

Abstract
The WRN helicase/exonuclease is mutated in Werner syndrome of genomic instability and premature aging. WRN-depleted fibroblasts, although remaining largely viable, have a reduced capacity to maintain replication forks active during a transient hydroxyurea-induced arrest. A strand exchange protein, RAD51, is also required for replication fork maintenance, and here we show that recruitment of RAD51 to stalled forks is reduced in the absence of WRN. We performed a siRNA screen for genes that are required for viability of WRN-depleted cells after hydroxyurea treatment, and identified HDAC1, a member of the class I histone deacetylase family. One of the functions of HDAC1, which it performs together with a close homolog HDAC2, is deacetylation of new histone H4 deposited at replication forks. We show that HDAC1 depletion exacerbates defects in fork reactivation and progression after hydroxyurea treatment observed in WRN- or RAD51-deficient cells. The additive WRN, HDAC1 loss-of-function phenotype is also observed with a catalytic mutant of HDAC1; however, it does not correlate with changes in histone H4 deacetylation at replication forks. On the other hand, inhibition of histone deacetylation by an inhibitor specific to HDACs 1–3, CI-994, correlates with increased processing of newly synthesized DNA strands in hydroxyurea-stalled forks. WRN co-precipitates with HDAC1 and HDAC2. Taken together, our findings indicate that WRN interacts with HDACs 1 and 2 to facilitate activity of stalled replication forks under conditions of replication stress.
Keywords: DNA helicase, DNA replication, histone acetylation, histone deacetylase 1 (HDAC1), human, small interfering RNA (siRNA), Werner syndrome protein, tissue culture
Introduction
Replication stress, defined as disturbances to normal progression rate, density, or distribution of replication forks, is a major driver of genomic instability and carcinogenesis (1–3). Replication stress caused by fluctuations in cellular pools of NTPs and dNTPs is highly relevant to the understanding of the mechanisms of oncogene-driven mutagenesis and chemosensitivity (1–3). Hydroxyurea (HU),4 a ribonucleotide reductase inhibitor, depletes dNTP pools in a dose-dependent manner to cause a reversible global reduction in replication fork progression rate. Slowing or stalling of forks in HU and subsequent reactivation of normal fork progression after HU are highly regulated processes, which protect forks from inactivation and ensure faithful and complete replication of the genome. This includes preserving the ability of forks to resume DNA synthesis after conditions normalize as well as preventing excessive truncation of nascent DNA strands at the fork and involves coordinated activities of many proteins, including checkpoint effectors and mediators, exonucleases, helicases, ATPases, low fidelity DNA polymerases, and proteins of homologous recombination machinery (4, 5). Nonetheless, prolonged stalling eventually leads to development of double strand breaks and activation of the DNA damage response (6–8).
We and others have shown that the human RECQ helicases WRN and BLM are among the proteins that are important for normal progression of replication forks as well as for the recovery of stalled forks after a transient HU arrest (9–15). Mutations in the BLM (16) and WRN (17) genes cause, respectively, Bloom syndrome (BS) and Werner syndrome, two heritable human genomic instability disorders characterized by developmental abnormalities (BLM) and premature aging (WRN), respectively (18, 19). Both syndromes are also associated with increased predisposition to specific types of cancer (20, 21). BLM and WRN are caretaker genes that maintain genomic stability through their roles in replication, repair, and telomere homeostasis (for review, see Refs. 6 and 22–24).
To delineate unique versus overlapping functions of WRN and BLM, we previously depleted WRN and/or BLM in SV40-transformed human fibroblasts and shown that these fibroblasts exhibit comparable defects in reactivation of replication forks after an HU-induced arrest (15). Despite comparable fork reactivation defects, WRN-depleted fibroblasts showed less HU cytotoxicity than BLM-depleted cells (25), enabling us to look for additional genes that may modify cytotoxicity induced by HU in WRN-deficient cells. Identification of such genes may provide novel insight into the mechanisms of resistance to replication stress as well as differences in the roles of WRN and BLM in the cell.
We thus conducted a siRNA screen for genes that were synthetic lethal with WRN deficiency in HU-treated human fibroblasts. We found that depletion of class I histone deacetylases HDAC1 or HDAC2 confers such a phenotype. Following up on this finding, we show that in WRN-depleted but not in WRN-proficient fibroblasts, HDAC1 is needed for efficient fork reactivation after HU. Moreover, we demonstrate co-immunoprecipitation of WRN with HDAC1 and HDAC2. Lastly, inhibition of deacetylase activity of HDAC1 and HDAC2 by a small molecule CI-994 leads to enhanced nascent strand processing at stalled forks. Based on the analysis of histone acetylation at stalled and moving forks and WRN, HDAC1, and RAD51 recruitment to forks, we propose new roles of histone deacetylases during the replication challenged with dNTP pool fluctuations. Our results highlight the importance of chromatin environment in mitigating disruptions to replication.
Experimental Procedures
Cells and Culture
The SV40-transformed human fibroblast GM639 fibroblast cell line and its pNeoA derivative GM639cc1 have been described before (14, 15, 25, 26). WV1 is an SV40-transformed Werner syndrome patient-derived fibroblast line that does not express any WRN protein (26). MCF10a spontaneously immortalized mammary epithelial and UW289.B1 brca1−/− (27) ovarian cancer cell lines were a gift of Drs. Piri Welcsh and Elizabeth Swisher (University of Washington). Embryonic rhabdomyosarcoma cell line RD was obtained from ATCC (ATCC CCL-136). Primary human fibroblast line HFF4 was described by us previously (28).
GM639cc1, RD, and HFF4 were grown in Dulbecco's modified minimal essential medium (DMEM) supplemented with l-glutamine, sodium pyruvate, 10% fetal bovine serum (Hyclone, Ogden, UT), and antibiotics, and MCF10a were grown in MGEM media (Lonza) supplemented with Single Quots (Lonza), 1% fetal bovine serum, and antibiotics. UW289.B1 cells were grown in 1:1 mixture of RPMI and MGEM supplemented with 3% FBS. All cell lines were kept in a humidified 5% CO2, 37 °C incubator.
Drugs and Other Reagents
A stock solution of 5-iododeoxyuridine (IdU) was at 2.5 mm in PBS, 5-chlorodeoxyuridine (CldU) was at 10 mm in PBS, and 5-ethynyldeoxyuridine (EdU) was at 10 mm in DMSO, and HU was at 1 m in PBS. IdU, CldU, and HU were purchased from Sigma, and EdU was from Life Technologies and Sigma. IdU and CldU were used at a concentration of 50 μm, and EdU was used at 10 μm. A stock solution of CI-994 (LC Laboratories) was made at 10 mm in DMSO and used at 3–4 μm unless specified otherwise. All reagent stocks were stored at −20 °C.
High Throughput Screen
We screened a library of siRNAs listed in supplemental Table S1 at the University of Washington Quellos Screening Core. WRN-depleted and mock-depleted GM639cc1 fibroblasts were generated for this screen by infection with a pLKO.1-based WRN shRNA construct (14, 15) or empty pLKO.1, respectively. Infected cells were selected by resistance to puromycin, and depletion was verified by Western blotting. siRNA transfection conditions were optimized before the screen using universal siRNA control as a transfection toxicity readout and siRNA to an essential gene, KIF11, as a transfection efficiency readout. Of interest, WRN-depleted cells were less sensitive to the nonspecific toxicity associated with transfection. Conditions were adjusted to keep transfection-associated toxicity in the two cell lines at the same level. Cells were then plated for transfection into 384-well plates, and the next day each cell line was transfected with library siRNAs (one gene per well in triplicate for treatment and control arms, four siRNAs per gene) as well as universal siRNA control and Kif11 siRNA controls per each plate. The next day wells in the treatment arm were incubated with 2 mm hydroxyurea for 6 h. Cell number and metabolism were evaluated after 3 days using CellTiter Glo dye (Promega) per manufacturer's instructions, and plates were scanned to quantify signal.
RNAi-mediated Depletion of WRN and RAD51
Short hairpin RNA (shRNA) constructs for depletion of WRN and RAD51 are described (14, 15, 25). Scrambled shRNA is 5′-CTCCATATCGAACAGTTGG-3′. siRNAs against HDAC1, HDAC2, WRN, and a negative control non-targeting siRNA were purchased from Qiagen (HDAC1-5 SI02634149, HDAC1-6 SI02663472, HDAC2-1 SI00434952, HDAC2-3 SI00434966, WRN-6 SI02663759). AllStars Cell death control siRNA (Qiagen) was used as transfection efficiency control. Doxycycline-inducible shRNA construct against WRN expresses the same hairpin as the previously described shRNA construct (14).
Inducible shRNA and siRNAs were used in HDAC+ and HDAC1 knock-out (KO) RD cells because in these lines lentiviral infection combined with immediate onset of WRN RNAi triggered cessation of cell division by days 8–10 post-infection. In HDAC1 KO RD cells transduced with HDAC1 gene constructs, siRNAs were used because they achieved a more rapid depletion of the protein than a combination of transduction followed by a recovery period and then by induction of RNAi with doxycycline.
CRISPR-Cas9-mediated Gene Deletion
Gene knockouts were performed in the RD embryonic rhabdomyosarcoma cell line. The HDAC1 knock-out clone (#17) used in Figs. 4, 5C, and 8D was generated by transduction of a pLenti-CRISPRv1 lentiviral vector (29) expressing Cas9 and a single HDAC1 gRNA followed by puromycin selection and isolation of individual clones. The knock-out was sequence- and Western blot-verified. The wild type isogenic control for this clone is parent RD line expressing GFP. For HDAC1 or HDAC2 deletion RD cell lines used in Figs. 5, D-F, two gRNAs per gene were used to enhance targeting efficiency. The gRNA sequences were GGACTGTCCAGTATTCGA and GGCTCAGACTCCCTATCT for HDAC1 and GGAATACTTTCCTGGCAC and GGTCATGCGGATTCTATG for HDAC2. These were cloned into a modified pLenti-CRISPRv1 (Addgene # 49535) viral vector containing TagRFPt in place of Cas9 (29). Cas9 was expressed from a separate modified pLenti-CRISPRv1 lacking a gRNA cassette (29). Virus particles were generated using standard protocols and used for transduction of RD cells. Transduced mass cultures were selected with puromycin, and >80% efficient knock-out of HDAC1 or HDAC2 was verified by Western blotting. The isogenic wild type control for these cells is the parent RD line expressing only Cas9. Individual clones were subsequently derived from these mass cultures and Western blot-verified. These clones (#2, 4, and 13) were used in the results shown in Figs. 6 and 7 and their supporting (not shown) data.
FIGURE 4.

HDAC1 and WRN bind nascent DNA. A, a schematic of iPOND. B, Western blot verification of loss of expression of HDAC1 in the HDAC1 knock-out clone (#17) of RD cells. Nucleolin (NCL) and WRN expression are shown for reference. C, a Western blot of niPOND precipitates from GM639cc1 cells labeled by EdU for 30 min, harvested, and subjected to a Click-iT reaction with and without biotin-azide before lysis and incubation with streptavidin beads. D, Western blots of niPOND performed with GM639cc1 and WV1, WRN-deficient fibroblasts. WV1 cells were labeled with EdU for 30 or 90 min. For EdU, serial 2- or 3-fold dilutions from 0.001% to 0.015% of lysates were loaded on a dot blot and visualized with HRP-conjugated anti-biotin antibody. EdU panels show a representative dilution within a linear range of EdU signal. E, a Western blot of niPOND performed with GM639cc1 cells labeled with EdU for 30 min and harvested immediately or chased for 2 h before harvest. F, a Western blot of niPOND performed with RD cells with intact (WT) or knocked-out (HDAC1 KO) HDAC1 gene. Cells were harvested immediately after an EdU pulse or after a 2.5-h chase. G, a Western blot of niPONDs performed with RD cells labeled with EdU for 30 min or for 15 min followed by a 6-h arrest with 2 mm HU in the presence of EdU. Panels framed in separate boxes come from independent experiments as simultaneous detection of HDAC1 and HDAC2 on one Western blot is not optimal due to their close molecular weights. More RD HDAC1 KO than WT cells were used in panels F and G in order to compensate for their lower percentage of replicating cells. Residual bands larger and smaller than expected for HDAC1 seen in the HDAC1 KO, no-HU lane may be nonspecific or represent cross-reactivity with HDAC2. Asterisks in E and F indicate time points of sample harvest.
FIGURE 5.

Co-immunoprecipitation of HDAC1, HDAC2, and WRN. A, a Western blot showing levels of WRN and HDAC1 in GM639cc1 cell nuclear extracts prior (input) and after (s/n) IP and in immunoprecipitates of an α-HDAC1 or an isotype-matching α-HA antibody. B, a Western blot showing levels of WRN and HDAC1 in nuclear extracts and α-HDAC1 immunoprecipitates from GM639cc1 and WV1 (WRN-deficient) cells. C, a Western blot showing WRN and HDAC1 levels in nuclear extracts and α-HDAC1 immunoprecipitates from control (WT) and clone #17 HDAC1 knock-out (HDAC1 KO) RD cells. D, a Western blot of WRN, HDAC1, HDAC2, and nucleolin (NCL, loading control) in whole-cell lysates of mass cultures of RD cells after CRISPR-Cas9-mediated knock-out of HDAC1 (HDAC1 KO) or HDAC2 (HDAC2 KO). WT cells express Cas9 only. Expression of HDAC1 or HDAC2 is quantified as percent of the levels seen in WT control. E, a Western blot of nuclear extracts and α-HDAC1 immunoprecipitates from control (WT), HDAC1, or HDAC2 knock-out RD cells introduced in D. F, a Western blot of nuclear extracts and α-HDAC2 immunoprecipitates from control (WT) or HDAC2 knock-out RD cells introduced in D. In E and F WRN levels in IPs are normalized to inputs and shown relative to these levels in WT controls.
FIGURE 8.

HDAC deacetylase activity inhibition by CI-994 increases the level of lysine 12-acetylated histone H4 at ongoing and stalled forks but does not affect fork reactivation after HU. A, GM639cc1 fibroblasts were maintained overnight with and without 3 μm CI-994, then labeled with EdU as indicated with or without 2 mm HU. Where indicated, CI-994 was present throughout the experiment. Regular iPOND (with formaldehyde cross-linking) was performed, and samples were analyzed by Western blotting. Input lanes contain 2.5% of cell lysates. EdU panels show representative levels in inputs derived as described in Fig. 4. B and C, quantitation of two (B), four (C, control cells), and two (C, WRN-depleted cells) independent iPOND experiments similar to the one shown in A. Levels of H4K12ac in pulldown assays were normalized to the EdU levels in inputs (EdU in., averaged over at least three values falling within a linear range of EdU signals in serial dilutions on a dot blot; EdU pd., EdU pulldown). A normalized level of H4K12ac in untreated controls was set as the baseline, and the rest of the values were expressed relative to it. Error bars are S.D. D, Replication fork-associated Lysine 12-acetylated histone H4 is not elevated in HDAC1 knock-out RD cells. A Western blot of iPOND samples treated with 2 mm HU as in A. Input lanes contain 2% of lysates. E–H, maRTA analysis of WRN-depleted GM639cc1 fibroblasts labeled with thymidine analogs (1st label, EdU; 2nd label, IdU) and treated with 2 mm HU for 6 h in the presence or absence of CI-994. CI-994 treatment was as in A–C above. Plotted values are derived as described for Fig. 3, C–E. In F, two independent experiments were averaged, and error bars are S.D. Datasets from each experiment contained 400–900 track measurements per sample. p value in a one-sided t test equaled 0.077. The G panel shows distributions of lengths of 1st (white) and 2nd (gray) label segments in ongoing forks derived from two experimental replicates. Numbers of tracks analyzed are shown below the graph, and significance was determined in Wilcoxon tests. **, p ≤ 5 × 10−03. H, ratios of 2nd to 1st label segment lengths in each ongoing fork derived from an additional experiment with a side-by-side comparison of HDAC1-depleted versus negative control and CI-994-treated versus untreated cells. All samples are WRN-depleted and were treated with HU. Data were box-plotted to evaluate consistency of fork progression.
FIGURE 6.
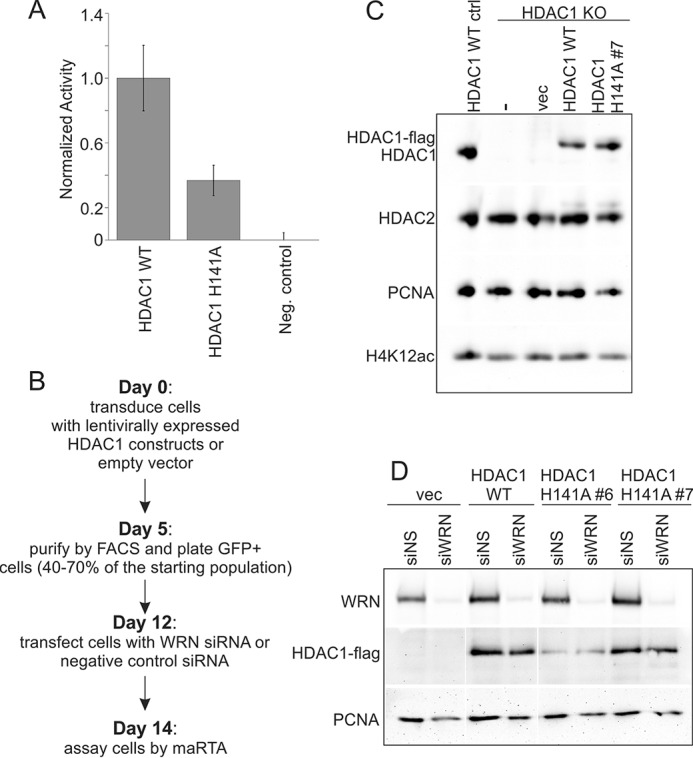
Expression and activity of H141A mutant of HDAC1. A, a bar graph comparing deacetylase activity of the wild type and H141A mutant HDAC1-FLAG proteins expressed from plasmids transiently transfected into HDAC1 KO RD cells (clone #17) and immunoprecipitated with α-FLAG antibody before the assay. Negative control is untransfected HDAC1 KO RD cells. B, a work flow diagram for generation of HDAC1 transgene-expressing, WRN-depleted HDAC1 KO cells. Shown is the fastest timeline available. C, a Western blot measuring expression of wild type and H141A mutant HDAC1-FLAG within 10 days of transduction with pLenti-EFS-T2A-emGFP constructs in HDAC1 KO RD cells (clone #4). D, a Western blot measuring expression of WRN and of wild type and two isolates of H141A mutant HDAC1-FLAG after flow-sorting to purify HDAC1-expressing cells, their outgrowth, and siRNA transfection.
FIGURE 7.

H141A mutant of HDAC1 shows a distinct phenotype of replication fork progression. A, a labeling scheme for maRTA. HU was used at 4 mm, and 1st and 2nd labels were CldU and IdU, respectively. B, percentages of ongoing forks in WRN-depleted untreated and HU-treated (HU) HDAC1 KO RD cells (clone #4) expressing the indicated HDAC1 transgenes or empty vector (vec). The bar graph shows average values derived from two experimental replicates, and error bars are S.D. C, lengths of 1st label (white) and 2nd label (gray) segments in two-label tracks of ongoing forks were box-plotted to evaluate fork progression. The length distribution data were derived from two experimental replicas. Statistical significance was calculated in Wilcoxon tests, and p value designations are as in Fig. 3D. NS, nonspecific. D and E, ratios of the 2nd to 1st label segment lengths in each ongoing fork in untreated (D) and HU-treated (E) cells were box-plotted to evaluate consistency of fork progression. The data used are from the set shown in C. In C–E the statistically significant differences are marked with asterisks standing for p values. p value designations are as in Fig. 3D, and all p values of the orders of magnitude at or below 5 × 10−06 are labeled as *****. Numbers of tracks analyzed are shown below the graph.
HDAC1 Mutant Construction and Expression
A plasmid with a HDAC1 ORF bearing a C-terminal FLAG tag and expressed under the control of the CMV promoter was a gift from Eric Verdin (Addgene #13820). A His to Ala mutation (CAT to GCT) at position 141 of the protein sequence was introduced using the New England BioLabs base changer site-directed mutagenesis protocol. Expression of wild type and mutant HDAC1-FLAG and their deacetylase activity were checked in transient transfections. For stable expression, HDAC1 ORFs were cloned into a T2A peptide-linked emGFP lentiviral expression vector (pLenti-EFS-T2A-emGFP). Virus particles were prepared using standard protocols and used for transduction of HDAC1 KO RD cells. Transduction efficiency and expression of HDAC1 proteins was verified by flow cytometry of GFP+ cells and Western blotting with the α-HDAC1 antibody, respectively. Transduced cells were subsequently purified by flow cytometric cell sorting based on GFP expression.
Deacetylation activity of wild type and mutant HDAC1 was determined in vitro using the Histone Deacetylase Activity kit (Active Motif), according to the manufacturer's recommendations.
Western Blotting
Western blotting of WRN was done as described (14, 25). Antibodies were as follows: mouse antibodies α-WRN 195C catalog no. W0393 (Sigma), α-HDAC1 catalog no. 5356 (Cell Signaling), α-HDAC2 catalog no. 5113 (Cell Signaling), α-phosphoH2AX (Ser-39) catalog no. 05-636 (Millipore), α-PCNA catalog no. sc-56 (Santa Cruz), α-nucleolin catalog no. 396400 (Life Technologies), α-RAD51 catalog no. 05-530 (Millipore), α-CHK1 catalog no. sc-8408 (Santa Cruz), α-FLAG catalog no. MA1–91878-BTIN (Thermo Fisher), and a rabbit antibody α-H4K12ac, catalog no. 39165 (Active Motif).
All proteins were visualized on Western blots by ECL (Thermo Scientific) and quantified using Storm phosphorimaging (Molecular Dynamics) or FluorChem Imager (Alpha Innotech). For presentation, images were saved in TIFF format, adjusted for brightness/contrast, and cropped using Adobe Photoshop or CorelPhotoPaint, then assembled into figures in CorelDraw. Image brightness/contrast adjustments were made to the whole Western blot images. In some cases lane order was changed, and extra lanes were deleted.
Microchannel Fabrication, DNA Fiber Stretching, and Replication Track Analysis
These procedures were done as described (14, 28, 30). Microscopy of stretched DNAs was performed on the Zeiss Axiovert microscope with a 40× objective, and images were captured with the Zeiss AxioCam HRm camera. Lengths of tracks were measured in raw merged images (jpegs) using Zeiss AxioVision software. Fluorochromes were Texas Red for EdU, Alexa594 for CldU, and Alexa488 for IdU. Details of statistical analysis are described in figure legends.
Immunoprecipitation of nascent DNA (iPOND) and Native iPOND (niPOND)
iPOND was performed as described (31), with modifications detailed in Kehrli and Sidorova (28). Briefly, aprotinin and leupeptin were added to permeabilization buffer (0.25% Triton X-100 in PBS) to a final concentration of 1 μg/ml each. The buffer used for sonication and lysis of cells was 50 mm Tris·HCl, pH 8.0, 0.5% SDS, 0.1% sodium deoxycholate, 0.25% Triton X-100, 1 μg/ml aprotinin, and 1 μg/ml leupeptin. niPOND was performed as described in Leung et al. (32). All steps were performed with ice-cold reagents. Briefly, nuclei were isolated in nuclei extraction buffer (20 mm HEPES, pH 7.2, 50 mm NaCl, 3 mm MgCl2, 300 mm sucrose, 0.5% Nonidet P-40 Substitute (Sigma), protease inhibitor mixture (Roche Applied Science)), washed in PBS, subjected to Click-iT reaction, and extracted twice with B1 buffer (25 mm NaCl, 2 mm EDTA, 50 mm Tris-HCl, pH 8.0, 1% Nonidet P-40 Substitute, protease inhibitors) by incubating for 15 min on ice followed by sonicating at 20 watts for 10 s in a cup-horn sonicator (Misonix) and pelleting in order to enrich for chromatin-associated proteins. Lysates were generated by sonication in B1 buffer for 6 min total in 30 s on, 30 s off pulses at a 40-watt setting. Lysates were cleared by 16,100 × g centrifugation for 10 min, then diluted 1:1 with B2 buffer (150 mm NaCl, 2 mm EDTA, 50 mm Tris-HCl, pH 8.0, 0.5% Nonidet P-40 Substitute, protease inhibitors). EdU+ DNA and associated proteins were captured by overnight incubation with streptavidin-agarose beads (Novagen).
To measure EdU amounts in lysates, serial 1:2 dilutions of each lysate in PBS (typically from 0.2 to 0.02 μl of lysate in 0.5 μl total volume) were loaded onto a nitrocellulose membrane, dried for 1–2 h, blocked in 5% BSA in TBST-Tween (TBST), incubated overnight with 1:150 dilution of HRP-conjugated α-biotin antibody catalog no. 7075 (Cell Signaling) in 5% BSA, TBST at 4 °C, washed and subjected to ECL, and quantified using FluorChem Imager. Values obtained for two-three serial dilutions that were within linear range of signal were averaged. EdU was also measured in some pulldown samples by the same procedure: 1 μl of pulldown sample was diluted in 50 μl of PBS, then loaded as a 2-fold dilution series after volume-adjusting to 0.5 μl. Normalizing results by EdU levels in starting lysates or in pulldown assays demonstrated similar results.
Immunoprecipitation
Immunoprecipitation was performed with nuclear extracts prepared as described above for niPOND except extraction of soluble proteins from permeabilized nuclei was performed only once and typically did not include mild sonication before high-speed centrifugation. Chromatin lysates were incubated for 2–3 h with α-HDAC1 (catalog no. 5356, Cell Signaling) antibody used at 1:400 dilution or α-HDAC2 (catalog no. 5113, Cell Signaling) antibody used at 1:200 dilution and with protein G Dynabeads (Invitrogen). Immunoprecipitates were washed four times with niPOND B2 buffer and analyzed in SDS-PAGE gels.
Results
High-throughput siRNA Screen Identifies HDAC1 and HDAC2 as Genes That Enhance HU Cytotoxicity in a WRN-deficient Background
We assembled a siRNA library targeting 320 DNA damage response, repair, and replication genes (supplemental Table S1), where each gene was targeted by a mixture of 4 siRNAs. Cell lines used in transfection were GM639cc1 SV40 fibroblasts that were either mock-depleted with an empty lentiviral vector (controls) or depleted of ≥75% of WRN by the same vector expressing shRNA against WRN (14, 15). Cells were plated in 384-well plates, transfected with the library of siRNAs, and allowed to grow for 3 days with or without a 6-h treatment with 2 mm HU on day 1 of outgrowth.
Cell growth and viability was evaluated in a CellTiter Glo luminescence assay, and growth values were normalized to the average of mock-transfected (Mock) controls for each cell line and condition (Fig. 1). Two-tailed p values were calculated in pairwise comparisons of untreated versus treated normalized growth for each gene, and only the measurements with p values ≤0.05 were considered. ATR and CHEK1 siRNAs were controls that were expected to show HU-dependent cell lethality. Of note, CHEK1 siRNA suppressed growth in untreated WRN-depleted but not control cells (Fig. 1B). This is consistent with the idea that WRN-depleted cells experience constitutive replication stress that requires ongoing CHK1-mediated checkpoint surveillance.
FIGURE 1.

A high throughput screen for genes that sensitize WRN-depleted cells to HU. A, a scatter plot of growth ratio values with and without HU (HU/no HU) for WRN-depleted (y axis) versus control, mock-depleted (x axis) cells. Note that the x axis begins at the value of 0.4. Shown are only the genes for which (i) the growth difference between HU and no HU conditions in WRN-depleted cells is statistically significant (p < 0.005) and (ii) the growth ratio values (HU/no HU) are at 0.5 or lower for WRN-depleted cells and at 0.5 or higher for controls cells. B, normalized mean growth values (as percent of growth of mock-transfected cells) are shown for a subset of individual genes for WRN-depleted and control cells. UNI, universal (nonspecific) control siRNA. Error bars are S.D.
We identified four classes of growth-suppressing knockdown phenotypes resulting from siRNA targeting: 1) constitutive, 2) WRN-dependent, 3) HU-dependent, and 4) WRN and HU-dependent. In the current study we were interested in the candidate genes identified in the category 4 and specifically in those that, when silenced by siRNAs, suppressed growth only mildly in the absence of HU (by <40% in WRN-depleted or control cells). Among these, we focused on the genes that, when silenced together with HU treatment, caused a >2-fold growth reduction only in WRN-depleted cells. No candidates from the library showed strong (>80%) growth inhibition in WRN-depleted cells while leaving control cells unaffected (Fig. 1A). Of the genes that displayed the most dramatic HU- and WRN-dependent reduction of growth, at least some (TOPBP1, EME1) proved to be HU-dependent but not WRN-dependent growth suppressors upon retesting (data not shown).
siRNA-mediated depletion of HDAC1 histone deacetylase conferred a phenotype that ranked within our criteria, causing a greater reduction of viability in HU-treated WRN-depleted cells compared with control cells (Fig. 1). Notably, HDAC2, a close homolog and binding partner of HDAC1 (33, 34), also exhibited some WRN and HU selectivity (Fig. 1). We validated these phenotypes of HDAC1 and HDAC2 using two independent siRNAs against these genes in population outgrowth assays. HDAC1 siRNA HDAC1-6 and HDAC2 siRNA siHDAC2-3 suppressed growth additively with WRN depletion and HU treatment (Fig. 2A), and, respectively, depleted 70–80% of HDAC1 and 40–50% of HDAC2 (Fig. 2, B and C). The strong growth suppression elicited by the moderate depletion of HDAC2 levels suggested that complete depletion of HDAC2 in the GM639cc1 WRN-deficient background may be lethal. Additionally, moderate depletion of HDAC2 by siHDAC2-3 and even by siHDAC2-1 (which depleted only 10–30% of HDAC2, Fig. 2C) was associated with a compensatory increase in the HDAC1 levels (Fig. 2B and 3A). These findings prompted us to focus on HDAC1, which we could deplete effectively and without concurrent changes in HDAC2 levels, as a priority for our follow-up studies.
FIGURE 2.

Depletion of HDAC1 or HDAC2 in WRN-depleted cells exacerbates the growth-suppressive effect of HU. A, population doubling levels of WRN-depleted or control (with scrambled shRNA) GM639cc1 SV40-transformed fibroblasts that were transfected with nonspecific siRNA (NS), HDAC1, or HDAC2 siRNAs (day 0) plated in triplicate into multiwell dishes (day 1) with or without treatment with 2 mm HU for 6 h on day 2 and allowed to grow for another 3 days. Cell counts in wells were averaged, and population doubling levels were determined using a formula ln(average count on dayn+1/average count on dayn)/0.693. Population doubling levels were plotted to derive linear trend lines. B and C, Western blots showing levels of HDAC1 and HDAC2 in scrambled shRNA control and WRN-depleted GM639cc1 fibroblasts transfected with nonspecific (NS) siRNA or siRNAs against HDAC1 or -2. HDAC1 or -2 levels are expressed as percentages of levels seen in cells transfected with NS siRNA. PCNA is a loading control.
FIGURE 3.

Co-depletion of WRN and HDAC1 additively reduces efficiency of replication fork reactivation after a transient arrest by HU. A, a Western blot comparing levels of HDAC1, HDAC2, and H4K12ac in control GM639cc1 fibroblasts transfected with nonspecific (NS), HDAC1, or HDAC2 siRNAs or incubated with the indicated doses of CI-994 overnight before cell harvest. B, a labeling scheme for maRTA includes 2 consecutive 30-min pulses of thymidine analogs (e.g. EdU and IdU) with or without the addition of 2 mm HU for 6 h at the end of the pulse of the first label. C, percent of ongoing forks was measured as a fraction of tracks labeled with two consecutive labels among all tracks containing the first label. Between 400 and 900 tracks were analyzed in each sample. Relative fork reactivation was determined as the percent of ongoing forks in HU-treated samples normalized to percent of ongoing forks in untreated samples for each cell line. The bar graph shows average values derived from two (scrambled shRNA control) or three (WRN shRNA) experimental replicates. Error bars are S.D., and significance was determined in a one-sided t test (p = 0.014), i.e. * indicates p < 0.05. D, lengths of 1st label (white) and 2nd label (gray) segments in two-label tracks of ongoing forks were box-plotted to evaluate fork progression. Shown is a plot of a representative set of samples with numbers of analyzed tracks (n) shown below the graph. Statistical significance was determined in Wilcoxon tests, and significant differences are marked on the graph; **, p ≤ 5 × 10−03, ****, p ≤ 5 × 10−05, etc.; all p values of the orders of magnitude at or below 5 × 10−06 are labeled as *****. E, ratios of 2nd- to 1st-label segment lengths in each ongoing fork were box-plotted to evaluate consistency of fork progression. The data are for untreated samples of the experiment shown in D. No statistically significant differences were detected. F, Western blots of depletion of WRN and HDAC1 in MCF10a spontaneously immortalized mammary epithelial cells. G, relative fork reactivation in MCF10a cells introduced in F after 6 h of 2 mm HU. Average values from two experimental replicas are plotted, and error bars are S.D. ***, one-sided t test p = 0.00017.
HDAC1 Is Important for Replication Fork Activity in WRN-depleted Cells
One of the functions of HDAC1 (and HDAC2) is to remove acetyl groups on de novo synthesized histones incorporated into chromatin during DNA replication, notably acetyls on histone H4 lysine residues 5 and 12, i.e. H4K5ac and H4K12ac (35). Both HDAC1 and -2 were detected at a replication fork (36). Fork progression rates are reduced in the absence or upon inhibition of both HDACs (35), although no phenotype specific to individual HDAC or to recovery after HU treatment has been reported yet. To address the interplay between HDAC1 and WRN during normal and HU-perturbed DNA replication, we performed microfluidic-assisted replication track analysis (maRTA) (30) on GM639cc1 fibroblasts depleted of WRN and/or HDAC1. In accordance with standard approaches (15, 30), reactivation of replication forks after a 6-h arrest by HU was measured by counting forks that resumed DNA synthesis within the first 30 min after removal of HU (i.e. incorporated both 1st and 2nd label) versus those that failed to reactivate (i.e. incorporated only the 1st label, Fig. 3B). Also, progression of replication forks before, during, or after HU was assessed by measuring lengths of 1st and 2nd label segments in two-label tracks. We previously showed that the fraction of reactivating forks drops in cells depleted of 80% or more of the WRN protein (15). In addition, we showed that in WRN-depleted cells reactivated forks progress slower than in WRN-proficient cells (14, 15). We will subsequently use the term fork recovery to describe both of these phenotypes of WRN loss.
When we co-depleted HDAC1 and WRN, fork reactivation was additively impaired (Fig. 3C). In control cells HDAC1 depletion had a minimal to no effect on the percentage of fork reactivation (Fig. 3C). Also, depletion of WRN or HDAC1 each reduced progression of forks in the absence of HU as well as after HU treatment, as evidenced by shorter track lengths, and notably, double depletion had an additive negative effect (Fig. 3D). In all cases, comparison of progression through 1st versus 2nd labeling periods for each ongoing fork revealed that it was reduced similarly; that is, the ratios of 2nd to 1st label segment lengths remained virtually unchanged for all cells and averaged close to 1 in untreated cells (Fig. 3E) and below 0.5 in HU-treated cells (data not shown). Together, the results in Fig. 3, D and E, are more consistent with a uniform fork rate reduction in HDAC1- and/or WRN-depleted cells rather than increased premature termination. Lastly, the additive fork reactivation phenotype was reproduced in a cell line of a different (epithelial) lineage, MCF10a (Fig. 3, F and G). Overall, the data suggest an additive reduction of fork recovery resulting from WRN and HDAC1 depletion, which is consistent with reduced cell growth post-HU treatment of these co-depleted cells.
WRN and HDAC1 Are Present on Newly Replicated DNA
To assess whether HDAC1 and WRN associate with replication forks or newly replicated DNA, we preformed native iPOND (niPOND, or native immune precipitation of nascent DNA). niPOND (32) pulls down proteins associated with EdU-labeled DNA via Click-iT-mediated conjugation of biotin-azide to EdU in permeabilized cells followed by lysis and incubation with streptavidin beads (Fig. 4A). Unlike regular iPOND (31), niPOND does not employ formaldehyde cross-linking of proteins to DNA, facilitating recovery of large proteins, particularly as large as WRN (∼167 kDa). As these experiments require large starting cell numbers, we used a WRN null, patient-derived SV40 fibroblast line WV1 and RD rhabdomyosarcoma lines with either a CRISPR-Cas9-mediated HDAC1 KO or a GFP-expressing empty vector (WT, Fig. 4B). RD HDAC1 KO clones grew more slowly that the control (for the clone shown in Fig. 4, the average population doubling was 0.64 ± 0.33/day versus 0.75 ± 0.26/day in the control). Also, we found lower levels of WRN expression in two independently derived HDAC1 KO clones compared with HDAC1 wild type controls (Fig. 4B and data not shown), which may indicate effects of HDAC1 on the WRN gene expression.
Using niPOND with nuclear lysates enriched in chromatin-bound proteins (see “Experimental Procedures”), we confirmed previous observations (36, 37) that HDAC1 was pulled down with EdU-labeled genomic DNA that corresponds to replication forks (Fig. 4C). We also detected the presence of H4K12ac, which corresponds to the acetylated histone H4 newly deposited at replication forks (Fig. 4, C and E). We demonstrated association of WRN with replication forks (Fig. 4C). Identification of WRN in EdU pulldown assays was further confirmed by using a WRN null fibroblast line WV1 (Fig. 4D).
The levels of PCNA and H4K12ac on EdU+ DNA decreased after a 2-h-long chase after EdU pulse, indicating that the DNA replication machinery has moved away from the EdU-labeled DNA segments, and the nascent chromatin has matured (Fig. 4, E and F). By contrast, the association of HDAC1 and WRN with EdU+ DNA was more long-lived, lasting at least 2.5 h after EdU pulse (Fig. 4F).
WRN recruitment to EdU+ DNA was observed in WT and HDAC1 KO cells (Fig. 4F). This result was also observed in another, independently derived HDAC1 KO clone (data not shown). Apparent lower abundance of WRN on EdU+ DNA paralleled its lower expression in HDAC1 KO RD cells. Interestingly, in the WRN null line recruitment of HDAC1 to new DNA was reduced (though not eliminated) compared with the non-isogenic WRN+ counterpart (Fig. 4D). This may be explained in part by the lower S-phase fraction in WRN null cells compared with WRN+ cells; however, a WRN-specific effect cannot be ruled out.
Incubation of HDAC1 KO or control cells with HU for 6 (Fig. 4G) or 12 h (data not shown) after the EdU pulse did not lead to dramatic changes in WRN or HDAC2 association with EdU+ DNA. As expected, HU reduced both total nuclear (chromatin-associated) and nascent DNA-associated PCNA (Fig. 4G). Overall, these results demonstrate that both HDAC1 and WRN can be found on newly replicated DNA. WRN association with DNA is not abolished in the absence of HDAC1 and vice versa. However, in at least one WRN null patient-derived fibroblast line we see a reduction in HDAC1 associated with new DNA.
WRN Co-immunoprecipitates with HDAC1 and HDAC2
To further investigate the interaction between WRN and HDAC1/2, we performed immunoprecipitations (IP) from nuclear extracts of GM639cc1 and WV1 fibroblasts or RD cells (Fig. 5). With an HDAC1 antibody, but not an isotype-matched HA control antibody, we were able to pull down most of the HDAC1 present in lysates together with 0.5–2% of WRN (Fig. 5, A and B). Precipitation of WRN by the HDAC1 antibody was reduced by 50% but not eliminated in HDAC1 KO cells (Fig. 5C). This residual WRN may co-precipitate with the highly homologous HDAC2, which was also detectable in HDAC1 antibody pulldown assays from HDAC1 KO cells (Fig. 5C). To address this further, we generated mass cultures of RD cells newly transduced with CRISPR-Cas9 constructs expressing two gRNAs each against HDAC1 or HDAC2 to facilitate deletion of these genes. These cultures were puromycin-selected to eliminate untransduced cells, but we did not propagate individual clones, thus preempting positive selection of cells with in-frame HDAC mutations. Western blotting with HDAC1 and HDAC2 antibodies showed that expression of HDAC1 or HDAC2 was reduced by >80% in these cultures (Fig. 5D), making them suitable for immunoprecipitation studies. Notably, as with RNAi against these HDACs (Fig. 3A), HDAC1 expression increased substantially upon reduction of HDAC2 expression, suggesting a compensatory feedback. This response was reciprocal, albeit to a lesser degree, as HDAC2 went up only by 36% in cells with reduced HDAC1.
We next performed IPs with HDAC1 or HDAC2 antibodies from nuclear extracts of these HDAC1 KO or HDAC2 KO cell cultures. As before, precipitation of WRN was reduced by about half if HDAC1 level in the extract was reduced (Fig. 5E). In contrast, when HDAC2 level was down, precipitation of WRN was substantially reduced (by 80%, Fig. 5F). These results suggest that WRN may physically associate with HDAC2, and via HDAC2, with HDAC1.
Effect of HDAC1 on Fork Recovery Is Modified by an H141A Mutation That Reduces Its Deacetylase Activity
As mentioned above, HDAC1 and -2 deacetylate histones incorporated de novo into nascent DNA, inviting a hypothesis that histone hyperacetylation at the fork may be responsible for the negative effect of HDAC1 deficiency on fork activity. However, our study (Fig. 3A) and previous work (35, 38) indicate that depletion of HDAC1 alone is not sufficient to change histone H4 acetylation levels in the cell due to likely redundancy with HDAC2 and HDAC3. We, therefore, asked whether deacetylase activity of HDAC1 is important for its cooperation with WRN at forks.
We introduced a H141A mutation into a FLAG-tagged HDAC1 (39), a change that was shown to inhibit deacetylase activity of the protein while preserving its associations with such proteins as RBBP4 and mSin3A (40). The defect in the HDAC1 H141A deacetylase activity was confirmed in vitro using α-FLAG antibody immunoprecipitates of the wild type and mutant proteins transiently expressed in HDAC1 KO RD cells (Fig. 6A). The genes were subsequently cloned into pLenti-EFS-T2A-emGFP vector and transduced into HDAC1 KO RD clones. HDAC1 expression in GFP+ cells was verified by Western blotting. No increase in whole cell levels of H4K12ac was observed, as before (compare Figs. 6C and 3A). Notably, at least two HDAC1 KO clones down-regulated expression of more than one isolate of HDAC1 H141A transgene over time compared with the wild type HDAC1 (data not shown and Fig. 6D), suggesting a possible toxicity of the mutant protein. We used siRNA (Fig. 6, B and D) to rapidly deplete WRN in these cells within a 2-week window in which the levels of at least some HDAC1 H141A isolates were comparable with the wild type protein (see “Experimental Procedures” for more detail). In separate experiments, WRN was also depleted in parental HDAC1 KO and HDAC1+ control cells.
All cell lines showed only minor differences in cell cycle distribution and proliferation index on the day of maRTA assays (data not shown). Using our labeling and HU treatment protocol (Fig. 7A), we found that WRN depletion had only a minor effect on fork reactivation in HDAC1+ cells and no effect in two untransduced HDAC1 KO clones (data not shown). Also, WRN depletion did not reduce a fraction of the reactivated forks in HDAC1 KO cells transduced with wild type HDAC1 versus HDAC1 H141A or empty vector (Fig. 7B). However, in all cases tested, WRN depletion elicited its phenotype of slowed fork progression, and this phenotype was exacerbated when combined with HDAC1 deficiency. Moreover, the HDAC1 H141A mutant showed a phenotype distinct from and more severe than HDAC1 KO.
In particular (Fig. 7C), depletion of WRN reduced fork progression without HU in vector control compared with HDAC1-expressing cells. Also, WRN depletion slowed fork progression after HU in vector control and HDAC1 H141A but not in HDAC1 WT-expressing HDAC1 KO cells. Further insight was gained by analyzing consistency of progression of each individual ongoing fork by plotting ratios of lengths of its 1st and 2nd label segments. In untreated cells, only the HDAC1 H141A mutant demonstrated excessive shortening of 2nd label segments versus 1st label segments (Fig. 7, C and D), suggesting premature fork termination. Furthermore, in HU-treated cells HDAC1 H141A mutant showed discordant impact on fork progression before/during HU (1st label) versus after HU (2nd label), which was significantly exacerbated by WRN depletion (Fig. 7E). Overall, these findings indicate that HDAC1 H141A deacetylase activity mutant displays a replication phenotype and that WRN-depleted, HDAC1 H141A-expressing cells show an additive defect in fork progression upon HU treatment.
HDACs 1–3 Inhibition and Histone H4 Hyperacetylation at Forks Do Not Mimic the Effect of HDAC1 Deletion on Fork Recovery
A complementary approach to address the impact of hyperacetylated histones on replication fork activity is to chemically inhibit more than one HDAC to achieve hyperacetylation and assess its effect on forks. The small molecule CI-994 inhibits HDACs 1, 2, and 3 (41), and we demonstrated its dose-dependent effect on whole cell levels of HDAC1 and -2 substrate, lysine 12-acetylated histone H4 (H4K12ac) as a readout (35, 36) (Fig. 3A). We next wanted to demonstrate the effect of CI-994 on fork-associated H4K12ac and employed iPOND analysis of proteins associated with nascent EdU-labeled DNA captured by formaldehyde cross-linking before cell lysis and precipitation of EdU+ DNA (31).
Protein levels at forks were quantified relative to the amounts of EdU in input samples (Fig. 8, A–C). As seen previously (36), HU arrest resulted in reduction of H4K12ac at forks compared with no-HU controls (Fig. 8, A and B). The addition of CI-994 increased the level of H4K12ac globally and at ongoing and HU-stalled forks (Fig. 8, A and B). Importantly, both in control and WRN-depleted cells, CI-994 treatment led to a comparable, ∼4-fold increase in H4K12ac levels at forks in HU (Fig. 8, A and C), indicating effective inhibition of histone deacetylase activity by CI-994. At the same time, iPOND analysis of RD wild type and HDAC1 KO cells showed that H4K12ac levels on EdU+ DNA appeared virtually the same in replicating cells and were not higher in HDAC1 KO compared with WT cells in HU arrest (Fig. 8D).
Having demonstrated a significant increase in fork-associated acetylated histone H4 in CI-994-treated cells, we asked whether this affected the ability of forks to reactivate upon release from HU (Fig. 8, E–H). In contrast to the additive deficiency elicited when HDAC1 was co-depleted with WRN in GM639cc1 fibroblasts (Fig. 3), CI-994 treatment of the same cells resulted only in a minor decrease in fork reactivation in HU-arrested WRN-depleted cells that was not statistically significant (Fig. 8F). A lack of effect of CI-994 on fork reactivation was also reproduced in an epithelial cell line MCF10a (data not shown). CI-994 suppressed fork progression rates in fibroblasts (Fig. 8G); however, forks in CI-994-treated, WRN-depleted fibroblasts did not show the evidence of premature termination or excessive slowing that we observed for HDAC1 H141A mutant (compare Figs. 8H and 7, D and E). As expected, in WRN-proficient cells, CI-994 had virtually no effect on fork reactivation (data not shown). Lastly, consistent with these data, CI-994 did not synergize with HU in suppressing growth of WRN-depleted or control cells (data not shown).
These results demonstrate that inhibition of HDAC1-3 deacetylase activity does not affect replication fork reactivation and progression in the same way as that observed in cells with a knockdown or H141A mutation of HDAC1. Also, hyperacetylation at stalled replication forks, at least at the levels achieved with CI-994, does not impede fork reactivation.
WRN Affects Recruitment of RAD51 to Stalled Forks in a Pathway Parallel to HDAC1
Our previous studies (15) and findings by Su et al. (42) suggest that WRN may affect fork recovery by functioning upstream of RAD51. HDAC1 (and -2) are thought to affect recruitment of RAD51 in double strand break repair (43). Thus we asked whether WRN modulates the association of RAD51 with stalled forks and whether HDAC1 may affect RAD51 function independently of WRN.
Using iPOND, we demonstrated recruitment of RAD51 to HU-arrested forks as well as accumulation of γH2AX and loss of PCNA from stalled forks (Fig. 9A, also see Fig. 8 for PCNA). We also determined that WRN-depleted GM639cc1 had ∼40% less RAD51 associated with HU-arrested forks than isogenic control cells (Fig. 9B).
FIGURE 9.

RAD51 recruitment to HU-stalled forks is reduced by depletion of WRN or inhibition of histone deacetylase activity of HDACs 1, 2, and 3. A, iPOND measurement of levels of RAD51, PCNA, and γH2AX in mock-depleted and WRN-depleted GM639cc1 fibroblasts pulse-labeled with EdU and treated with 2 mm HU as indicated. B, quantitation of two independent experiments performed as in A. Levels of RAD51 in pulldown assays were normalized to EdU levels in input samples (EdU in.) as in Fig. 8, B and C. Normalized RAD51 levels in HU-treated control cells were set as the baseline, and the rest of the values were expressed relative to it. C and D, maRTA analysis of RAD51-depleted GM639cc1 fibroblasts labeled with EdU and IdU as 1st and 2nd labels, respectively, and treated with 2 mm HU as indicated. C, maRTA labeling scheme and a Western blot of RAD51 depletion in GM639cc1 cells. CHK1 is the internal control. D, a bar graph of relative fork reactivation, derived as in Fig. 3C from two replicate experiments with 500–800 track measurements per sample in each experiment. Significance was determined in a one-tailed t test (*, p = 0.035). E, iPOND measurement of RAD51 and H4K12ac recruited to replication forks in mock-depleted GM639cc1 fibroblasts treated with 3 μm CI-994 overnight before and during the experiment. 2 mm HU addition and EdU pulse-labeling are as in A. F, quantitation of two independent iPOND experiments performed as in E. Quantitation and plotting are as in B, except RAD51 levels in pulldown assays were normalized to EdU levels in pulldown assays (EdU pd., lanes 1 and 2) or in inputs (EdU in., lanes 3 and 4) for comparison.
To determine whether HDAC1 depletion required RAD51 to affect activity of stalled forks, we measured the frequency of stalled fork reactivation in HDAC1- and RAD51-depleted cells (Fig. 9C). Co-depletion of HDAC1 and RAD51 reduced fork reactivation after HU compared with RAD51-depleted cells with active HDAC1 (Fig. 9D). In contrast, the addition of CI-994 had no effect on fork reactivation in RAD51-depleted cells, although both CI-994 treatment and HDAC1 depletion reduced fork progression rates in these cells with or without HU treatment (data not shown). Thus, RAD51-depleted cells displayed the same additive phenotype with HDAC1 loss, as did WRN-depleted cells. As we previously showed that combined loss of WRN and RAD51 was not additive with respect to fork reactivation compared with the individual effects of these genes (15), our new results suggest that HDAC1 may facilitate fork recovery after HU-induced stress via a pathway parallel to the WRN/RAD51-dependent pathway.
HDAC1-3 Inhibition May Modulate RAD51-mediated Daughter Strand Truncation at Stalled Forks
Despite the fact that CI-994 treatment of fibroblasts did not reduce fork reactivation after HU, by iPOND we found that it reduced the association of RAD51 with stalled forks upon HU-induced arrest (Fig. 9, E and F). Thus, we were interested in investigating what function of RAD51 at stalled forks other than reactivation may be affected by CI-994.
RAD51 is central to the pathway of protection of newly synthesized DNA strands at HU-stalled forks uncovered by Schlacher et al. (44, 45), which notably does not affect the ability of forks to reactivate replication after HU. In this pathway, nascent strands can be truncated (likely, resected or digested by endonucleases) at HU-stalled forks, and this process is limited by loading of RAD51 onto DNA. Truncation of nascent DNA at stalled forks is up-regulated if RAD51, BRCA1, or BRCA2 is defective (44, 46). Thus we predicted that by inhibiting association of RAD51 with stalled forks, CI-994 may enhance truncation of newly synthesized, daughter DNA strands.
Increased truncation of daughter DNA strands can be readily detected by maRTA and similar techniques as a shortening of replication tracks corresponding to forks that are stalled by HU (44, 46). To observe this, cells were pulsed with the 1st label followed by a high dose of HU that ensured complete stalling of forks and was added in the absence of label; then cells were released from HU into the 2nd label, allowing forks to resume replication. Control cells were pulsed with 1st and 2nd labels separated by a no-label gap (Fig. 10A).
FIGURE 10.

CI-994 treatment enhances nascent strand truncation. A, a labeling scheme for maRTA analysis of nascent strand truncation and examples of tracks seen in UW289.B1 BRCA1-deficient ovarian carcinoma cells. Pulses of 1st label (IdU) and 2nd label (EdU) are separated by a 6-h interval with no label and either with or without 5 mm HU. Nascent strand truncation is revealed as shortening of 1st label tracks upon incubation with HU. B, quantitation of 1st label track lengths from two experimental replicas performed with UW289.B1 cells as depicted in A. CI-994 was added at 3 μm overnight before and during the experiments. Significance was measured in Wilcoxon tests, and p value designations are as in Figs. 3, 7, and 8, i.e. *, p ≤ 5 × 10−2; **, p ≤ 5 × 10−3; etc. Box fill: white, 1st label tracks in untreated cells; horizontal stripes, 1st label-only tracks (inactivated forks); vertical stripes, 1st label segments of two-label tracks (reactivated forks). Note that without HU (white boxes), the absolute majority of tracks are either 1st label- or 2nd label-only, although they can belong to either ongoing or inactivated forks. C, quantitation of 1st label track lengths from two experimental replicas performed with primary human fibroblasts as depicted in A. Designations and statistics are as in B. CI-994 was added at 6 μm overnight before and during the experiments.
Without HU, the vast majority of 1st and 2nd label tracks were non-adjacent. With HU, some tracks had adjacent 1st and 2nd label segments, representing forks that stalled in HU and then reactivated after HU removal, and other tracks had the 1st label only, representing forks that were unable to reactivate (Fig. 10A). In accordance with previous studies (46), comparison of the lengths of tracks of the 1st label in HU-treated versus untreated BRCA1 null cells revealed a highly significant shortening of these tracks upon HU treatment. Specifically, both 1st label-only tracks (Fig. 10B, compare lanes 1 and 2) and, to an even greater extent, 1st label segments in two-label tracks (Fig. 10B, compare lanes 1 and 3) were shorter in HU-treated cells compared with 1st label tracks of untreated cells. This demonstrates resection during HU arrest and reveals that both the forks that were unable to reactivate (lane 2), and the ones able to do so (lane 3) have undergone resection. Moreover, treatment with CI-994 enhanced resection in both of these classes of forks (compare lanes 2 and 3 and lanes 5 and 6 in Fig. 10B).
We next used primary fibroblasts to determine if CI-994 can elicit a similar phenotype in a wild type background that is not sensitized to daughter strand truncation (Fig. 10C). HU alone had no effect on 1st label-only tracks corresponding to inactivated forks (Fig. 10C, compare lanes 1 and 2) and had only a slight, if any, effect on 1st label tracks of reactivated forks (lanes 1 and 3). However, in CI-994-treated primary fibroblasts, we observed a highly significant HU-dependent shortening of 1st-label tracks in both inactivated and reactivated forks (Fig. 10C, lanes 4–6). This effect was in addition to the HU-independent shortening of tracks upon CI-994 treatment (Fig. 10C, lanes 1 and 4), which was also observed in BRCA1 null cells (Fig. 10B) and in the experiments described earlier (Figs. 8 and 9). Together, the results indicate that CI-994 can up-regulate daughter strand truncation in HU. This is consistent with the fact that CI-994 reduces association of RAD51 with stalled forks, which would be expected to upset the balance between truncation and protection of nascent DNA strands. Moreover, the results suggest that forks that reactivate after HU actually undergo more, not less, strand truncation during HU arrest than forks that fail to activate.
Discussion
HDAC1 and -2 are class I histone deacetylases with roles in gene expression, cell signaling, and homeostasis as well as DNA repair and replication (33, 34). HDAC1 and HDAC2 are close homologs and frequently function as a homo- or heterodimer. Consistent with previous work (36), our study has demonstrated that HDAC1 and -2 are both present at the replication fork. We found that deficiency in HDAC1 alone can affect progression rate of replication forks. We also found that HDAC1 and HDAC2 facilitate survival of WRN-depleted human cells treated with the replication inhibitor hydroxyurea.
WRN facilitates normal fork progression as well as fork reactivation and progression after HU-induced replication arrest (12–15, 47). Our results indicate that in fibroblasts and mammary epithelial cell lines, these phenotypes of WRN deficiency are exacerbated by depletion of HDAC1. We also established, in the embryonic rhabdosarcoma cells line with a deletion of the endogenous HDAC1, that a deacetylase activity mutant HDAC1, H141A, combined with depletion of WRN reduced fork progression after HU treatment more dramatically than HDAC1 null. In contrast, inhibiting HDAC deacetylase activity with CI-994, a small molecule inhibitor with the highest activity against HDACs 1–3 (41), does not affect fork reactivation and progression after HU. Moreover, unlike HDAC1 depletion (Figs. 1–3), HDAC inhibition did not sensitize WRN-depleted fibroblasts to HU.
To explain the role of HDAC1 at HU-arrested forks, we first turned to a known, prominent substrate of HDAC1/2 at the fork–hyperacetylated histones incorporated into newly replicated DNA. However, we observed no simple correlation between HDAC1 inactivation, histone H4 hyperacetylation, and fork reactivation after HU treatment. In fibroblasts and epithelial cells, additive defect of fork reactivation caused by co-depletion of HDAC1 and WRN was observed without any change in total level of H4K12ac, and total and fork-associated levels of H4K12ac were not higher in HDAC1 knock-out or mutant clones of embryonal rhabdomyosarcoma cells. Conversely, chemical inhibition of HDACs 1–3 in WRN-depleted fibroblasts and epithelial cells had a modest or undetectable fork reactivation phenotype but led to a marked increase in H4K12ac. From these data, we conclude that, first, HDAC1 is redundant with HDAC2 and perhaps HDAC3 for histone H4 deacetylation at ongoing and stalled forks. Second, that cooperation of HDAC1 with WRN at the fork may involve histone or non-histone substrate(s) specific to HDAC1 and not HDAC2 or -3. Third, co-inhibition of HDACs 2 and 3 together with HDAC1 or the resulting hyperacetylation at the fork in and of itself may counteract or modify the function of HDAC1. Further studies, including comparison of the effects of mutation or chemical inhibition of HDAC1 on its enzymatic and, possibly, nonenzymatic activities at forks will be needed to fully understand the underlying mechanisms.
Association of WRN and HDAC1 with Newly Replicated DNA
Using native iPOND we found that HDAC1 is present at replication forks and remains on newly replicated DNA for a while after fork passage. HDAC2 behaves similarly and associates with DNA regardless of whether or not HDAC1 is present. We also observed direct association of WRN with newly replicated DNA at forks and after fork passage. This finding may explain why WRN was not identified as a fork-associated protein in proteomic studies that combined iPOND with mass spectrometry (48, 49).
Our studies showed that a fraction of WRN co-immunoprecipitates with HDAC2 and HDAC1 (Fig. 5), which would be consistent with the presence of the three proteins on nascent DNA. The functional significance and molecular details of this interaction require further investigation. WRN is detectable on DNA in the absence of HDAC1 and vice versa (Fig. 4), which is not surprising given that HDACs and WRN interact with numerous proteins, many of which can recruit them to DNA. Nevertheless, it is possible that deficiency in HDAC1 can result in reduced recruitment of co-factors associated with WRN, causing exacerbation of WRN-depleted replication fork phenotype induced by HU treatment.
In rhabdomyosarcoma cells association of WRN with forks was not substantially changed by the addition of HU. This is consistent with our and others' previous findings of WRN involvement both in HU-arrested and unperturbed DNA replication (9, 15). Alternatively, this may reflect a unique response of these neoplastic cells to HU treatment, and in fact we observed multiple differences between RD cells and SV40-immortalized fibroblasts in normal and HU-stressed replication. On the other hand, iPOND-MS studies reported enrichment of WRN on HU-arrested or HU and ATR (ATM and Rad3-related) inhibitor-collapsed replication forks, i.e. forks that developed double strand breaks (48, 49). Further dissection of cell type/cell fate-specific differences in fork metabolism and fork collapse versus protection responses will be required to understand the reasons for these discrepancies.
Lastly, we showed that in patient-derived WRN null fibroblasts less HDAC1 was bound to newly replicated DNA compared with unrelated wild type fibroblasts. This may be a result of a long term adaptation to growth without WRN rather than a direct effect of WRN loss. Nevertheless, if confirmed in other cell lines, this phenotype may provide insight into the effects of WRN and HDAC1 on maintenance of heterochromatin (50, 51).
Roles of WRN and RAD51 in Fork Reactivation
We showed that HDAC1 depletion is additive not only with WRN but also with RAD51 depletion in suppressing replication fork reactivation after HU treatment (Fig. 9). We and others have previously shown that RAD51, as WRN, is important for fork reactivation after HU (15, 52). Co-depleting WRN and RAD51 was epistatic for fork reactivation, suggesting that the two proteins act in the same functional pathway (15). Consistent with these results, our new data demonstrate that WRN depletion reduces the association of RAD51 with stalled forks. This reduction could reflect decreased recruitment to or increased loss of RAD51 from forks and is consistent with the phenotype recently reported in camptothecin-treated cells where chromatin-bound RAD51 was reduced in the absence of WRN (42). However, other recent findings suggest that RAD51 may function upstream of WRN (47, 53) and provide the WRN/DNA2 complex with a regressed fork as a substrate (54).
A model that can reconcile these observations is that WRN and RAD51 function in a feedback loop; regression of a fork by RAD51 generates a substrate for WRN/DNA2, which generate single-stranded DNA (ssDNA); binding of RAD51 to this ssDNA limits WRN/DNA2 activity.
Deacetylase Activity of HDACs 1 and 2 and RAD51-dependent Pathway of Fork Protection
De novo synthesized histones H3 and H4 that are incorporated into nascent chromatin concurrent with DNA replication are hyperacetylated, and these acetyl groups are removed as chromatin matures. Thus, replication forks typically operate in a hyperacetylated chromatin environment. Interestingly, deacetylation of new histones, at least on the lysine residues Lys-5 and Lys-12 of histone H4, proceeds regardless of whether replication forks are progressing or are stalled by HU (Ref. 36 and Fig. 8). Therefore, stalled forks become increasingly hypoacetylated as a function of HU arrest time. We were interested to determine whether this affects the ability of forks to reactivate after HU. We found that preventing histone H4 deacetylation at stalled forks by inhibiting HDACs 1–3 with a small molecule CI-994 did not affect fork reactivation (Figs. 8 and 9) but reduced the association of RAD51 with stalled forks (Fig. 9) and enhanced truncation of newly synthesized daughter DNA strands (Fig. 10). Importantly, this daughter strand truncation does not prevent fork reactivation, in agreement with previous findings (44, 45). Taken together, the data suggest that although histone deacetylase activity is not required for replication fork reactivation, it may be important for modulating RAD51 activity to maintain the integrity of nascent strands of stalled forks.
We propose that, under normal conditions, hyperacetylated chromatin around replication forks antagonizes the binding of RAD51 and other proteins that participate in DNA strand processing to facilitate fork stability (4). Loss of acetylation associated with the prolonged fork stalling creates a more permissive environment for fork remodeling. Overriding the loss of acetylation at a stalled fork may, therefore, disrupt the homeostasis of fork remodeling, leading to increased strand truncation.
In conclusion, our findings provide new insights into the role of epigenetic mechanisms in preserving genomic stability. Further studies will be required to delineate the molecular mechanisms by which HDAC1 contributes to the maintenance of non-histone and histone architecture at stalled replication forks to support efficient and faithful DNA replication and cell viability.
Author Contributions
K. K. performed and analyzed the experiments shown in Figs. 2–7. M. P. designed CRISPR/Cas9 constructs for deletion of HDAC1 and HDAC2, generated HDAC1-deleted and HDAC2-deleted cell lines, and cloned pLenti-EFS-T2A-emGFP constructs for HDAC1 expression. P. L. provided technical assistance and contributed to the data analyses for Fig. 10. E. C. supervised M. P., conceived some of the experiments, performed histone deacetylase assays, and co-wrote the manuscript. R. M. Jr. conceived and designed the siRNA library, conceived the study, and critically revised the manuscript. J. M. S. conceived and coordinated the study, designed, performed (for Figs. 4 and 5–7) and analyzed the experiments, and co-wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to the members of the Monnat and Chen laboratories for support and helpful discussions and to James Annis, Dr. Tim Martins, and Dr. Carla Grandori of the Quellos High Throughput Core for assistance in conducting siRNA screening. We thank Dr. Alessio Ligabue for advice on site-directed mutagenesis and Dr. Weiliang Tang for a gift of doxycycline-inducible WRN shRNA construct. We thank Dr. Piri Welcsh and Dr. Elizabeth Swisher for the gifts of MCF10a and UWB1.289 cells.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM115482 (to J. M. S.), P01CA77852 (to R. M., Jr.), and K08 AR063165-03 and R01CA196882 (to E. C.). This work was also supported by St. Baldrick's Scholar Award, Sarcoma Foundation and Rally Foundation grants (to E. C.), and by the University of Washington Royalty Research Fund and Seattle Cancer Consortium Breast NCI SPORE (to J. M. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table 1.
- HU
- hydroxyurea
- HDAC
- histone deacetylase
- iPOND
- immunoprecipitation of nascent DNA
- niPOND
- native immunoprecipitation of nascent DNA
- maRTA
- microfluidic-assisted replication track analysis
- BS
- Bloom syndrome
- IdU
- 5-iododeoxyuridine
- CldU
- 5-chlorodeoxyuridine
- EdU
- 5-ethynyldeoxyuridine
- IP
- immunoprecipitation
- PCNA
- proliferating cell nuclear antigen
- gRNA
- guide ribonucleic acid.
References
- 1. Zeman M. K., and Cimprich K. A. (2014) Causes and consequences of replication stress. Nat. Cell Biol. 16, 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hills S. A., and Diffley J. F. (2014) DNA replication and oncogene-induced replicative stress. Curr. Biol. 24, R435–R444 [DOI] [PubMed] [Google Scholar]
- 3. Magdalou I., Lopez B. S., Pasero P., and Lambert S. A. (2014) The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 30, 154–164 [DOI] [PubMed] [Google Scholar]
- 4. Neelsen K. J., and Lopes M. (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 16, 207–220 [DOI] [PubMed] [Google Scholar]
- 5. Leman A. R., and Noguchi E. (2013) The replication fork: understanding the eukaryotic replication machinery and the challenges to genome duplication. Genes 4, 1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sidorova J. M. (2008) Roles of the Werner syndrome RecQ helicase in DNA replication. DNA Repair 7, 1776–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petermann E., and Helleday T. (2010) Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 11, 683–687 [DOI] [PubMed] [Google Scholar]
- 8. Nam E. A., and Cortez D. (2011) ATR signalling: more than meeting at the fork. Biochem. J. 436, 527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rodríguez-López A. M., Jackson D. A., Iborra F., and Cox L. S. (2002) Asymmetry of DNA replication fork progression in Werner's syndrome. Aging Cell 1, 30–39 [DOI] [PubMed] [Google Scholar]
- 10. Rao V. A., Conti C., Guirouilh-Barbat J., Nakamura A., Miao Z.-H., Davies S. L., Saccá B., Hickson I. D., Bensimon A., and Pommier Y. (2007) Endogenous γ-H2AX-ATM-Chk2 checkpoint activation in Bloom's syndrome helicase-deficient cells is related to DNA replication arrested forks. Mol. Cancer Res. 5, 713–724 [DOI] [PubMed] [Google Scholar]
- 11. Davies S. L., North P. S., and Hickson I. D. (2007) Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 14, 677–679 [DOI] [PubMed] [Google Scholar]
- 12. Franchitto A., Pirzio L. M., Prosperi E., Sapora O., Bignami M., and Pichierri P. (2008) Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 183, 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ammazzalorso F., Pirzio L. M., Bignami M., Franchitto A., and Pichierri P. (2010) ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 29, 3156–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sidorova J. M., Li N., Folch A., and Monnat R. J. Jr. (2008) The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 7, 796–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sidorova J. M., Kehrli K., Mao F., and Monnat R. Jr. (2013) Distinct functions of human RECQ helicases WRN and BLM in replication fork recovery and progression after hydroxyurea-induced stalling. DNA Repair 12, 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ellis N. A., Groden J., Ye T.-Z., Straughen J., Lennon D. J., Ciocci S., Proytcheva M., and German J. (1995) The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83, 655–666 [DOI] [PubMed] [Google Scholar]
- 17. Yu C. E., Oshima J., Wijsman E. M., Nakura J., Miki T., Piussan C., Matthews S., Fu Y. H., Mulligan J., Martin G. M., and Schellenberg G. D. (1997) Mutations in the consensus helicase domains of the Werner syndrome gene: Werner's Syndrome Collaborative Group. Am. J. Hum. Genet. 60, 330–341 [PMC free article] [PubMed] [Google Scholar]
- 18. Monnat R. J., Jr. (2010) Human RECQ helicases: Roles in DNA metabolism, mutagenesis and cancer biology. Semin. Cancer Biol. 20, 329–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Croteau D. L., Popuri V., Opresko P. L., and Bohr V. A. (2014) Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 83, 519–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. German J. (1997) Bloom's syndrome. XX. The first 100 cancers. Cancer Genet. Cytogenet. 93, 100–106 [DOI] [PubMed] [Google Scholar]
- 21. Lauper J. M., Krause A., Vaughan T. L., and Monnat R. J. Jr. (2013) Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PLoS ONE 8, e59709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rossi M. L., Ghosh A. K., and Bohr V. A. (2010) Roles of Werner syndrome protein in protection of genome integrity. DNA Repair 9, 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chu W. K., and Hickson I. D. (2009) RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer 9, 644–654 [DOI] [PubMed] [Google Scholar]
- 24. Sidorova J. M., and Monnat R. J. Jr. (2015) Human RECQ helicases: roles in cancer, aging, and inherited disease. Adv. Genomics Genet. 5, 19–33 [Google Scholar]
- 25. Mao F. J., Sidorova J. M., Lauper J. M., Emond M. J., and Monnat R. J. (2010) The human WRN and BLM RecQ helicases differentially regulate cell proliferation and survival after chemotherapeutic DNA damage. Cancer Res. 70, 6548–6555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Swanson C., Saintigny Y., Emond M. J., and Monnat R. J. Jr. (2004) The Werner syndrome protein has separable recombination and survival functions. DNA Repair 3, 475–482 [DOI] [PubMed] [Google Scholar]
- 27. DelloRusso C., Welcsh P. L., Wang W., Garcia R. L., King M.-C., and Swisher E. M. (2007) Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol. Cancer Res. 5, 35–45 [DOI] [PubMed] [Google Scholar]
- 28. Kehrli K., and Sidorova J. M. (2014) Mitomycin C reduces abundance of replication forks but not rates of fork progression in primary and transformed human cells. Oncoscience 1, 540–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelsen T. S., Heckl D., Ebert B. L., Root D. E., Doench J. G., and Zhang F. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sidorova J. M., Li N., Schwartz D. C., Folch A., and Monnat R. J. Jr. (2009) Microfluidic-assisted analysis of replicating DNA molecules. Nat. Protoc. 4, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sirbu B. M., Couch F. B., and Cortez D. (2012) Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 7, 594–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leung K. H., Abou El Hassan M., and Bremner R. (2013) A rapid and efficient method to purify proteins at replication forks under native conditions. Biotechniques 55, 204–206 [DOI] [PubMed] [Google Scholar]
- 33. Kelly R. D., and Cowley S. M. (2013) The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem. Soc. Trans. 41, 741–749 [DOI] [PubMed] [Google Scholar]
- 34. Moser M. A., Hagelkruys A., and Seiser C. (2014) Transcription and beyond: the role of mammalian class I lysine deacetylases. Chromosoma 123, 67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bhaskara S., Jacques V., Rusche J. R., Olson E. N., Cairns B. R., and Chandrasekharan M. B. (2013) Histone deacetylases 1 and 2 maintain S-phase chromatin and DNA replication fork progression. Epigenetics Chromatin 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sirbu B. M., Couch F. B., Feigerle J. T., Bhaskara S., Hiebert S. W., and Cortez D. (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 25, 1320–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aranda S., Rutishauser D., and Ernfors P. (2014) Identification of a large protein network involved in epigenetic transmission in replicating DNA of embryonic stem cells. Nucleic Acids Res. 42, 6972–6986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller K. M., Tjeertes J. V., Coates J., Legube G., Polo S. E., Britton S., and Jackson S. P. (2010) Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 17, 1144–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emiliani S., Fischle W., Van Lint C., Al-Abed Y., and Verdin E. (1998) Characterization of a human RPD3 ortholog, HDAC3. Proc. Natl. Acad. Sci. U.S.A. 95, 2795–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hassig C. A., Tong J. K., Fleischer T. C., Owa T., Grable P. G., Ayer D. E., and Schreiber S. L. (1998) A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc. Natl. Acad. Sci. U.S.A. 95, 3519–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bantscheff M., Hopf C., Savitski M. M., Dittmann A., Grandi P., Michon A.-M., Schlegl J., Abraham Y., Becher I., Bergamini G., Boesche M., Delling M., Dümpelfeld B., Eberhard D., Huthmacher C., et al. (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265 [DOI] [PubMed] [Google Scholar]
- 42. Su F., Mukherjee S., Yang Y., Mori E., Bhattacharya S., Kobayashi J., Yannone S. M., Chen D. J., and Asaithamby A. (2014) Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep. 9, 1387–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fukuda T., Wu W., Okada M., Maeda I., Kojima Y., Hayami R., Miyoshi Y., Tsugawa K., and Ohta T. (2015) Class I HDAC inhibitors inhibit the retention of BRCA1 and 53BP1 at the site of DNA damage. Cancer Sci. 106, 1050–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., and Jasin M. (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schlacher K., Wu H., and Jasin M. (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pathania S., Bade S., Le Guillou M., Burke K., Reed R., Bowman-Colin C., Su Y., Ting D. T., Polyak K., Richardson A. L., Feunteun J., Garber J. E., and Livingston D. M. (2014) BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat Commun 5, 5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thangavel S., Berti M., Levikova M., Pinto C., Gomathinayagam S., Vujanovic M., Zellweger R., Moore H., Lee E. H., Hendrickson E. A., Cejka P., Stewart S., Lopes M., and Vindigni A. (2015) DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 208, 545–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sirbu B. M., McDonald W. H., Dungrawala H., Badu-Nkansah A., Kavanaugh G. M., Chen Y., Tabb D. L., and Cortez D. (2013) Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 288, 31458–31467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dungrawala H., Rose K. L., Bhat K. P., Mohni K. N., Glick G. G., Couch F. B., and Cortez D. (2015) The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell 59, 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pegoraro G., Kubben N., Wickert U., Göhler H., Hoffmann K., and Misteli T. (2009) Ageing-related chromatin defects through loss of the NURD complex. Nat. Cell Biol. 11, 1261–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang W., Li J., Suzuki K., Qu J., Wang P., Zhou J., Liu X., Ren R., Xu X., Ocampo A., Yuan T., Yang J., Li Y., Shi L., Guan D., et al. (2015) A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Petermann E., Orta M. L., Issaeva N., Schultz N., and Helleday T. (2010) Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 37, 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Iannascoli C., Palermo V., Murfuni I., Franchitto A., and Pichierri P. (2015) The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 43, 9788–9803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J. A., Herrador R., Vindigni A., and Lopes M. (2015) Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 208, 563–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.