

Abstract
The correlation between aberrant DNA methylation with cancer promotion and progression has prompted an interest in discerning the associated regulatory mechanisms. Kaiso (ZBTB33) is a specialized transcription factor that selectively recognizes methylated CpG-containing sites as well as a sequence-specific DNA target. Increasing reports link ZBTB33 overexpression and transcriptional activities with metastatic potential and poor prognosis in cancer, although there is little mechanistic insight into how cells harness ZBTB33 transcriptional capabilities to promote and progress disease. Here we report mechanistic details for how ZBTB33 mediates cell-specific cell cycle regulation. By utilizing ZBTB33 depletion and overexpression studies, it was determined that in HeLa cells ZBTB33 directly occupies the promoters of cyclin D1 and cyclin E1, inducing proliferation by promoting retinoblastoma phosphorylation and allowing for E2F transcriptional activity that accelerates G1- to S-phase transition. Conversely, in HEK293 cells ZBTB33 indirectly regulates cyclin E abundance resulting in reduced retinoblastoma phosphorylation, decreased E2F activity, and decelerated G1 transition. Thus, we identified a novel mechanism by which ZBTB33 mediates the cyclin D1/cyclin E1/RB1/E2F pathway, controlling passage through the G1 restriction point and accelerating cancer cell proliferation.
Keywords: cell cycle, cyclin D1, DNA methylation, gene regulation, transcription, Kaiso (ZBTB33), cyclin E1
Introduction
Cytosine methylation in the CpG context (mCpG)2 is a prevalent and essential epigenetic modification required for the maintenance of genomic stability, control of gene expression, and the regulation of chromatin structure. Misappropriation of genomic DNA methylation patterns has consequently been associated with cancer promotion and progression (1, 2). Indeed, it has become increasingly evident that nearly all cancers exhibit aberrant alterations in DNA methylation preceding tumorigenesis. Furthermore, specific patterns of gene hyper-methylation leading to inappropriate transcriptional regulation appear to vary between tumor type and grade (3). In gene promoters, DNA methylation elicits transcriptional regulation by either preventing or promoting transcription factor binding. Specifically, mCpG sites can be high affinity targets for specialized transcription factors, termed methyl-CpG-binding proteins (MBPs), which mediate chromatin remodeling and transcriptional control (4).
ZBTB33 (also known as Kaiso) is the founding member of the zinc finger family of MBPs and has been shown to regulate gene expression that is causally linked to tumorigenesis (5–11). Importantly, ZBTB33 and its two orthologs, ZBTB4 and ZBTB38, are unique among other MBPs in that they exhibit bimodal DNA recognition, specifically targeting both methylated and sequence-specific non-methylated DNA sites (12–14). Intriguingly, ZBTB33 has been shown to recognize methylated DNA and its sequence-specific site, termed the Kaiso-binding site (KBS; TCCTGCNA), utilizing the same set of three Cys2His2 zinc fingers (12, 15). Furthermore, this bimodal DNA recognition has afforded ZBTB33 the capability of functioning as both a transcriptional repressor and activator depending on the sequence context and cellular phenotype (16–23).
There is mounting evidence that increased ZBTB33 expression and corresponding transcriptional activities are associated with cancer progression. In colon cancer cells, methyl-dependent repression of CDKN2A by ZBTB33 protects these cells from cell cycle arrest (19). Similarly, ZBTB33-null mice crossed with APCMIN/+ mice demonstrated resistance to intestinal cancer, implying a direct correlation between the presence of ZBTB33 and the disease state (9). ZBTB33 also displays a dynamic subcellular localization mediated by its binding partner p120 catenin (p120ctn) (24, 25) that appears to play a role in its disease-inducing potential. Specifically, a cytoplasmic-to-nuclear shift has been directly linked to the more aggressive phenotypes of patients with breast and prostate cancer (PCa) tumors (5–7, 10). Indeed, a high nuclear ZBTB33 presence correlated with the pathologies, histology, and grading in invasive cohorts of high grade and basal/triple-negative breast cancers (5, 7, 10). Similarly, in PCa an increase in ZBTB33 protein expression and nuclear localization correlated with higher tumor grades and Gleason scores, with the highest levels of ZBTB33 being found in metastatic PCas (6). However, the subcellular localization of ZBTB33 appears to be variable depending on the tumor type, as the presence of cytoplasmic ZBTB33 has been linked to poor prognosis in non-small cell lung cancer (26, 27) and pancreatic cancer (28). Combined, these findings suggest that various cancer types are able to modulate the expression levels, cellular localizations, and transcriptional responses of ZBTB33 to differentially provide a context-specific survival advantage. Nonetheless, there is little mechanistic insight into how various cellular phenotypes harness the transcriptional capabilities of ZBTB33 to differentially promote and progress the disease state.
Interestingly, a significant number of findings have implicated ZBTB33 in the regulation of cellular proliferation. ZBTB33 knock-out mice exhibited increased body weight and size, due to splenomegaly resulting from increased splenocyte proliferation (18), and a dramatic reduction of lateral ventricles indicative of increased embryonic neuronal stem cell proliferation (29). Conversely, the small intestinal crypt of transgenic mice overexpressing ZBTB33 exhibited decreased cell proliferation (30). Furthermore, various ZBTB33 depletion studies have shown a consequential enhancement of cellular proliferation in lung carcinomas (BE1, LTEP-A-2, and SPC-A-1) (26), HCT 116 cell colon carcinomas (16), SK-LMS-1 vulva leiomyosarcoma cells (31), HEK293 embryonic kidney fibroblasts (32), and K562 blast crisis chronic myeloid leukemia cells when additionally depleted of p120ctn (33). In contrast, various lines of evidence have also demonstrated a pro-proliferative function for ZBTB33. Indeed, ZBTB33 depletion sensitizes Colo320 and HCT 116 colon cancer cell lines to cell cycle arrest after release from serum starvation (19) and induces decreased cellular proliferation in PC3 PCa cells (11).
Given the evident role for ZBTB33 in regulating cellular proliferation in cancer, we initiated studies to mechanistically interrogate the differential cell cycle responses mediated by the transcriptional activities of ZBTB33 in two different cell lines, HeLa and HEK293, both of which have been used extensively for studies of the cell cycle. Collectively, our data demonstrate that ZBTB33 transcriptionally regulates the G1-phase transition, although ZBTB33 acts as a pro-proliferative factor in HeLa cells and an anti-proliferative in HEK293 cells. Specifically, we have determined that ZBTB33 directly occupies the promoter regions of cyclin D1 and cyclin E1 in a KBS and methyl-specific manner, respectively, to enhance cyclin expression in HeLa cells. This ensures appropriate retinoblastoma (RB1) phosphorylation and E2F transcriptional activity, facilitating an accelerated G1- to S-phase transition. In contrast, in HEK293 cells ZBTB33 indirectly regulates cyclin E abundance resulting in reduced RB1 hyper-phosphorylation leading to decreased E2F activity and a decelerated transition through the G1-phase.
Results
ZBTB33 Is Required for Proper HeLa Cell Proliferation but Has an Inhibitory Effect on HEK293 Cell Growth
ZBTB33 depletion studies were performed by using two different ZBTB33 targeting siRNA sequences or a scrambled (Scr) siRNA control in both HeLa and HEK293 cells. The efficiency of RNA transfection was measured and determined to be ∼70% and ∼96% in HeLa and HEK293 cells, respectively (Fig. 1, A and B). Despite the fact that HeLa cells have been transformed by the human papilloma virus E7 oncogene (34), they preserve an evident cell cycle progression regulation (35) for which their response to ZBTB33 depletion can be evaluated. Immunoblot analyses confirmed that appreciable ZBTB33 depletion was achieved at the protein level 48 h after siRNA transfection (Fig. 1, C and D). Subsequent fluorescence quantification of cell viability in ZBTB33-depleted HeLa cells showed a marked reduction (Fig. 1E). In contrast, but consistent with previous observations (32), ZBTB33-depleted HEK293 cells exhibited a significant increase in cell viability (Fig. 1F). Notably, for both cell lines the observed alterations in cell viability appeared to occur without deviations in the extent of cell death (Fig. 1, G and H), indicating that ZBTB33 depletion in HeLa cells leads to decelerated cell proliferation while stimulating HEK293 proliferation. As further confirmation, direct cell counting similarly showed that ZBTB33 depletion induced substantial deceleration of HeLa proliferation (Fig. 1I) and acceleration of HEK293 proliferation (Fig. 1J).
FIGURE 1.

ZBTB33 depletion induces differential proliferation trends in HeLa and HEK293 cells. A and B, FACS analysis of GFP expression in HeLa and HEK293 cells 48 h after GFP mRNA transfection. C and D, immunoblot analyses of ZBTB33 protein expression in HeLa and HEK293 cells after transfection with either a scrambled (Scr) or ZBTB33 targeting siRNAs. E and F, fluorometric quantitation of cell viability after ZBTB33 depletion. G and H, FACS analysis of apoptosis in ZBTB33-depleted HeLa and HEK293 cells after 48 h. I and J, growth curves of HeLa and HEK293 cells after ZBTB33 depletion. *, p < 0.05; **, p < 0.005 by Student's t test.
ZBTB33 Regulates the G1- to S-phase Transition
Next, we sought to identify which cell cycle checkpoint(s) is/are regulated by ZBTB33 through monitoring changes in ZBTB33 protein levels during each cell cycle phase. Double immunostaining for ZBTB33 and the M-phase marker PHH3 was conducted on HeLa and HEK293 cells in combination with a 30-min exposure to EdU for S-phase labeling (Fig. 2, A and C). Quantification of the mean fluorescence intensities in HeLa cells showed significant differences in ZBTB33 abundance between the various cell cycle phases (Fig. 2B), whereas in HEK293 cells the amount of ZBTB33 was nearly identical across all cell cycle phases (Fig. 2D). These findings indicate that in HeLa cells ZBTB33 protein expression levels are modulated throughout the cell cycle course and that ZBTB33 mainly plays a role in G1- and M-phase progression.
FIGURE 2.

ZBTB33 abundance correlates with the regulation of cell cycle progression in HeLa cells. A and C, HeLa and HEK293 cells, respectively, 30 min after EdU pulse under regular growth conditions to visualize cells in S-phase, were additionally stained for phospho-histone 3 (PHH3 blue, M-phase) and ZBTB33 (green). From these images, the mean fluorescence intensity (normalized with cell area) of ZBTB33 staining per cell in S-, G1-, and M-phase was plotted (B and D). Because the extent of G1 is by far longer than G2 duration, we approximated that PHH3 and EdU negative cells were overwhelmingly in G1. Two random fields were analyzed per cell line. Numbers in the plots reflect the number of cells measured within each cell cycle phase. Scale bars, 20 μm. E and G, FACS analysis of cell cycle phase distributions after ZBTB33 depletion in HeLa and HEK293 cells, respectively. From replicates of the FACS data, the percent populations of cells in each cell cycle phase were plotted (F and H). Error bars reflect the mean ± S.D.; *, p < 0.05; **, p < 0.005; ***, p < 0.0005 by Student's t test; ns, not significant.
To interrogate this further, cell distributions across the different cell cycle phases were determined by fluorescence-activated cell sorting (FACS) analysis after ZBTB33 depletion using propidium iodide staining. Significantly, HeLa cells exhibited a substantially increased G1-phase population at 48 h post-ZBTB33 depletion concomitant with a decreased S-phase relative to the control (Fig. 2, E and F), indicating that ZBTB33 depletion was sufficient to induce a G1 arrest in HeLa cells. ZBTB33-depleted HEK293 cells, however, showed a moderately significant decrease in G1-phase cell population relative to the control (Fig. 2, G and H). Thus, these observations suggested that ZBTB33 is necessary for proper G1-phase progression in HeLa cells, although it has a moderate inhibitory effect on the G1-phase in HEK293 cells.
Gene Expression Profiling in ZBTB33-depleted HeLa Cells
To begin delineating the mechanism by which ZBTB33 modulates HeLa cell growth, RNA-seq was utilized to compare global transcriptome alterations between normal and ZBTB33-depleted cells. In the absence of ZBTB33, a substantial number of genes (758) demonstrated a significant transcriptional alteration relative to the control. Interestingly, it can be seen that whereas ZBTB33 transcriptional activities are largely repressive in HeLa cells (512 genes), there are appreciable numbers of genes in which ZBTB33 also appears to mediate transcriptional activation (246 genes) (Fig. 3A). Thus, in the global context these RNA-seq results reaffirm that ZBTB33 indeed has the capability of being a bi-modal regulator of transcriptional activity in HeLa cells. To identify the most significant biological pathways associated with the transcriptionally altered genes in our RNA-seq data, we performed both ingenuity pathway analysis (IPA) and Gene Set Enrichment Analysis (GSEA) (Fig. 3, B–E, and Table 1). Both of these analyses indicated that as the immunofluorescence and FACS data predicted, ZBTB33 clearly modulates genes associated with regulation of the G1- to S-phase transition in HeLa cells.
FIGURE 3.

Identification of ZBTB33-regulated transcriptional signature in HeLa cells. A, unsupervised hierarchical clustering heatmap from RNA-seq data depicting gene transcripts exhibiting a 2-fold up-regulation (green) or down-regulation (red) after ZBTB33 depletion relative to the control. B, altered genes depicted in A were subjected to Ingenuity Pathway Analysis to identify biological pathways uniquely modulated by ZBTB33. Only the 12 most statistically significant (all p < 0.05) down-regulated pathways that were also identified in our GSEA analysis are depicted. C–E, GSEA correlation plots (55) depicting ZBTB33 regulation of cell cycle genes (C), genes associated with G1- to S-phase transition (D), and genes associated with DNA replication (E). The barcode plot indicates the position of the genes in each gene set; red and blue represent positive and negative Pearson correlations with ZBTB33 depletion, respectively; and FDR is false discovery rate. F, hierarchical clustering heatmap illustrating differential expression of genes affecting cell cycle in ZBTB33-depleted HeLa cells. The heatmap color represents Z-score normalized isotig number for each gene. Blue indicates high gene expression, and red indicates low. Genes labeled with an asterisk are established E2F targets (51).
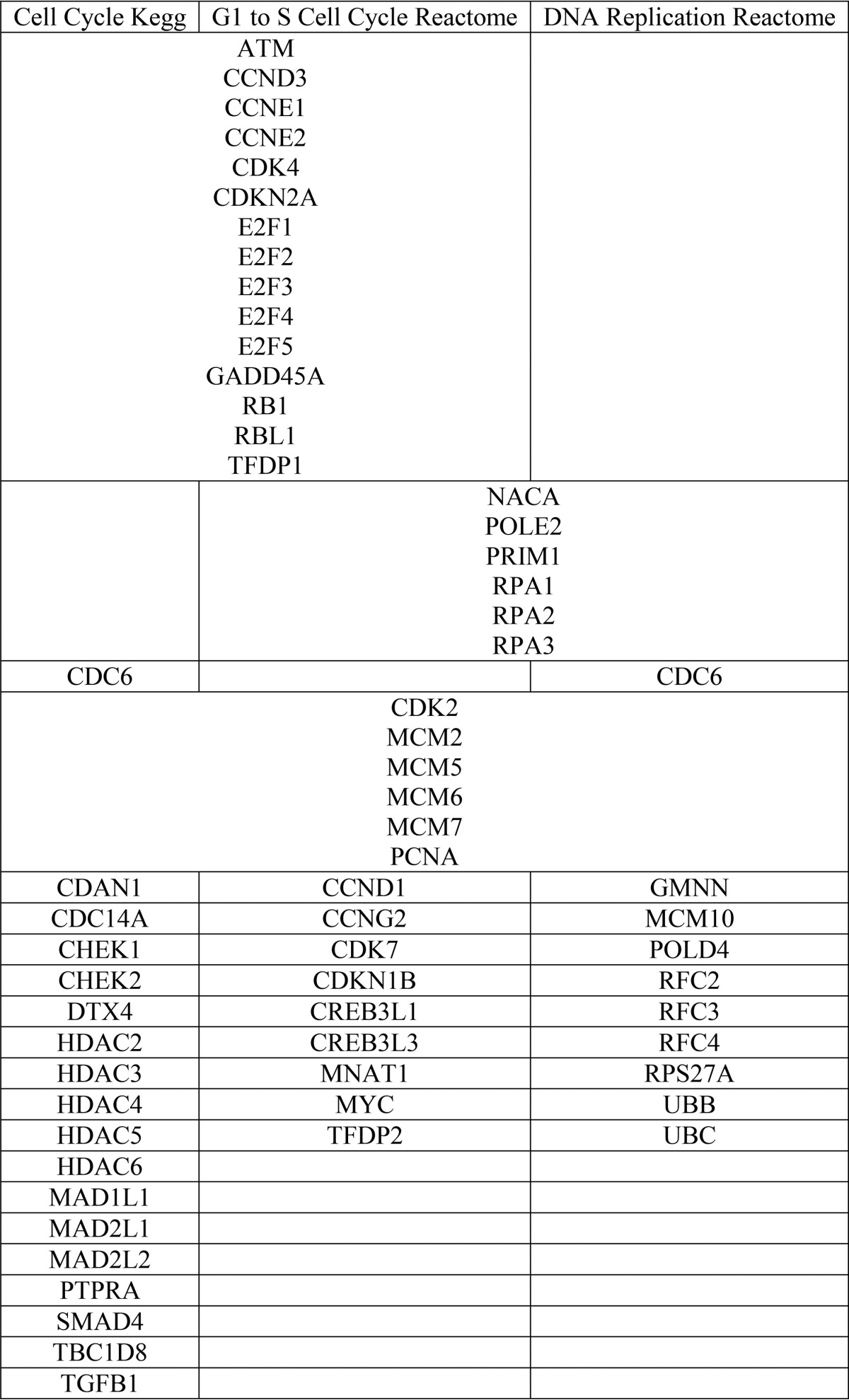
TABLE 1.
Gene lists from the GSEA correlation plots depicted in Fig. 3, C–E
ZBTB33 Regulates Cyclin D1 and Cyclin E1 Expression
Having established a definitive causal link between ZBTB33 transcriptional activities and the G1- to S-phase transition in HeLa cells, we sought to identify the cell cycle regulators that are most likely direct ZBTB33 gene targets. Thus, a list of candidate cell cycle regulator genes was mined from the RNA-seq data using the above discussed GSEA results. From this analysis, 30 candidate genes were found to have a statistically significant differential expression between the control and ZBTB33-depleted cells (Fig. 3F). Of particular note, two master regulators of G1- to S-phase transition, cyclin D1 and cyclin E1 (36), were among the most significantly down-regulated genes upon ZBTB33 depletion in HeLa cells. Direct repression of cyclin D1 by ZBTB33 has been reported previously in a number of cancerous cell lines (16, 17); however, our findings here suggest that ZBTB33 is transcriptionally activating cyclin D1 in HeLa cells. Similarly, in A549 and SPC cells, ZBTB33 transcriptional activities have been associated with repression of cyclin E, although it has not been definitively discerned as to whether this is a direct or indirect consequence (17).
To compare our HeLa cell findings with HEK293 cells and to investigate whether the observed changes in mRNA transcript levels translate at the protein level, we performed comparative semi-quantitative RT-PCR (sq-RT-PCR) and immunoblot analyses from lysates of control and ZBTB33-depleted cells. In HeLa cells, the quantities of cyclin D1 and cyclin E1 at both the mRNA and protein levels were significantly lower in ZBTB33-depleted cells (Fig. 4, A–D). In contrast, transcript levels of cyclin D1 and cyclin E1 were seemingly unaffected by ZBTB33 depletion in HEK293 cells (Fig. 4A). Interestingly, at the protein level cyclin D1 also exhibited no change after ZBTB33 depletion (Fig. 4, B and F); however, cyclin E displayed a significant 2-fold increase in HEK293 cells (Fig. 4, B and E). Cyclin E cellular abundance is regulated predominantly at the levels of gene transcription and ubiquitin-dependent proteolysis (37). Accordingly, the discrepancy between increased cyclin E protein levels and unaffected mRNA expression suggests that post-transcriptional ubiquitin-dependent regulation of this protein is activated in ZBTB33-depleted HEK293 cells. Together, these findings indicate that in HeLa cells ZBTB33 plays a direct role in transcriptionally mediating cellular levels of cyclin D1 and cyclin E1, whereas in HEK293 cells transcription of these genes appears to be ZBTB33-independent.
FIGURE 4.

ZBTB33 regulates cyclin E1 and cyclin D1 expression. A, relative mRNA expression levels of ZBTB33, cyclin E1, and cyclin D1 in HeLa and HEK293 cells determined after ZBTB33 depletion by sq-RT-PCR. B, immunoblot analysis for ZBTB33, cyclin E, and cyclin D1 in HeLa and HEK293 cells after ZBTB33 depletion. The low molecular weight cyclin E bands are due to protease or alternative splicing processing (50, 60). C–F, band intensities from three independent immunoblots were quantitated and normalized to β-actin to determine the protein expression levels of cyclin E and cyclin D1 after ZBTB33 depletion relative to the Scr control in both HeLa and HEK293 cells. *, p < 0.05; **, p < 0.01; ***, p < 0.005 by Student's t test; ns, not significant.
ZBTB33 Directly Occupies Both Cyclin D1 and Cyclin E1 Promoter Regions in HeLa Cells
It was shown previously that repression of the cyclin D1 gene in MCF-7 and HCT 116 cells required dual occupation by ZBTB33 at both a −1067 location that contains the KBS as well as an mCpG-containing site at the +69 position (16). It was further confirmed that occupation at the +69 position is methyl-dependent. To resolve the perceived discrepancy by which ZBTB33 mediates transcriptional activation of cyclin D1 in HeLa cells and repression in MCF-7 and HCT 116 cells, we performed ChIP-qPCR at the −1067 and +69 loci and analyzed these findings in the context of global methylome mapping in HeLa cells. From ChIP-qPCR, we observed a significant occupation of ZBTB33 at the −1067 KBS-containing site, but no occupation at the CpG-containing +69 site (Fig. 5, A and B). Interestingly, global methylome analysis demonstrated that in HeLa cells the cyclin D1 promoter harbors a low methylation incidence (Fig. 5A). This finding is consistent with the lack of ZBTB33 occupation at the +69 CpG site and reaffirms that ZBTB33 indeed binds this region in a methyl-dependent manner.
FIGURE 5.

ZBTB33 directly occupies and regulates cyclin D1 and cyclin E1 gene promoters. A and D, WGSBS read coverage tracks depicting percent DNA methylation (mCpG) levels at cyclin D1 and cyclin E1 promoters. Regions within each promoter amplified by ChIP-qPCR are indicated (gray bars). B, ChIP-qPCR analysis at the cyclin D1 promoter in HeLa and HEK293 cells at known ZBTB33 occupation sites, including a KBS-containing location at −1067 and a CpG-containing site at the +69 position. C, scheme depicting design of the wild type (WT) and KBS-mutated minimal cyclin D1 promoter-luciferase reporter plasmids (top). The minimal WT or KBS-mutated cyclin D1 promoter-luciferase reporter plasmids were co-transfected with ZBTBT33 siRNAs, and luciferase activity was quantitated (bottom). Luciferase reads were normalized to Renilla activity. E, ChIP-qPCR analysis for ZBTB33 occupation of the cyclin E1 promoter in HeLa and HEK293 cells at various CpG-containing regions. The PSMD5 gene promoter was selected as a positive control in HEK293 cells based on publicly available ZBTB33 ChIP-seq data (GSM1334009 and GSM803504). Error bars reflect mean ± S.D.; *, p < 0.02 by Student's t test.
To corroborate these findings, we assessed the ability of ZBTB33 to regulate luciferase expression under control of either a minimal wild type (WT) or mutated cyclin D1 promoter that considerably altered the −1069 KBS core (Fig. 5C). As anticipated, luciferase activity of the mutated cyclin D1 promoter was significantly reduced relative to the WT (Fig. 5C). Moreover, co-transfection of either the WT or mutant cyclin D1 promoter-reporter plasmid with ZBTB33 siRNAs resulted in a similar decrease in luciferase activity (Fig. 5C). Combined, these data substantiate that the ability of ZBTB33 to transcriptionally activate cyclin D1 in HeLa cells is dependent on the integrity of the −1067 KBS loci.
Finally, we sought to determine whether ZBTB33 also directly occupies the cyclin E1 promoter. Unlike the cyclin D1 promoter, cyclin E1 does not have a KBS site within approximately −1000 bp of the TSS, but from our methylome analysis it does have regions of high CpG methylation (Fig. 5D). ChIP-qPCR analysis for ZBTB33 binding in several of these regions demonstrated that it indeed appears to preferentially occupy mCpG sites in the promoter but not within intergenic regions (Fig. 5E). Moreover, occupation at this site is methyl-dependent as treatment of HeLa cells with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine abrogates ZBTB33 binding (Fig. 5E). Although promoter methylation is typically associated with gene silencing, recent large scale DNA methylation profiling studies have shown that depending on the cellular context, hyper-methylated promoters are also found near highly transcribed genes (38–40). Thus, our findings suggest that ZBTB33 has the ability to function as a methyl-dependent activator of gene activity. To the best of our knowledge, this is the first evidence that ZBTB33 directly occupies and transcriptionally modulates cyclin E1 expression to affect cell cycle progression. Together, our findings demonstrate that in HeLa cells ZBTB33 is a direct transcriptional regulator of two key cell cycle genes that function in the G1- to S-phase transition. Finally, ChIP-qPCR analysis of cyclin D1 and cyclin E1 promoter occupancy by ZBTB33 in HEK293 cells reveals a lack of ZBTB33 association with these promoters (Fig. 5, B and E), further confirming that ZBTB33 does not regulate cyclin D1 transcription and indirectly regulates cyclin E abundance in HEK293 cells.
Aberrant RB1-E2F Activity in ZBTB33-depleted Cells
To establish a causal relationship between ZBTB33 regulation of cyclin D1 and cyclin E1 transcription, HeLa cell proliferation, and G1- to S-phase transition, we assessed phosphorylation of RB1 and subsequent E2F activity in ZBTB33-depleted cells. The cyclin D-cyclin E-RB1-E2F pathway functions as a bi-stable switch regulating G1-phase transition upon cellular commitment to proliferation (41). At the beginning of G1, E2F is bound and repressed by RB1. With sufficient stimulation, activities of cyclin D-Cdk4,6 and cyclin E-Cdk2 complexes are successively induced to phosphorylate RB1 and relieve its repression of E2F (36, 42, 43). Of note, it has been previously determined that the human papilloma virus E7 oncoprotein-binding site on RB1 is separable from the regions of RB1 required to impose cell cycle arrest. As evidence, mutant RB1 proteins deficient in E7 binding retain their ability to induce cell cycle arrest and repress transcription of E2F target genes (44).
As a first analysis, we utilized a pan-RB1 antibody to discern whether global levels of RB1 protein were affected in either HeLa or HEK293 cells after ZBTB33 depletion. HEK293 cells harbor a mixture of both hyper- and hypo-phosphorylated RB1 isoforms (Fig. 6A) (45). By comparison, HeLa cells typically show a predominant single RB1 band (46, 47), consistent with the hyper-phosphorylated form as observed in these studies (Fig. 6A). Notably, Fig. 6A clearly shows that in neither cell line are there any detectably significant changes in total pan-RB1 protein levels indicating that ZBTB33 depletion did not sequester RB1 for proteolysis or reduce RB1 degradation in HEK293 cells.
FIGURE 6.

ZBTB33 regulates the RB1-E2F pathway. A, representative immunoblots for whole (pan) and phosphorylated RB1 in HeLa and HEK293 cells (100 and 12 μg of total protein per lane, respectively) after ZBTB33 depletion. Note, HeLa and HEK293 β-actin immunoblots were analyzed using different exposure intensities. B and C, 6×E2F-luciferase reporter was transfected into control and ZBTB33-depleted HeLa and HEK293 cells, and luciferase activity was quantitated. Luciferase reads, normalized to Renilla activity, represent endogenous E2F activity. D, immunoblot analysis for ZBTB33 in HeLa cells transfected with ZBTB33 overexpression plasmids under control of either a 2-kb minimal endogenous ZBTB33 promoter or a highly active CMV promoter. E and F, 6×E2F-luciferase reporter was transfected into mock control or ZBTB33-overexpressing cells demonstrating dose-dependent E2F activity in HeLa cells and a decreased E2F activity in HEK293 cells. G, representative immunoblots for E2F1–5 proteins in HeLa and HEK293 cells after ZBTB33 depletion. *, p < 0.05; **, p < 0.01; ***, p < 0.005 by Student's t test.
Next, an antibody-specific to RB1 phosphorylated at Ser-780 (pRB1Ser-780) was selected as this site is exclusively phosphorylated by the cyclin D-Cdk(4/6) complexes (48). Similarly, an antibody specific to phosphorylated Thr-821 (pRB1Thr-821) was used to evaluate cyclin E-Cdk2 activity. Consistent with the observed decrease in cyclin D1 levels (Fig. 4, B and D), ZBTB33-depleted HeLa cells exhibited reduced pRB1Ser-780 (Fig. 6A). Although ZBTB33-depleted HeLa cells harbored reduced cyclin E levels (Fig. 4, B and C), they did not show a corresponding significant decrease in pRB1Thr-821 (Fig. 6A). These unexpected results may be attributed to compensatory activity of the cyclin A-Cdk2 complex, which can also act to phosphorylate Thr-821 (48).
Proper activation of the E2F family of transcription factors is essential for adequate G1- to S-phase transition. At early G1-phase, E2F proteins are bound to and repressed by hypo-phosphorylated RB1 (Fig. 7). With sufficient stimulation, the activity of cyclin D-Cdk(4,6) and cyclin E-Cdk2 complexes are induced to initiate RB1 hyper-phosphorylation, relieving E2F repression (49). Given that ZBTB33 depletion was demonstrated to alter RB1 phosphorylation, we sought to determine the effect of ZBTB33 depletion and overexpression on E2F activity utilizing a 6×E2F luciferase reporter assay (50). As expected, ZBTB33-depleted HeLa cells displayed significantly reduced endogenous E2F activity (Fig. 6B) and, concomitantly, a concentration-dependent increase in E2F activity with ZBTB33 overexpression (Fig. 6, D and E). Notably, the observed increase in cyclin E abundance in ZBTB33-depleted HEK293 cells (Fig. 4, B and E) resulted in an increase of pRB1Thr-821 (Fig. 6A). Accordingly, ZBTB33 depletion in HEK293 cells resulted in increased E2F activity (Fig. 6C), while ZBTB33 overexpression inhibited E2F function (Fig. 6F). Interestingly, many of the genes identified by RNA-seq analysis to be dysregulated by ZBTB33 depletion in HeLa cells (Fig. 3F) are also established E2F targets (51). Furthermore, RNA-seq analysis also showed differential expression of several E2F transcription factors upon ZBTB33 depletion in HeLa cells (Fig. 3F). In general, immunoblot analyses confirmed these observations at the protein level, particularly showing increased E2F1 and decreased E2F2 and E2F3 proteins after reduction of ZBTB33 (Fig. 6G). Nonetheless, a direct ZBTB33 regulation of E2F gene transcription is difficult to discern given that the E2F proteins also regulate their own expression (51). Although this provides some complexity in interpreting the E2F luciferase reporter assay, it is clear that the net E2F activity is differentially modulated by ZBTB33 between HeLa and HEK293 cells.
FIGURE 7.

Diagram illustrating the canonical cyclin D1/cyclin E1/RB1/E2F pathway in regulating G1- to S-phase progression (top) in comparison with proposed models for the consequences of ZBTB33 depletion on these pathways in HeLa (left) and HEK293 (right) cells.
Discussion
Dysregulated expression of G1-phase cyclins has been correlated with the genesis of a large proportion of human malignancies (52). In particular, tumor up-regulation of cyclin D1 and cyclin E is generally correlated with poor prognosis. Indeed, the activation of Cdk4 and Cdk6 by cyclin D1 along with the association of cyclin E with Cdk2, function as gatekeepers at the G1 restriction checkpoint (R-point) to regulate G1- to S-phase transition and initiation of DNA replication. Consequently, overexpression of these genes has been implicated in the acceleration of cell cycle transition through the R-point that promotes genetic instability and uncontrolled cell proliferation by eliminating p53/p21 regulatory constraints (52).
Interestingly, the observed increase in ZBTB33 expression and nuclear localization associated with high grade and poor prognosis in human breast and prostate tumors appears to correlate with tumor phenotypes harboring increased cyclin D1 and cyclin E levels. ZBTB33 has been reported to play a critical role in TGFβ signaling regulation of metastasis in breast cancer, such that increasing levels of ZBTB33 are associated with decreased breast cancer metastasis-free survival times (5, 7, 10). Likewise, cyclin D1 is one of the most commonly amplified genes and frequently overexpressed proteins in breast cancer (53), and the level of cyclin E expression is used as a powerful clinical predictor of survival status in patients with breast cancer (54). Elevated levels of either ZBTB33 or cyclin D1 in primary prostate tumors have also been shown to coincide with increased tumor aggressivity and Gleason score (6, 56, 57). Furthermore, cyclin D1 and cyclin E have been identified as critical co-activators of androgen-dependent transcription and cell cycle progression in PCa cells, suggesting that their aberrant expression in tumors may contribute to persistent activation of androgen receptor function, even during androgen ablation therapy (56, 58).
Although it has been determined that ZBTB33 can transcriptionally modulate cyclin D1 and cyclin E in several cell lines (16, 17), it has thus far been shown to directly or indirectly transcriptionally repress these cyclins and subsequently reduce their proliferation capabilities. However, these findings are contradictory to the established pro-proliferative characteristic of G1 cyclins observed in high grade tumors. Interestingly, similar to our observations in HeLa cells, ZBTB33 depletion induced a decreased cellular proliferation in PC3 cells, a cell line model for high grade PCa (11). Given the increasing evidence that various cancer phenotypes are able to differentially modulate the expression levels, cellular localizations and transcriptional responses of ZBTB33, and the correlation between ZBTB33 activities in the more aggressive forms of breast and prostate tumors, it stands to reason that in addition to its established role in transcriptionally inducing epithelial-to-mesenchymal transition programming in high grade breast and prostate tumors (6, 7, 10, 59), ZBTB33 may also mediate cellular proliferation through up-regulation of G1 cyclins. As evidence for a possible mechanism by which ZBTB33 may mediate pro-proliferative activities in cancer, we show here for the first time a positive causal correlation between cell proliferation and ZBTB33, cyclin D1 and cyclin E expression levels that appears to be cell type-specific (Fig. 7).
Repression of cyclin D1 was previously shown to require ZBTB33 co-occupation at a −1067 KBS-containing loci as well as an mCpG site located at +69 (16). In HeLa cells we observed that activation of cyclin D1 was facilitated through occupation at the −1067 KBS region and that occupation of the +69 was abrogated by the lack of DNA methylation at this site. Thus, these findings suggest that altering the methylation status at the +69 site may provide a novel mechanism by which different cell types can direct the transcriptional activities of ZBTB33 to be repressive or activating at the cyclin D1 promoter. In contrast, activation of the cyclin E1 promoter appears to be reliant on methyl-dependent CpG recognition. Furthermore, we demonstrated in HeLa cells that ZBTB33 is absolutely necessary for proper cyclin D1 and cyclin E expression and that it subsequently plays a pro-proliferative role by positively regulating the RB1-E2F pathway (Fig. 7). Conversely, ZBTB33 depletion in HEK293 cells resulted in an accelerated cell proliferation due to an indirect increase in cyclin E abundance (Fig. 7), suggesting that in HEK293 cells ZBTB33 normally functions to restrain cyclin E protein levels. Thus, together we have provided novel mechanistic insight into how various cell phenotypes may be able to differentially tune the transcriptional activities of ZBTB33 at cyclin D1 and cyclin E1 promoters to alter the rates of cellular proliferation in cancer.
Experimental Procedures
Cell Culturing
HeLa and HEK293 cells were cultured in DMEM supplemented with 4.5 g/liters glucose, 2 mm glutamine, and 10% FBS and maintained at 37 °C and 5% CO2. Cell counting and viability analysis were performed on a Countess Automated Cell Counter (Thermo Fisher Scientific). Cell line authentication to confirm lack of cross-contamination was routinely verified by short tandem repeat DNA profiling.
Plasmids and Luciferase Reporter Assays
The ZBTB33 open reading frame (ORF), obtained from GenScript, was transferred into the pcDNA3.1 mammalian expression vector (Thermo Fisher Scientific), pCMV-ZBTB33. To put ZBTB33 under control of its endogenous promoter, the original CMV promoter was replaced with a DNA fragment PCR-amplified from genomic human DNA encompassing 2 kb of DNA upstream of the ZBTB33 TSS, pZBTB332K-ZBTB33. To generate the cyclin D1-luciferase reporter plasmids, an 1150-bp DNA fragment upstream of the cyclin D1 TSS was PCR-amplified from genomic human DNA and cloned into the pGL3-Basic vector (Promega). Mutation of the −1067 KBS was completed utilizing standard cloning strategies. The E2F transcription reporter, 6×E2F-luciferase (50) was co-transfected with ZBTB33 siRNAs or overexpressing vectors. The Dual-Luciferase reporter assays (Promega) were performed 48 h after transfection according to the manufacturer's directions and measured on a luminometer. A CMV-Renilla construct was used for normalizing the efficiency of the transfection.
RNA Interference and RNA/Plasmid Transfection
For all ZBTB33 depletion experiments, HeLa or HEK293 cells were transfected with either 30 pmol per well (6-well plate) of a scrambled siRNA or one of two siRNAs designed against the ZBTB33 transcript (Thermo Fisher Scientific) using Lipofectamine RNAiMAX (Thermo Fisher Scientific). Depending on the experiment, RNA or protein was extracted from the cells at either 24 or 48 h after transfection as detailed below. For mRNA transfection, 30 pmol of GFP mRNA (TRILINK Biotechnologies) was transfected into a 6-well plate using Lipofectamine RNAiMAX. Cells were collected 48 h after transfection, and GFP expression was followed by FACS analysis. Plasmid transfections in HeLa cells were performed by either using the Neon Transfection System (Thermo Fisher Scientific) following the manufacturer's protocol (2× pulse voltage 1005 V, pulse width 35 ms) or Lipofectamine 3000 (Thermo Fisher Scientific). For HEK293 cells, plasmid transfections were completed either utilizing a calcium phosphate procedure as described previously (50) or Lipofectamine 3000 (Thermo Fisher Scientific).
Immunoblotting
Cells were collected in ice-cold PBS and then lysed and sonicated in 1× RIPA buffer (10 mm Tris-HCl (pH 7.2), 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholic acid, and 5 mm EDTA) supplemented with an EDTA-free protease inhibitor mixture (Roche Applied Science). Samples were separated by gel electrophoresis, transferred to nitrocellulose or PVDF membranes, and immunoblotted.
Antibodies (Abs)
For primary antibodies, the following were used: β-actin (Santa Cruz Biotechnology, sc-47778); cyclin D1 (Cell Signaling, 2926); cyclin E (Thermo Fisher, MA5-14336); E2F1 (Abgent, AP7593b); E2F2 (Assay Biotechnology, C11025); E2F3 (Abgent, AP14598c); E2F4 (Bethyl Laboratories, A302-134A); E2F5 (Elabscience, ENT1445); PHH3 (Millipore, 06-570); RB1 (BD Biosciences, 554136); RB1 pT821 (Abcam, ab32015); RB1 pS780 (Abcam, ab173289); and ZBTB33 (Santa Cruz Biotechnology, sc-23871). For secondary antibodies, the following were used: goat anti-rabbit DyLight 800 (Rockland); goat anti-mouse IRDye 680LT (LI-COR); goat anti-mouse IgG (H+L) Alexa Fluor 488 (Thermo Fisher, A-11001); and goat anti-rabbit IgG (H+L) Alexa Fluor 546 (Thermo Fisher, A-11035).
Semi-quantitative RT-PCR (sq-RT-PCR)
Transfected cells were washed with PBS. Total RNA was isolated by TRIzol (Thermo Fisher Scientific) and reverse-transcribed using the high capacity cDNA reverse transcription kit (Thermo Fisher Scientific) following the manufacturer's protocols. The PCR products were analyzed on a 2% agarose gel. Relevant bands were excised, purified, and sequenced for validation. A primer list is available in Table 2.
TABLE 2.
Primers utilized for sq-RT-PCR
| Gene | Sense | Antisense |
|---|---|---|
| β-Actin | agctacgagctgcctgacgg | gatccacacggagtacttgcg |
| Cyclin D1 | gtgcagaaggaggtcctgcc | gcttgttcaccaggagcagc |
| Cyclin E1 | gaggaaggcaaacgtgaccg | gctcaagaaagtgctgatccc |
| ZBTB33 | tggagcgacgtttaaagaaggg | ctgaaagaatattcttgtgagcc |
Immunofluorescence, Microscopy, and Quantitative Fluorescence Intensity Analysis
Cells were fed with 10 mm EdU under regular growth conditions for 30 min and then fixed in 3.7% ice-cold formaldehyde in PBS for 15 min followed by a 5-min incubation with 100% MeOH at −30 °C. Cells were permeabilized with PBST (0.5% Triton X-100 in PBS) for 20 min at room temperature before S-phase detection using the Click-iT EdU Alexa Fluor 488 Imaging kit (Molecular Probes), following the manufacturer's protocol. Subsequent to adequate washings in 3% BSA in PBS, cells were blocked with PBST containing 10% normal goat serum for 1 h at 4 °C. Primary Abs were incubated overnight at 4 °C in PBST with 5% normal goat serum, and secondary Abs were incubated in the same buffer for 1 h at room temperature. Images were acquired using a Zeiss Axiovert 200 M microscope and captured with SlideBook software (Intelligent Imaging Innovations). Quantifications of ZBTB33 mean fluorescence intensity were performed on two randomly collected fields per cell line using ImageJ software as described previously (50).
Cell Proliferation and Apoptosis
For both fluorometric proliferation and apoptotic assays, cells were harvested 48 h post-siRNA transfection. Fluorometric cell proliferation and apoptosis analyses were performed following the manufacturer's protocols for the CyQuant NF cell proliferation assay and CellEventTM Caspase-3/7 Green flow Cytometry assay kits, respectively (Thermo Fisher Scientific). Cell growth curves were performed on siRNA-transfected cells that were grown in 6-cm plates for 48 h prior to transferring to 24-well culture dishes (HeLa, 5 × 104 cells/well; HEK293, 1.5 × 104 cells/well) and then cultured for an additional 0–4 days. Cell numbers were counted daily using a Countess Automated Cell Counter (Thermo Fisher Scientific).
Cell Cycle Analysis by FACS
Cells were harvested 48 h post-siRNA transfection and fixed/permeabilized in ice-cold 1:1 PBS/MeOH-acetone (4:1 (v/v)) while vortexing. Subsequent to adequate washings in FACS buffer (0.5% BSA, 0.05% NaN3 in PBS), cells were incubated overnight with 0.1% Triton X-100, 50 mg/ml RNase A, and 50 mg/ml propidium iodide (Sigma) in FACS buffer at 4 °C. Cells were analyzed with a FACScan flow cytometer (BD Biosciences), and cell cycle gating was examined using FLOWJO software.
Statistical Analysis
All above-mentioned experiments were repeated three to four times independently of each other. Data were analyzed with Excel software using Student's t test (Microsoft). p values (two-tailed) of 0.05 were considered statistically significant. For multiple group analyses, χ2 values were applied.
ChIP-qPCR
HeLa cells were fixed with 1% formaldehyde for 10 min to cross-link protein-DNA complexes, followed by a quench with 125 mm glycine. The cell lysis, chromatin isolation, and immunoprecipitation reaction procedures were performed using materials from the commercially available Zymo-Spin ChIP kit (Zymo Research) following the manufacturer's instructions. In short, after lysis, washing, and chromatin isolation, a Diagenode Bioruptor Standard sonication device (run at maximum amplitude five times for 15 min in ice water) was used to shear the cross-linked DNA down to 100–400-bp fragments. The immunoprecipitation was carried out using an Ab against ZBTB33 (6F8, Santa Cruz Biotechnology). After quantitating the amount of genomic DNA by PicoGreen assay, equal amounts of ChIP amplicons for each sample were prepared with the SYBR Green real time PCR master mix (Thermo Fisher Scientific) and analyzed by qPCR on a QuantStudio 6 Flex real time PCR system (Thermo Fisher Scientific). The fold enrichment was determined based on the cycle differences after normalization to input DNA. HeLa cells were grown in the absence or presence of 5-aza-2′-deoxycytidine (15 μm, Sigma) for 48 h. Medium was renewed daily. A region from the ELL3 gene was also amplified as a negative control. The qPCR primers are listed in Table 3. Sequencing validated the authenticity of the cyclin E1 promoter PCR products.
TABLE 3.
Primers used for ChIP-qPCR
| Gene | Sense | Antisense |
|---|---|---|
| CCND1 −1067a (Fig. 5, A and B, a) | tttacatctgcttaagtttgcg | ttagaatttgccctgggact |
| CCND1 + 69a (Fig. 5, A and B, b) | cacacggactacaggggagtt | ctcggctctcgcttctgctg |
| CCNE1 (Fig. 5, D and E, c) | cttggtccaggcactatgcc | ggctccacaggacctgaccc |
| CCNE1 (Fig. 5, D and E, d) | gggtcaggtcctgtggagcc | cccgcaagcccttcctggc |
| CCNE1 (Fig. 5, D and E, e) | cagaaaggtcttcagagagcc | cgccaggcacgccctccc |
| CCNE1 (Fig. 5, D and E, f) | cgccgtgtttacattccac | gacgcgggagaagtctg |
| CCNE1 (Fig. 5, D and E, g) | caaacgtgaccgttgtgagta | gatcaggttatgagctccgttc |
| ELL3 (Fig. 5, B and E) | tcactcagggagcgcctcattatt | atagcctcctgtgttctgccact |
| PSMD5 (Fig. 5E) | gatctgatcacagcctccttg | caaccactggctacagttga |
a Data are according to Ref. 16.
RNA-seq
To achieve ZBTB33 depletion, 3 × 106 HeLa cells per replicate were transfected with either ZBTB33-specific or control siRNAs as discussed above. 24 h after transfection, cells were washed with 0.01 m PBS (pH 7.4) and resuspended in TRIzol (Thermo Fisher Scientific) prior to RNA extraction with the Direct-zol RNA kit (Zymo Research). Prior to sequencing analysis, RNA aliquots were reverse-transcribed, and the amount of ZBTB33 was determined by qPCR as described above, utilizing HPRT1 as a normalization control, to ensure that sufficient ZBTB33 knockdown was achieved. For RNA-seq, RNA quality control measurements, purification, library construction, and sequencing were all performed by the High-Throughput Genomics Core within the University of Utah Huntsman Cancer Institute. In short, RNA quality was measured on a Bioanalyzer RNA 6000 Nano Chip. Small and long directional RNA-seq libraries were constructed using the Illumina TruSeq Standard mRNA sample preparation kit v2 with poly(A) selection. Libraries were sequenced with a 50-bp single-end run on the Illumina HiSeq 2000 platform.
Whole Genome Shotgun Bisulfite Sequencing (WGSBS)
Genomic DNA was isolated from 20 × 106 HeLa cells using the DNeasy Blood and Tissue kit (Qiagen). DNA quality control measurements, bisulfite (BS) conversion, purification, library construction, and sequencing were all performed by the High-Throughput Genomics Core within the University of Utah Huntsman Cancer Institute. The BS conversion and library construction were performed using the EpiGnome/TruSeq DNA methylation kit (Illumina). Briefly, genomic DNA was denatured, and BS was converted in a reaction containing unmethylated λ DNA (Promega) as a control. Following purification, adapter-ligated DNA was generated using EpiGnome polymerase. Adapter-ligated DNA molecules were enriched by 10 cycles of PCR, and concentration was normalized and sequenced with a 125-cycle paired-end run on the Illumina HiSeq 2000 platform.
Sequencing Data Analyses
RNA-seq and WGSBS fastq files were aligned to the human genome (hg19) using Novoalign (Novocraft, Inc.) and peak called with the USeq suite (62). RNA-seq reads were aligned with all known and theoretical splice junctions using the following parameters: -r All 50 -t 40 -o SAM 90 -k. The USeq NovoalignParser application was then used to parse the alignment files into binary point data by setting the posterior probability to 0 and alignment score threshold to 60. The MultipleReplicaDefinedRegionScanseqs USeq application, which utilizes the DESeq R package (63), identified statistically significant differentially expressed genes between cells treated with the scrambled and ZBTB33 siRNAs. The WGSBS fastq data were aligned in Novoalign bisulfite mode using the following parameters: -oSAM -rRandom -t240 -h120 -b2. The SAM alignments were sorted with the Picard SortSam.jar script, and all duplicates were removed using the Picard MarkDuplicates.jar script for WGSBS datasets. The results were converted into .bam files and indexed using SAMtools (64). Peak calling was performed with the USeq NovoalignBisulfiteParser to parse the alignments into four binary PointData sets containing the number of observed converted Cyt and non-converted Cyt at each reference Cyt site sequenced in the genome for both the plus and minus strands.
Bioinformatics Analysis
IPA (www.ingenuity.com) and GSEA (55) analysis tools were used to compute significant biological pathways enriched in the depleted ZBTB33 RNA-seq dataset relative to the control. For IPA analysis, only genes with ≥2-fold expression differences and p values lower than 0.01 calculated by USeq (see above) were used. The obtained canonical pathways with FDR values lower than 0.01 were considered significant. GSEA was run to identify significant up- and down-regulated expression patterns using the c2.all.v3.0.symbols.gmt gene set collection from the Broad Institute Molecular Signatures Database. Only gene sets with a minimum size of 10 in the MsigDB version 3.0 c2 curated database were selected. The Kolmogorov-Smirnov statistics with a cumulative null distribution of 1000 permutations were used to calculate the enrichment scores of each gene set. Furthermore, all gene sets with a Benjamini-Hochberg and GSEA FDR values lower than 0.01 were deemed significant.
Author Contributions
A. P. and B. A. B.-K. conceived of the project idea, designed the study, and wrote the manuscript with T. W. T. A. P. conducted the experiments and analyzed the results. T. W. T. performed the ChIP-qPCR experiments, prepared samples for RNA-seq and WGSBS experiments, and performed bioinformatics analyses on these data sets. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank B. Dalley for assistance with the high-throughput sequencing and T. Mosbruger for assistance with bioinformatics processing.
This work was supported by the Department of Chemistry, University of Utah, Grant RSG-14-185-01-DMC from the American Cancer Society, and National Institutes of Health Award P30CA042014 from the NCI for Huntsman Cancer Institute core facilities. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The reported ZBTB33 RNA-seq and WGSBS HeLa cell data have been deposited within the NCBI Gene Expression Omnibus (accession number GSE81784).
- mCpG
- methylated CpG
- MBP
- methyl-CpG-binding protein
- KBS
- Kaiso-binding site
- PCa
- prostate cancer
- Scr
- scrambled siRNA
- IPA
- ingenuity pathway analysis
- GSEA
- gene set enrichment analysis
- TSS
- transcription start site
- WGSBS
- whole genome shotgun bisulfite sequencing
- sq-RT-PCR
- semi-quantitative RT-PCR
- qPCR
- quantitative PCR
- FDR
- false discovery rate
- EdU
- ethynyl deoxyuridine
- BS
- bisulfite
- Ab
- antibody.
References
- 1. Esteller M. (2005) Aberrent DNA methylation as a cancer-inducing mechanism. Annu. Rev. Pharmacol. Toxicol. 45, 629–656 [DOI] [PubMed] [Google Scholar]
- 2. Jones P. A., and Baylin S. B. (2007) The epigenomics of cancer. Cell 128, 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Costello J. F., Frühwald M. C., Smiraglia D. J., Rush L. J., Robertson G. P., Gao X., Wright F. A., Feramisco J. D., Peltomäki P., Lang J. C., Schuller D. E., Yu L., Bloomfield C. D., Caligiuri M. A., Yates A., et al. (2000) Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat. Genet. 24, 132–138 [DOI] [PubMed] [Google Scholar]
- 4. Klose R. J., and Bird A. P. (2006) Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97 [DOI] [PubMed] [Google Scholar]
- 5. Bassey-Archibong B. I., Kwiecien J. M., Milosavljevic S. B., Hallett R. M., Rayner L. G., Erb M. J., Crawford-Brown C. J., Stephenson K. B., Bédard P. A., Hassell J. A., and Daniel J. M. (2016) Kaiso depletion attenuates transforming growth factor-β signaling and metastatic activity of triple-negative breast cancer cells. Oncogenesis 5, e208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones J., Wang H., Zhou J., Hardy S., Turner T., Austin D., He Q., Wells A., Grizzle W. E., and Yates C. (2012) Nuclear Kaiso indicates aggressive prostate cancers and promotes migration and invasiveness of prostate cancer cells. Am. J. Pathol. 181, 1836–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones J., Wang H., Karanam B., Theodore S., Dean-Colomb W., Welch D. R., Grizzle W., and Yates C. (2014) Nuclear localization of Kaiso promotes the poorly differentiated phenotype and EMT in infiltrating ductal carcinomas. Clin. Exp. Metastasis 31, 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pierre C. C., Longo J., Mavor M., Milosavljevic S. B., Chaudhary R., Gilbreath E., Yates C., and Daniel J. M. (2015) Kaiso overexpresison promotes intestinal inflammation and potentiates intestinal tumorigenesis in ApcMin/+ mice. Biochim. Biophys. Acta 1852, 1846–1855 [DOI] [PubMed] [Google Scholar]
- 9. Prokhortchouk A., Sansom O., Selfridge J., Caballero I. M., Salozhin S., Aithozhina D., Cerchietti L., Meng F. G., Augenlicht L. H., Mariadason J. M., Hendrich B., Melnick A., Prokhortchouk E., Clarke A., and Bird A. (2006) Kaiso-deficient mice show resistance to intestinal cancer. Mol. Cell. Biol. 26, 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vermeulen J. F., van de Ven R. A., Ercan C., van der Groep P., van der Wall E., Bult P., Christgen M., Lehmann U., Daniel J., van Diest P. J., and Derksen P. W. (2012) Nuclear Kaiso expression is associated with high grade and triple-negative invasive breast cancer. PLoS ONE 7, e37864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H., Liu W., Black S., Turner O., Daniel J. M., Dean-Colomb W., He Q. P., Davis M., and Yates C. (2016) Kaiso, a transcriptional repressor, promotes cell migration and invasion of prostate cancer cells through regulation of miR-31 expression. Oncotarget 7, 5677–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buck-Koehntop B. A., Stanfield R. L., Ekiert D. C., Martinez-Yamout M. A., Dyson H. J., Wilson I. A., and Wright P. E. (2012) Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc. Natl. Acad. Sci. U.S.A. 109, 15229–15234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Daniel J. M., Spring C. M., Crawford H. C., Reynolds A. B., and Baig A. (2002) The p120(ctn)-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res. 30, 2911–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Filion G. J., Zhenilo S., Salozhin S., Yamada D., Prokhortchouk E., and Defossez P. A. (2006) A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell. Biol. 26, 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buck-Koehntop B. A., Martinez-Yamout M. A., Dyson H. J., and Wright P. E. (2012) Kaiso uses all three zinc fingers and adjacent sequence motifs for high affinity binding to sequence-specific and methyl-CpG DNA targets. FEBS Lett. 586, 734–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Donaldson N. S., Pierre C. C., Anstey M. I., Robinson S. C., Weerawardane S. M., and Daniel J. M. (2012) Kaiso represses the cell cycle gene cyclin D1 via sequence-specific and methyl-CpG-dependent mechanisms. PLoS ONE 7, e50398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang G., Wang Y., Dai S., Liu Y., Stoecker M., Wang E., and Wang E. (2012) P120-catenin isoforms 1 and 3 regulate proliferation and cell cycle of lung cancer cells via βcatenin and Kaiso respectively. PLoS ONE 7, e30303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koh D. I., Yoon J. H., Kim M. K., An H., Kim M. Y., and Hur M. W. (2013) Kaiso is a key regulator of spleen germinal center formation by repressing Bcl6 expression in splenocytes. Biochem. Biophys. Res. Commun. 442, 177–182 [DOI] [PubMed] [Google Scholar]
- 19. Lopes E. C., Valls E., Figueroa M. E., Mazur A., Meng F.-G., Chiosis G., Laird P. W., Schreiber-Agus N., Greally J. M., Prokhortchouk E., and Melnick A. (2008) Kaiso contributes to DNA methylation-dependent silencing of tumor suppressor genes in colon cancer cell lines. Cancer Res. 68, 7258–7263 [DOI] [PubMed] [Google Scholar]
- 20. Pierre C. C., Longo J., Bassey-Archibong B. I., Hallett R. M., Milosavljevic S., Beatty L., Hassell J. A., and Daniel J. M. (2015) Methylation-dependent regulation of hypoxia inducible factor-1 α gene expression by the transcription factor Kaiso. Biochim. Biophys. Acta 1849, 1432–1441 [DOI] [PubMed] [Google Scholar]
- 21. Prokhortchouk A., Hendrich B., Jørgensen H., Ruzov A., Wilm M., Georgiev G., Bird A., and Prokhortchouk E. (2001) The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 15, 1613–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rodova M., Kelly K. F., VanSaun M., Daniel J. M., and Werle M. J. (2004) Regulation of the Rapsyn promoter by Kaiso and d-catenin. Mol. Cell. Biol. 24, 7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spring C. M., Kelly K. F., O'Kelly I., Graham M., Crawford H. C., and Daniel J. M. (2005) The catenin p120ctn inhibits Kaiso-mediated transcriptional repression of the b-catenin/TCF target gene matrilysin. Exp. Cell Res. 305, 253–265 [DOI] [PubMed] [Google Scholar]
- 24. Daniel J. M., and Reynolds A. B. (1999) The catenin p120ctn interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol. Cell. Biol. 19, 3614–3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelly K. F., Spring C. M., Otchere A. A., and Daniel J. M. (2004) NLS-dependent nuclear localization of p120ctn is necessary to relieve Kaiso-mediated transcriptional repression. J. Cell Sci. 117, 2675–2686 [DOI] [PubMed] [Google Scholar]
- 26. Dai S.-D., Wang Y., Miao Y., Zhao Y., Zhang Y., Jiang G.-Y., Zhang P.-X., Yang Z.-Q., and Wang E.-H. (2009) Cytoplasmic Kaiso is associated with poor prognosis in non-small cell lung cancer. BMC Cancer 9, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dai S.-D., Wang Y., Jiang G.-Y., Zhang P.-X., Dong X.-J., Wei Q., Xu H.-T., Li Q.-C., Zhao C., and Wang E.-H. (2010) Kaiso is expressed in lung cancer: its expression and localization is affected by p120ctn. Lung Cancer 67, 205–215 [DOI] [PubMed] [Google Scholar]
- 28. Jones J., Mukherjee A., Karanam B., Davis M., Jaynes J., Reams R. R., Dean-Colomb W., and Yates C. (2016) African Americans with pancreatic ductal adenocarcinoma exhibit gender differences in Kaiso expression. Cancer Lett. 380, 513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kulikov A. V., Korostina V. S., Kulikova E. A., Fursenko D. V., Akulov A. E., Moshkin M. P., and Prokhortchouk E. B. (2016) Knockout Zbtb33 gene results in an increased locomotion, exploration and pre-pulse inhibition in mice. Behav. Brain Res. 297, 76–83 [DOI] [PubMed] [Google Scholar]
- 30. Chaudhary R., Pierre C. C., Nanan K., Wojtal D., Morone S., Pinelli C., Wood G. A., Robine S., and Daniel J. M. (2013) The POZ-ZF transcription factor Kaiso (ZBTB33) induces inflammation and progenitor cell differentiation in the murine intestine. PLoS ONE 8, e74160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soubry A., Staes K., Parthoens E., Noppen S., Stove C., Bogaert P., van Hengel J., and van Roy F. (2010) The transcriptional repressor Kaiso localizes at the mitotic spindle and is a constituent of the pericentriolar material. PLoS ONE 5, e9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koh D.-I., Han D., Ryu H., Choi W.-I., Jeon B.-N., Kim M.-K., Kim Y., Kim J. Y., Parry L., Clarke A. R., Reynolds A. B., and Hur M.-W. (2014) KAISO, a critical regulator of p53-mediated transcription of CDKN1A and apoptotic genes. Proc. Natl. Acad. Sci. U.S.A. 111, 15078–15083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cofre J., Menezes J. R., Pizzatti L., and Abdelhay E. (2012) Knock-down of Kaiso induces proliferation and blocks granulocytic differentiation in blast crisis of chronic myeloid leukemia. Cancer Cell Int. 12, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helt A. M., and Galloway D. A. (2003) Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 24, 159–169 [DOI] [PubMed] [Google Scholar]
- 35. Dorner D., Vlcek S., Foeger N., Gajewski A., Makolm C., Gotzmann J., Hutchison C. J., and Foisner R. (2006) Lamina-associated polypeptide 2α regulates cell cycle progression and differentiation via the retinoblastoma-E2F pathway. J. Cell Biol. 173, 83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sherr C. J. (1994) G1 phase progression: cycling on cue. Cell 79, 551–555 [DOI] [PubMed] [Google Scholar]
- 37. Clurman B. E., Sheaff R. J., Thress K., Groudine M., and Roberts J. M. (1996) Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 10, 1979–1990 [DOI] [PubMed] [Google Scholar]
- 38. Spruijt C. G., and Vermeulen M. (2014) DNA methylation: old dog, new tricks? Nat. Struct. Mol. Biol. 21, 949–954 [DOI] [PubMed] [Google Scholar]
- 39. Iurlaro M., Ficz G., Oxley D., Raiber E. A., Bachman M., Booth M. J., Andrews S., Balasubramanian S., and Reik W. (2013) A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 14, R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu S., Wan J., Su Y., Song Q., Zeng Y., Nguyen H. N., Shin J., Cox E., Rho H. S., Woodard C., Xia S., Liu S., Lyu H., Ming G. L., Wade H., et al. (2013) DNA methylation presents distinct binding sites for human transcription factors. Elife 2, e00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yao G., Lee T. J., Mori S., Nevins J. R., and You L. (2008) A bistable Rb-E2F switch underlies the restriction point. Nat. Cell Biol. 10, 476–482 [DOI] [PubMed] [Google Scholar]
- 42. Sherr C. J., and Roberts J. M. (2004) Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 18, 2699–2711 [DOI] [PubMed] [Google Scholar]
- 43. Bartek J., Bartkova J., and Lukas J. (1996) The retinoblastoma protein pathway and the restriction point. Curr. Opin. Cell Biol. 8, 805–814 [DOI] [PubMed] [Google Scholar]
- 44. Dick F. A., Sailhamer E., and Dyson N. J. (2000) Mutagenesis of the pRB pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins. Mol. Cell. Biol. 20, 3715–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chano T., Saji M., Inoue H., Minami K., Kobayashi T., Hino O., and Okabe H. (2006) Neuromuscular abundance of RB1CC1 contributes to the non-proliferating enlarged cell phenotype through both RB1 maintenance and TSC1 degradation. Int. J. Mol. Med. 18, 425–432 [PubMed] [Google Scholar]
- 46. Vance K. W., Shaw H. M., Rodriguez M., Ott S., and Goding C. R. (2010) The retinoblastoma protein modulates Tbx2 functional specificity. Mol. Biol. Cell 21, 2770–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goodwin E. C., and DiMaio D. (2000) Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. U.S.A. 97, 12513–12518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zarkowska T., and Mittnacht S. (1997) Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem. 272, 12738–12746 [DOI] [PubMed] [Google Scholar]
- 49. Classon M., and Harlow E. (2002) The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2, 910–917 [DOI] [PubMed] [Google Scholar]
- 50. Capecchi M. R., and Pozner A. (2015) ASPM regulates symmetric stem cell division by tuning cyclin E ubiquitination. Nat. Commun. 6, 8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bracken A. P., Ciro M., Cocito A., and Helin K. (2004) E2F target genes: unraveling the biology. Trends Biochem. Sci. 29, 409–417 [DOI] [PubMed] [Google Scholar]
- 52. Malumbres M., and Barbacid M. (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 [DOI] [PubMed] [Google Scholar]
- 53. Roy P. G., and Thompson A. M. (2006) Cyclin D1 and breast cancer. Breast 15, 718–727 [DOI] [PubMed] [Google Scholar]
- 54. Lents N. H., and Baldassare J. J. (2004) CDK2 and cyclin E knockout mice: lessons from breast cancer. Trends Endocrinol. Metab. 15, 1–3 [DOI] [PubMed] [Google Scholar]
- 55. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Comstock C. E., Revelo M. P., Buncher C. R., and Knudsen K. E. (2007) Impact of differential cyclin D1 expression and localisation in prostate cancer. Br. J. Cancer 96, 970–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ozbek E., Mizrak B., Ozbek M., Buyukberber S., and Davarci M. (2000) Cyclin-D1 protooncogene expression in prostate cancer. Turkish J. Cancer 30, 15–23 [Google Scholar]
- 58. Yamamoto A., Hashimoto Y., Kohri K., Ogata E., Kato S., Ikeda K., and Nakanishi M. (2000) cyclin E as a coactivator of the androgen receptor. J. Cell Biol. 150, 873–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bassey-Archibong B. I., Kwiecien J. M., Milosavljevic S. B., Hallett R. M., Rayner L. G., Erb M. J., Crawford-Brown C. J., Stephenson K. B., Bédard P. A., Hassell J. A., and Daniel J. M. (2016) Kaiso depletion attenuates transforming growth factor-beta signaling and metastatic activity of triple-negative breast cancer cells. Oncogenesis 5, e208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang X. D., Rosales J. L., Magliocco A., Gnanakumar R., and Lee K. Y. (2003) cyclin E in breast tumors is cleaved into its low molecular weight forms by calpain. Oncogene 22, 769–774 [DOI] [PubMed] [Google Scholar]
- 61. Deleted in proof
- 62. Nix D. A., Courdy S. J., and Boucher K. M. (2008) Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics 9, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Love M. I., Huber W., and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup. (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]