation of IFT-A machinery by phosphorylation of these IFT components.
Based on the results presented in this paper and previous reports, we propose a model of the functional mechanism of ift122 in photoreceptor survival (Fig. 8, A and B). In the wild-type retina at 4 dpf, photoreceptors form outer segments, and opsins are transported from cell bodies to the outer segments through cilia by the IFT machinery (Fig. 8A, upper panel). In the IFT-B mutant retina (Fig. 8A, lower panel), cilia formation is severely disrupted due to the absence of anterograde IFT machinery, resulting in the complete absence of outer segment formation. This causes acute opsin accumulation in the cell body, thereby triggering early onset photoreceptor degeneration. In the ift122 mutant retina, however, loss of IFT-A causes a partial loss of anterograde IFT machinery (Fig. 8A, middle panel), resulting in formation of smaller photoreceptor outer segments. As in the ift122 mutant retina, opsins are partially transported to the outer segments; opsin accumulation in the cell body is less severe; and photoreceptor degeneration is late in onset and moderate in the ift122 mutant retina, compared with that in the IFT-B-deficient retina. In fact, we found that opsins are transported, at least partially, through cilia into the outer segments in the ift122 mutant. Thus, our findings support the idea that the main function of IFT-A in opsin transport is recycling of IFT-B, which is essential for cilia formation and opsin transport into outer segments (Fig. 8B). Importantly, our results suggest that zebrafish IFT-A mutants are excellent models to study slow onset human retinal degenerative diseases.
FIGURE 8.
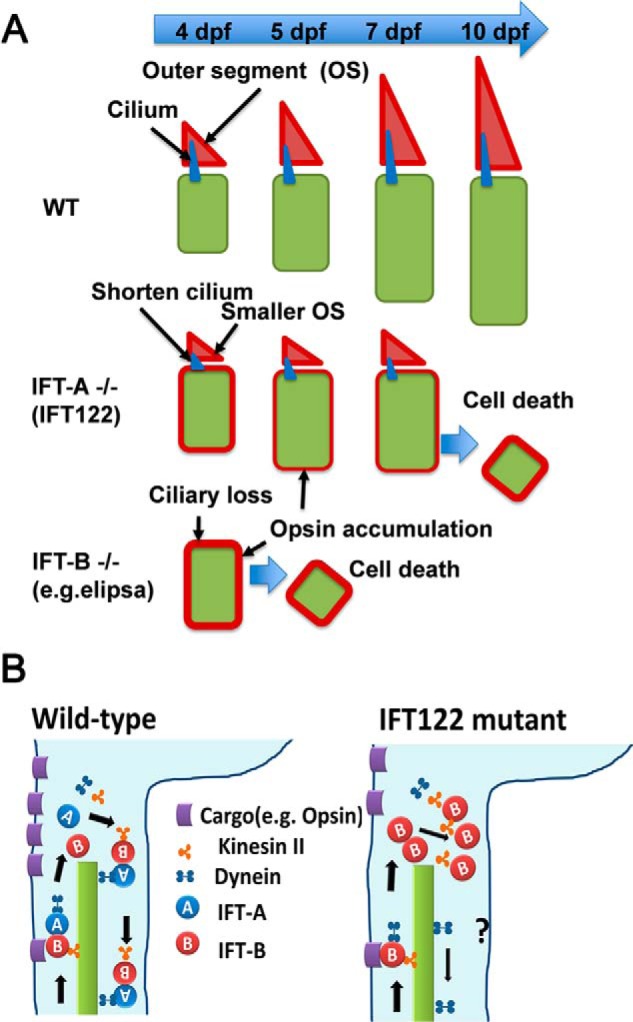
Hypothetical model of ift122 function in photoreceptor cell. A, upper panel, at 4 dpf elongation of photoreceptor OS begins in the wild-type retina. Opsin produced in photoreceptor cell bodies is transported to the OS (red triangles) and accumulated in the OS. Middle panel, in ift122 mutant photoreceptors, smaller OS and shorter cilia were observed at 4 dpf. A certain amount of opsin is transported to the OS; however, probably because of insufficient transport capacity of IFT, opsins are accumulated in the photoreceptor cell bodies. The moderate ectopic opsin accumulation causes the late onset photoreceptor cell death around 7–10 dpf. Lower panel, in the IFT complex B mutant retina (e.g. elipsa) photoreceptors, cilia, and OS are not formed, because the anterograde IFT system is completely disrupted. Opsin accumulates in the photoreceptor cell bodies in the IFT-B mutant retina. The ectopic opsin accumulation causes early photoreceptor cell death around 5 dpf. B, left panel, in wild-type photoreceptor cells, the IFT complex B mediates efficient cargo transport from inner segments to outer segments along ciliary axonemes and are recycled by retrograde transport machinery. Right panel, in ift122 mutant photoreceptors, the IFT complex B is accumulated in the tip of axonemes of connecting cilia due to the loss of IFT recycling. Even under such conditions, certain amount of cargos are transported to outer segments and small outer segments are formed in ift122 mutant photoreceptors.
Experimental Procedures
Zebrafish Maintenance and Breeding
The maintenance and breeding of zebrafish strains as well as the staging of embryonic development were performed as described previously (23, 41, 42). All procedures were approved by the Institutional Safety Committee on Recombinant DNA Experiments (approval ID 3380-4) and the Animal Experimental Committees of the Institute for Protein Research (approval ID 24-05-1), Osaka University, and were performed in compliance with the institutional guidelines.
Mapping and Phenotypic Rescue
A map cross was set up between heterozygous carriers of the ift122jj263 allele (AB genetic background) and wild-type WIK strain homozygotes. To determine the segregation pattern of genomic polymorphisms, F2 embryos were genotyped as described previously (20). To map the ift122 allele, we used the following primers: ift122p17, 5′-ACTTACCATCTTTGGCGTAAGCGA-3′, and ift122p18, 5′-TCATCGTCGCTGCTGGAAACAGA-3′. Mutations were identified by direct sequencing of PCR products amplified from both genomic DNA and cDNA. To rescue the mutant phenotype, the full-length human IFT122 was cloned into the pXT7 vector (19), and in vitro transcription was performed using mMessage mMachine kit (Ambion Inc.), according to the manufacturer's instructions. Approximately 40 pg of RNA was injected into embryos at the one-cell stage.
Immunohistochemistry
Immunohistochemical analysis was performed as described previously (42). Zebrafish larvae were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min to 3 h at room temperature. Fixed larvae were cryoprotected using 30% sucrose in PBS, embedded in TissueTek OCT compound 4583 (Sakura), frozen, and sectioned at 20 μm thickness. Sections were placed on slides, dried for 30 min at room temperature, rehydrated in PBS for 5 min, incubated in blocking solution (5% normal goat serum and 0.5% Triton X-100 in PBS) for 1 h, and incubated with primary antibody in blocking solution for 4 h at room temperature. Slides were washed with PBS three times for 10 min each and incubated with secondary antibodies for 2 h at room temperature. Specimens were observed using a confocal microscope (LSM700, Carl Zeiss). We used the following primary antibodies: mouse monoclonal anti-acetylated α-tubulin (6-11B-1, 1:1500, Sigma) and anti-Arrestin 3a (Zpr1, 1:250, ZIRC); rabbit polyclonal anti-M-opsin (AB5405, 1:1000, Millipore); and anti-Ift88 (1:500, gift from Dr. G. J. Pazour) (43). For actin staining, we used rhodamine phalloidin (PHDR1, 1:500, Cytoskeleton). To obtain the data in Fig. 5, Q and R, the signal intensity of ift88 staining (green channel) in selected areas was measured using the ImageJ software from the confocal images. The length of cilia stained for acetylated α-tubulin (Fig. 5,Q and R, red channel) was measured using the Photoshop CS6 software from confocal images. To obtain Fig. 4M data, photoreceptor layer size was measured as an angle between lines extending from the center of the lens to the upper and lower edges of the area occupied by photoreceptors. The region of the photoreceptor cell layer affected by degeneration was measured in the same way. Measurements were performed on confocal images of each retinal section using the Photoshop software. Size of unaffected region was calculated by dividing the angle of unaffected region by the angle of entire photoreceptor region.
Opsin Transport Assay
We performed a pulse-chase experiment using GFP fused to 44 C-terminal amino acids of zebrafish opsin (GFP-opsin), which are sufficient for outer segment targeting as described previously (32). GFP fusion constructs were injected into embryos at the one-cell stage. Embryos were heat-shocked at 96 hpf for 30 min at 37 °C. Larvae were fixed at 4 and 24 h after heat shock and cryosectioned. Sections were counter-stained with rhodamine-phalloidin and imaged using a confocal microscope (LSM700, Carl Zeiss). Fluorescence signal intensities of GFP in photoreceptor cell bodies and entire cells, including outer segments, were measured in the green channel using ImageJ software. Signal intensity in cell bodies was measured between the outer limiting membrane and the outer plexiform layer. Percentages of photoreceptor cell body signal intensity were calculated dividing the signal intensity in the cell body by the signal intensity in the entire cell.
Electron Microscopy
For electron microscopy, zebrafish larvae were whole-fixed for 3 h in 2.5% glutaraldehyde and 2.5% paraformaldehyde buffered with sodium phosphate. After rinsing, samples were post-fixed in 2% osmium tetraoxide containing 8% sucrose for 2 h. To prevent osmium precipitation, 8% sucrose was added. Samples were rinsed again and dehydrated in an ethanol series (50–100%, 10 min each) and then processed for transmission electron microscopy or scanning electron microscopy. For transmission electron microscopy, dehydrated specimens were incubated in QY-1 (Nissin EM Co., Japan) and embedded in a mixture of Epok812 (44). Ultrathin sections (70 nm) were cut and stained with 3% uranyl acetate and Sato's lead stain (45). Sections were observed using an H-9500SD transmission electron microscope (Hitachi High-Tech, Japan) fitted with a slow scan CCD camera (TemCam-F224HD; TVIPS, Germany) and EMMENU4.0 software. For scanning electron microscopy observations, dehydrated specimens were incubated in hexamethydisilazane for 30 min and dried. After osmium coating, specimens were imaged in an S-5200 (Hitachi High-Tech, Japan) at 15 kV.
RT-PCR Analysis
RNA was extracted from wild-type embryos with TRIzol (Invitrogen) following the manufacturer's instructions. cDNA synthesis was performed using the SuperScript II reverse transcriptase (Invitrogen) followed by PCR amplification. Sequences of primers are as follows: for ift122, 5′-AAGGATTTCCTGGGCTCAGCGGAC-3′ and 5′-CACTGCATTGACAGCATCCAGTAG-3′; for gapdh, 5′-GGATTGCCGTTCATCCATCTTTGAC-3′ and 5′-CGGTTGCTGTAACCGAACTCATTG-3′.
Statistical Analysis
Data are presented as means ± S.D. as indicated in the figure legends. Statistical significance was evaluated using Student's t test, and *, p < 0.05 was taken to be statistically significant.
Author Contributions
Y. O. designed the project. M. B., T. C., A. U., H. H., and Y. O. carried out immunohistochemical, molecular genetic, molecular biological, and biochemical analysis. N. K. and R. K. performed electron microscopic analysis. M. B., T. C., A. U., H. H., T. F., and Y. O. analyzed the data. M. B. and Y. O. wrote the manuscript. J. M. organized the genetic screen in which the jj263 allele was originally isolated and helped to analyze the data from subsequent experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. G. J. Pazour (University of Massachusetts Medical School) for the anti-IFT88 antibody and M. Kadowaki, A. Tani, A. Ishimaru, Y. Tohjima, and H. Abe for technical assistance.
This work was supported by PRESTO from the Japan Science and Technology Agency (JST); Grant-in-aid for Scientific Research on Innovative Areas JP25113519, Grants-in-aid for Scientific Research (B) JP15H04669 and JP25293070; Grant-in-aid for Scientific Research (C) JP16K08583; Young Scientists (B) Grant JP15K18955 from Japan Society for the Promotion of Science (JSPS); Nanotechnology Platform Grant 12024046 from The Ministry of Education, Culture, Sports, Science and Technology (MEXT); The Takeda Science Foundation; The Uehara Memorial Foundation; The Novartis Foundation; The Mochida Memorial Foundation for Medical and Pharmaceutical Research; The Naito Foundation; The Senri Life Science Foundation; The Kato Memorial Bioscience Foundation; The Daiichi-Sankyo Foundation of Life Science; The Suzuken Memorial Foundation; The Osaka Community Foundation; NIG Collaborative Research Program Grant 2016B5; the initial stages of this project were supported by National Institutes of Health RO1 Award EY018176 from the NEI (to J. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GPCR
- G protein-coupled receptor
- IFT
- intraflagellar transport
- RP
- retinitis pigmentosa
- dpf
- days post-fertilization
- INL
- inner nuclear layer
- ONL
- outer nuclear layer
- OS
- outer segment
- Shh
- sonic hedgehog.
References
- 1. Fliegauf M., Benzing T., and Omran H. (2007) When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 8, 880–893 [DOI] [PubMed] [Google Scholar]
- 2. Gerdes J. M., Davis E. E., and Katsanis N. (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137, 32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nigg E. A., and Raff J. W. (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139, 663–678 [DOI] [PubMed] [Google Scholar]
- 4. Kennedy B., and Malicki J. (2009) What drives cell morphogenesis: a look inside the vertebrate photoreceptor. Dev. Dyn. 238, 2115–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosenbaum J. L., and Witman G. B. (2002) Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 3, 813–825 [DOI] [PubMed] [Google Scholar]
- 6. Tokuyasu K., and Yamada E. (1959) The fine structure of the retina studied with the electron microscope. IV. Morphogenesis of outer segments of retinal rods. J. Biophys. Biochem. Cytol. 6, 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hartong D. T., Berson E. L., and Dryja T. P. (2006) Retinitis pigmentosa. Lancet 368, 1795–1809 [DOI] [PubMed] [Google Scholar]
- 8. Sung C. H., and Leroux M. R. (2013) The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nat. Cell Biol. 15, 1387–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D. A., Emma F., Klingenberg C., Hennekam R. C., Torre G., Garshasbi M., Tzschach A., Szczepanska M., Krawczynski M., Zachwieja J., Zwolinska D., Beales P. L., et al. (2010) Cranioectodermal dysplasia, sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 86, 949–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mill P., Lockhart P. J., Fitzpatrick E., Mountford H. S., Hall E. A., Reijns M. A., Keighren M., Bahlo M., Bromhead C. J., Budd P., Aftimos S., Delatycki M. B., Savarirayan R., Jackson I. J., and Amor D. J. (2011) Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am. J. Hum. Genet. 88, 508–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gilissen C., Arts H. H., Hoischen A., Spruijt L., Mans D. A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S. G., Roepman R., Knoers N. V., Veltman J. A., and Brunner H. G. (2010) Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 87, 418–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis E. E., Zhang Q., Liu Q., Diplas B. H., Davey L. M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C. V., Muzny D. M., Young A. C., Wheeler D. A., Cruz P., Morgan M., et al. (2011) TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arts H. H., Bongers E. M., Mans D. A., van Beersum S. E., Oud M. M., Bolat E., Spruijt L., Cornelissen E. A., Schuurs-Hoeijmakers J. H., de Leeuw N., Cormier-Daire V., Brunner H. G., Knoers N. V., and Roepman R. (2011) C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet. 48, 390–395 [DOI] [PubMed] [Google Scholar]
- 14. Bredrup C., Saunier S., Oud M. M., Fiskerstrand T., Hoischen A., Brackman D., Leh S. M., Midtbø M., Filhol E., Bole-Feysot C., Nitschké P., Gilissen C., Haugen O. H., Sanders J. S., Stolte-Dijkstra I., et al. (2011) Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 89, 634–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cortellino S., Wang C., Wang B., Bassi M. R., Caretti E., Champeval D., Calmont A., Jarnik M., Burch J., Zaret K. S., Larue L., and Bellacosa A. (2009) Defective ciliogenesis, embryonic lethality; and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev. Biol. 325, 225–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin J., Lin Y., Norman R. X., Ko H. W., and Eggenschwiler J. T. (2011) Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc. Natl. Acad. Sci. U.S.A. 108, 1456–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tran P. V., Haycraft C. J., Besschetnova T. Y., Turbe-Doan A., Stottmann R. W., Herron B. J., Chesebro A. L., Qiu H., Scherz P. J., Shah J. V., Yoder B. K., and Beier D. R. (2008) THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 40, 403–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Behal R. H., Miller M. S., Qin H., Lucker B. F., Jones A., and Cole D. G. (2012) Subunit interactions and organization of the Chlamydomonas reinhardtii intraflagellar transport complex A proteins. J. Biol. Chem. 287, 11689–11703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Doerre G., and Malicki J. (2002) Genetic analysis of photoreceptor cell development in the zebrafish retina. Mech. Dev. 110, 125–138 [DOI] [PubMed] [Google Scholar]
- 20. Tsujikawa M., and Malicki J. (2004) Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron 42, 703–716 [DOI] [PubMed] [Google Scholar]
- 21. Li J., and Sun Z. (2011) Qilin is essential for cilia assembly and normal kidney development in zebrafish. PLoS ONE 6, e27365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pathak N., Obara T., Mangos S., Liu Y., and Drummond I. A. (2007) The zebrafish fleer gene encodes an essential regulator of cilia tubulin polyglutamylation. Mol. Biol. Cell 18, 4353–4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Omori Y., Zhao C., Saras A., Mukhopadhyay S., Kim W., Furukawa T., Sengupta P., Veraksa A., and Malicki J. (2008) Elipsa is an early determinant of ciliogenesis that links the IFT particle to membrane-associated small GTPase Rab8. Nat. Cell Biol. 10, 437–444 [DOI] [PubMed] [Google Scholar]
- 24. Sun Z., Amsterdam A., Pazour G. J., Cole D. G., Miller M. S., and Hopkins N. (2004) A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development 131, 4085–4093 [DOI] [PubMed] [Google Scholar]
- 25. Fadool J. M. (2003) Development of a rod photoreceptor mosaic revealed in transgenic zebrafish. Dev. Biol. 258, 277–290 [DOI] [PubMed] [Google Scholar]
- 26. Kramer-Zucker A. G., Olale F., Haycraft C. J., Yoder B. K., Schier A. F., and Drummond I. A. (2005) Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer's vesicle is required for normal organogenesis. Development 132, 1907–1921 [DOI] [PubMed] [Google Scholar]
- 27. Kane D. A., and Kimmel C. B. (1993) The zebrafish midblastula transition. Development 119, 447–456 [DOI] [PubMed] [Google Scholar]
- 28. Cao Y., Park A., and Sun Z. (2010) Intraflagellar transport proteins are essential for cilia formation and for planar cell polarity. J. Am. Soc. Nephrol. 21, 1326–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bisgrove B. W., Snarr B. S., Emrazian A., and Yost H. J. (2005) Polaris and Polycystin-2 in dorsal forerunner cells and Kupffer's vesicle are required for specification of the zebrafish left-right axis. Dev. Biol. 287, 274–288 [DOI] [PubMed] [Google Scholar]
- 30. Marszalek J. R., Liu X., Roberts E. A., Chui D., Marth J. D., Williams D. S., and Goldstein L. S. (2000) Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell 102, 175–187 [DOI] [PubMed] [Google Scholar]
- 31. Tam B. M., Moritz O. L., Hurd L. B., and Papermaster D. S. (2000) Identification of an outer segment targeting signal in the COOH terminus of rhodopsin using transgenic Xenopus laevis. J. Cell Biol. 151, 1369–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao C., and Malicki J. (2011) Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J. 30, 2532–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ocbina P. J., Eggenschwiler J. T., Moskowitz I., and Anderson K. V. (2011) Complex interactions between genes controlling trafficking in primary cilia. Nat. Genet. 43, 547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mukhopadhyay S., Wen X., Chih B., Nelson C. D., Lane W. S., Scales S. J., and Jackson P. K. (2010) TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 24, 2180–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krock B. L., Mills-Henry I., and Perkins B. D. (2009) Retrograde intraflagellar transport by cytoplasmic dynein-2 is required for outer segment extension in vertebrate photoreceptors but not arrestin translocation. Invest. Ophthalmol. Vis. Sci. 50, 5463–5471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Omori Y., Chaya T., Katoh K., Kajimura N., Sato S., Muraoka K., Ueno S., Koyasu T., Kondo M., and Furukawa T. (2010) Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc. Natl. Acad. Sci. U.S.A. 107, 22671–22676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ozgül R. K., Siemiatkowska A. M., Yücel D., Myers C. A., Collin R. W., Zonneveld M. N., Beryozkin A., Banin E., Hoyng C. B., van den Born L. I., European Retinal Disease Consortium, Bose R., Shen W., Sharon D., Cremers F. P., et al. (2011) Exome sequencing and cis-regulatory mapping identify mutations in MAK, a gene encoding a regulator of ciliary length, as a cause of retinitis pigmentosa. Am. J. Hum. Genet. 89, 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tucker B. A., Scheetz T. E., Mullins R. F., DeLuca A. P., Hoffmann J. M., Johnston R. M., Jacobson S. G., Sheffield V. C., and Stone E. M. (2011) Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 108, E569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burghoorn J., Dekkers M. P., Rademakers S., de Jong T., Willemsen R., and Jansen G. (2007) Mutation of the MAP kinase DYF-5 affects docking and undocking of kinesin-2 motors and reduces their speed in the cilia of Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 104, 7157–7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chaya T., Omori Y., Kuwahara R., and Furukawa T. (2014) ICK is essential for cell type-specific ciliogenesis and the regulation of ciliary transport. EMBO J. 33, 1227–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Malicki J., Jo H., Wei X., Hsiung M., and Pujic Z. (2002) Analysis of gene function in the zebrafish retina. Methods 28, 427–438 [DOI] [PubMed] [Google Scholar]
- 42. Omori Y., and Malicki J. (2006) oko meduzy and related crumbs genes are determinants of apical cell features in the vertebrate embryo. Curr. Biol. 16, 945–957 [DOI] [PubMed] [Google Scholar]
- 43. Pazour G. J., Baker S. A., Deane J. A., Cole D. G., Dickert B. L., Rosenbaum J. L., Witman G. B., and Besharse J. C. (2002) The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol. 157, 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sato S., Omori Y., Katoh K., Kondo M., Kanagawa M., Miyata K., Funabiki K., Koyasu T., Kajimura N., Miyoshi T., Sawai H., Kobayashi K., Tani A., Toda T., Usukura J., et al. (2008) Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat. Neurosci. 11, 923–931 [DOI] [PubMed] [Google Scholar]
- 45. Sato T. (1968) A modified method for lead staining of thin sections. Journal of Electron Microscopy 17, 158–159 [PubMed] [Google Scholar]