

Abstract
A defect in O-mannosyl glycan is the cause of α-dystroglycanopathy, a group of congenital muscular dystrophies caused by aberrant α-dystroglycan (α-DG) glycosylation. Recently, the entire structure of O-mannosyl glycan, [3GlcAβ1-3Xylα1]n-3GlcAβ1-4Xyl-Rbo5P-1Rbo5P-3GalNAcβ1-3GlcNAcβ1-4 (phospho-6)Manα1-, which is required for the binding of α-DG to extracellular matrix ligands, has been proposed. However, the linkage of the first Xyl residue to ribitol 5-phosphate (Rbo5P) is not clear. TMEM5 is a gene product responsible for α-dystroglycanopathy and was reported as a potential enzyme involved in this linkage formation, although the experimental evidence is still incomplete. Here, we report that TMEM5 is a xylosyltransferase that forms the Xylβ1-4Rbo5P linkage on O-mannosyl glycan. The anomeric configuration and linkage position of the product (β1,4 linkage) was determined by NMR analysis. The introduction of two missense mutations in TMEM5 found in α-dystroglycanopathy patients impaired xylosyltransferase activity. Furthermore, the disruption of the TMEM5 gene by CRISPR/Cas9 abrogated the elongation of the (-3GlcAβ1-3Xylα1-) unit on O-mannosyl glycan. Based on these results, we concluded that TMEM5 acts as a UDP-d-xylose:ribitol-5-phosphate β1,4-xylosyltransferase in the biosynthetic pathway of O-mannosyl glycan.
Keywords: dystroglycan, glycoconjugate, glycosylation, glycosyltransferase, muscular dystrophy, O-mannosyl glycan, TMEM5, xylosyltransferase
Introduction
O-Mannosyl glycan is a type of O-glycan in which the reducing terminal mannose is attached to proteins via Ser and Thr residues. A defect in O-mannosyl glycan of α-dystroglycan (α-DG)3 is the cause of α-dystroglycanopathy, a group of congenital muscular dystrophies with neuronal migration disorders (1, 2). Recent studies have revealed the various structures of O-mannosyl glycans, and these structures are classified into three types: core M1, GlcNAcβ1-2Man; core M2, GlcNAcβ1-2(GlcNAcβ1-6)Man; and core M3, GalNAcβ1-3GlcNAcβ1-4Man (1, 3–7). Many genes responsible for α-dystroglycanopathy were shown to be involved in core M3 processing (1, 2). Therefore, understanding the pathomechanism of α-dystroglycanopathy requires determining the complete structure and biosynthetic mechanism of this glycan.
Recently, several groups, including ours, proposed the entire glycan structure on core M3, (3GlcAβ1-3Xylα1)n-3GlcAβ1-4Xyl-Rbo5P-1Rbo5P-3GalNAcβ1-3GlcNAcβ1-4(phospho-6)Manα1-peptide/protein (Fig. 1) (8–13). Glycans containing ribitol 5-phosphate (Rbo5P) were first identified in mammals, and several reports confirmed the presence of Rbo5P in the glycan on α-DG (14, 15). The tandem Rho5P moiety extends from the GalNAc residue in the core M3 glycan to a GlcA/Xyl unit, which serves as a primer for a (-3GlcAβ1-3Xylα1-) polysaccharide chain. The (-3GlcAβ1-3Xylα1-) repeat is required for the binding of α-DG to extracellular matrix ligands such as laminin, and a defect in the (-3GlcAβ1-3Xylα1-) repeat was thought to cause α-dystroglycanopathy (7, 9). The (-3GlcAβ1-3Xylα1-) repeat is formed by the action of LARGE, which has both GlcA transferase activity and Xyl transferase activity (9).
FIGURE 1.

Biosynthesis of the core M3 structure of the O-mannosyl glycan and the structures of the acceptor substrates and products used in this study. P, phosphate; CDP-Rbo, CDP-ribitol.
The biosynthetic pathway of O-mannosyl glycan is initiated by the transfer of mannose to Ser or Thr via POMT1/POMT2 in the endoplasmic reticulum (8). Subsequently, the phosphocore M3 unit (GalNAcβ1-3GlcNAcβ1-4(phospho-6)Man-) is formed by GTDC2, B3GALNT2, and SGK196 in the endoplasmic reticulum (10), and the first and second Rbo5P are then transferred by fukutin and FKRP, respectively, in the Golgi (13). Then, a GlcA/Xyl unit is formed to serve as a primer for the (-3GlcAβ1-3Xylα1-) repeat. The first GlcA/Xyl unit is not formed by the action of LARGE (11, 12). Defects in this biosynthetic pathway seem to result in the loss of laminin binding and cause α-dystroglycanopathy. Recently, the GlcA in this unit was shown to be formed via a β1,4 linkage to Xyl by the action of B4GAT1 (11, 12). However, how the Xyl is linked to the tandem Rbo5P structure and the identity of the xylosyltransferase that forms this linkage remain unclear.
TMEM5 was thought to be a candidate enzyme involved in the LARGE-independent synthesis of the GlcA/Xyl unit, because the (-3GlcAβ1-3Xylα1-) repeat was absent in α-dystroglycanopathy patients with a mutation in the TMEM5 gene (16). TMEM5 was one of the proteins of uncertain function among the gene products responsible for α-dystroglycanopathy (16, 17). The latest report suggested that TMEM5 was a potential xylosyltransferase (15). However, there was no direct evidence that the Xyl was transferred to Rbo5P, and hence, the role of TMEM5 activity in glycan biosynthesis is still not fully characterized.
In the present study, we determined the enzymatic activity of TMEM5 and examined the effect of the mutations found in α-dystroglycanopathy patients on TMEM5 enzyme activity. The effect of CRISPR/Cas9-mediated TMEM5 gene deletion on core M3 processing was also analyzed. Finally, we proposed the complete glycan structure of functional α-DG.
Results
TMEM5 Is a Xylosyltransferase
To address the possibility that TMEM5 acts as a xylose transferase to form Xyl-Rbo5P, we prepared a soluble form of TMEM5 (sTMEM5) and examined its activity using a potential glycopeptide substrate that contained the full glycan structure of FKRP products, based on our previous report (Fig. 1) (13). Additionally, because the FKTN product has a Rbo5P residue on the non-reducing terminal of core M3 (Fig. 1), we also examined whether the FKTN product served as an acceptor substrate for TMEM5. sTMEM5 was expressed in HEK293T cells and immunoprecipitated with anti-c-Myc antibody-agarose. Immunoprecipitated sTMEM5 was detected at the appropriate molecular mass (55 kDa) by Western blotting (Fig. 2A). The FKRP or FKTN product was incubated with sTMEM5 and UDP-Xyl and separated by HPLC. However, the reaction product and/or unreacted substrate eluted as single peaks in all cases (Fig. 2B): the FKRP product peak eluted at 19.6 min with or without sTMEM5, and the FKTN product peak eluted at 19.9 min with sTMEM5. Because it is impossible to follow the enzyme reaction under these conditions, we decided to use UDP-[14C]Xyl as a donor substrate to examine whether Xyl was transferred to the acceptor substrates. We detected significant incorporation of radioactivity into the acceptor substrate only when sTMEM5 was reacted with the FKRP product but not with the FKTN product (Fig. 2C). MS analysis revealed that the TMEM5 product (m/z 2343.2, Fig. 2E) was increased by 132.1 Da (which corresponds to a Xyl residue) compared with the FKRP product (m/z 2211.1, Fig. 2D). These results showed that TMEM5 might recognize the second Rbo5P on the FKRP product as an acceptor substrate, but not the Rbo5P in the FKTN product. In addition, the MS data of the TMEM5 product (Fig. 2E) showed that the Xyl residue was quantitatively transferred to the acceptor substrate, and this fraction was subjected to NMR analysis.
FIGURE 2.

Assay of the xylosyltransferase activity of TMEM5. A, immunoprecipitation of the Myc-tagged proteins. Soluble Myc-tagged proteins in the culture supernatant were immunoprecipitated with anti-Myc-agarose (rabbit polyclonal) and subjected to Western blotting with an anti-Myc antibody (goat polyclonal). The migration positions of the molecular weight standards are shown on the left. B and C, assay for TMEM5 activity with the FKTN or FKRP product. After a 12-h reaction at 37 °C, the substrate and product were separated by HPLC, and the UV absorbance was detected at 215 nm (B); the [14C]Xyl-labeled product was detected via liquid scintillation counting (C). D and E, the MALDI-TOF MS spectra of the TMEM5 substrate (D) and its product (E) were prepared by the nonradioactive reaction (37 °C for 12 h). F, function of the TMEM5 mutant proteins. After a 12-h reaction at 37 °C, the substrate and product were separated by HPLC, and the [14C]Xyl-labeled product was detected.
NMR Analysis of the TMEM5 Product
We performed a detailed NMR analysis to determine the structure of the TMEM5 product. 1H, 13C, and 31P assignments of the TMEM5 product were obtained through a series of one- and two-dimensional NMR measurements, including one-dimensional 31P, two-dimensional 1H-13C heteronuclear single quantum correlation (HSQC), and two-dimensional 1H-31P heteronuclear multiple-bond correlation (HMBC). The assignments of the TMEM5 product and its substrate (FKRP product) are summarized in Table 1. Then, we compared the NMR spectra of the TMEM5 product with those of the substrate (Fig. 3). Significant chemical shift differences were observed for the second ribitol (Rbo′) signals but not for the other signals, suggesting that Xyl is attached to the Rbo′ residue. In particular, Rbo′ C4 showed a large 13C downfield shift (8 ppm). Additionally, an inter-residue NOE was observed between Xyl H1 and Rbo′ H4 (Fig. 4). Thus, we concluded that Xyl is linked to the C4 position of Rbo′. The anomericity of the Xyl residue was examined by the scalar coupling constants. 3J(H1,H2) and 1J(C1,H1) of the Xyl residue were 8.7 and 163 Hz, respectively (Fig. 5), clearly indicating a β-linkage. Therefore, TMEM5 is an enzyme that transfers Xyl from UDP-Xyl to the C4 position of the second Rbo′ of the core M3 glycan via a β-linkage.
TABLE 1.
1H and 13C chemical shifts of TMEM5 substrate and product
| Glycan | Residue | Chemical shift |
|||||
|---|---|---|---|---|---|---|---|
| H1 C1 | H2 C2 | H3 C3 | H4 C4 | H5 C5 | H6 C6 | ||
| ppm | |||||||
| TMEM5 substrate | Man | 4.92 | 3.87 | 3.87 | 3.90 | 3.84 | 4.0a |
| 103.83 | 72.44 | 71.76 | 78.97 | 73.02 | 65.91 | ||
| TMEM5 product | Man | 4.92 | 3.87 | 3.86 | 3.91 | 3.84 | 4.0a |
| 103.81 | 72.49 | 71.75 | 78.94 | 73.06 | 65.83 | ||
| TMEM5 substrate | GlcNAc | 4.6a | 3.86 | 3.79 | 3.56 | 3.56 | 3.77, 3.94 |
| 103.9a | 57.08 | 83.56 | 71.31 | 78.02 | 63.40 | ||
| TMEM5 product | GlcNAc | 4.6a | 3.86 | 3.79 | 3.56 | 3.56 | 3.77, 3.93 |
| 103.9a | 57.09 | 83.57 | 71.31 | 77.99 | 63.45 | ||
| TMEM5 substrate | GalNAc | 4.6a | 4.02 | 4.22 | 4.16 | 3.73 | 3.8a |
| 103.9a | 54.15 | 77.44 | 69.51 | 77.36 | 63.62 | ||
| TMEM5 product | GalNAc | 4.6a | 4.02 | 4.22 | 4.16 | 3.73 | 3.8a |
| 103.9a | 54.16 | 77.43 | 69.51 | 77.42 | 63.63 | ||
| TMEM5 substrate | Rbo | 4.0b, 4.1b | 4.0b | 3.79 | 4.0b | 4.0b, 4.1b | |
| 69.2b | 73.5b | 73.82 | 73.5b | 69.2b | |||
| TMEM5 product | Rbo | 4.0a, 4.1a | 4.0a | 3.79 | 4.0a | 4.0a, 4.1a | |
| 69.2a | 73.5a | 73.87 | 73.5a | 69.2a | |||
| TMEM5 substrate | Rbo′ | 3.65, 3.79 | 3.85 | 3.75 | 4.0b | 4.0b, 4.1b | |
| 64.98 | 74.74 | 74.34 | 73.5b | 69.2b | |||
| TMEM5 product | Rbo′ | 3.62, 3.81 | 3.80 | 3.87 | 4.14 | 4.07, 4.17 | |
| 65.30 | 74.32 | 74.27 | 81.60 | 67.12 | |||
| TMEM5 product | Xyl | 4.62 | 3.29 | 3.46 | 3.61 | 3.32, 3.95 | |
| 105.62 | 75.91 | 78.26 | 72.03 | 67.84 | |||
a Two peaks are overlapped.
b Three peaks are overlapped.
FIGURE 3.

Comparison of the NMR spectra of the TMEM5 substrate and its product. Upper panel, two-dimensional 1H-13C HSQC spectra of the TMEM5 substrate (black) and the TMEM5 product (red). Lower panel, two-dimensional 1H-31P HMBC spectrum of the TMEM5 product. Rbo, first ribitol; Rbo′, second ribitol; x, low molecular weight impurities. The NMR spectra were collected at 298 K.
FIGURE 4.
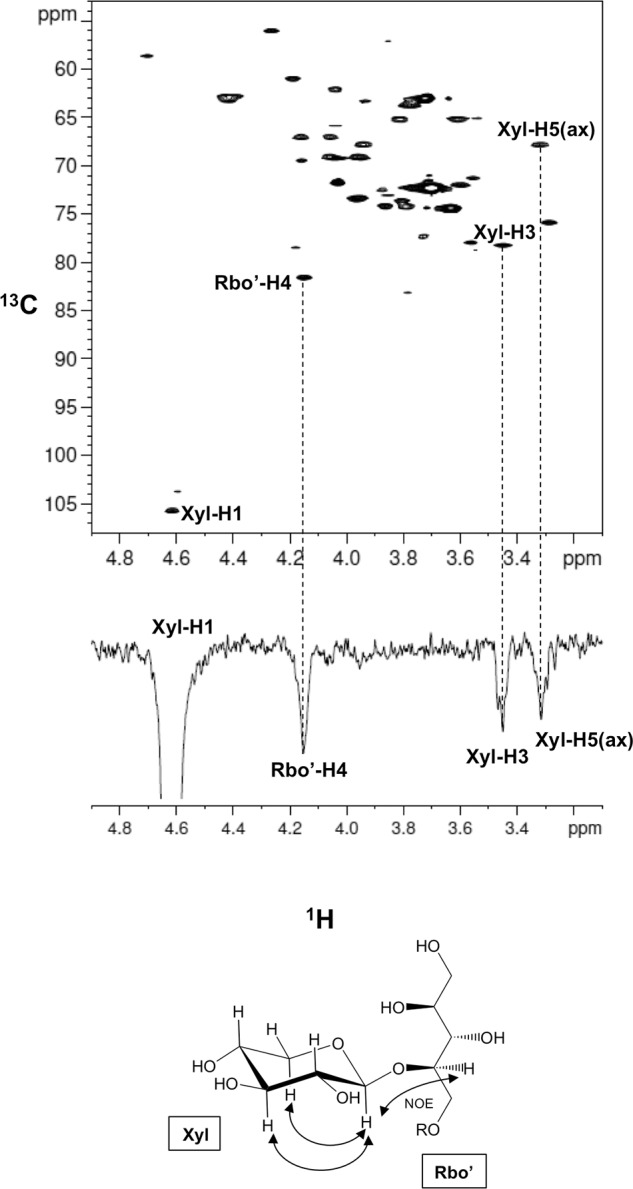
Xyl is linked to the C4 position of the second ribitol. Upper panel, two-dimensional 1H-13C HSQC spectrum of the TMEM5 product at 279 K. Lower panel, one-dimensional selective NOESY spectrum of the TMEM5 product at 279 K. The Xyl H1 signal was selectively inverted with a Gaussian-shaped pulse of 160 ms. The mixing time was set to 200 ms. Rbo′, second ribitol.
FIGURE 5.
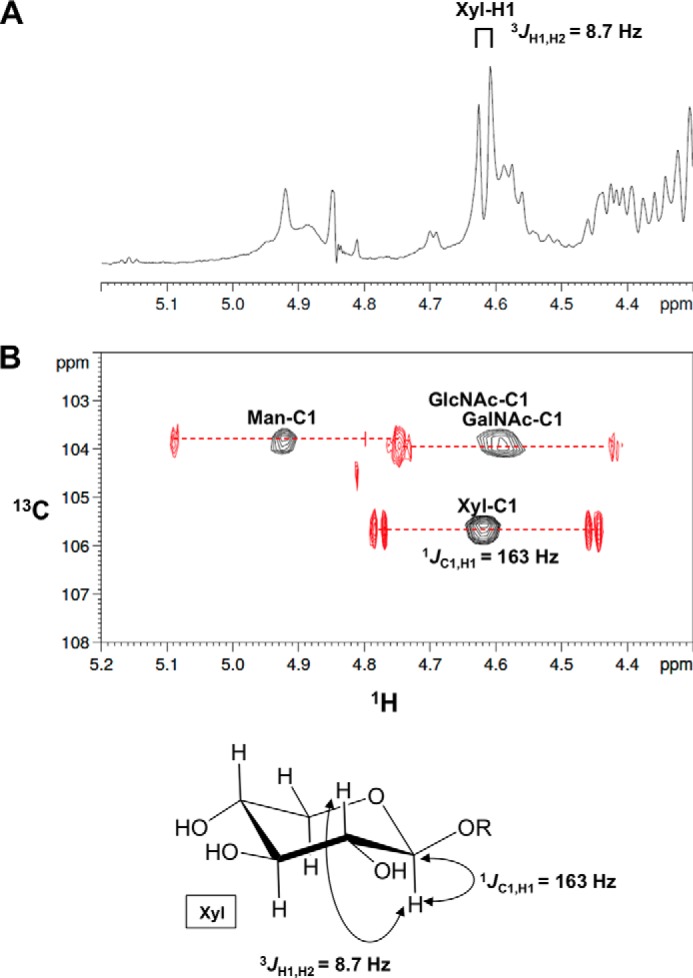
Xyl is linked to the second ribitol via a β-linkage. A, one-dimensional 1H NMR spectrum (anomeric region) of the TMEM5 product. B, overlay of the two-dimensional 1H-13C HSQC spectrum of the TMEM5 product (anomeric region) with 13C-decoupling during acquisition (shown in “black”) and two-dimensional 1H-13C HSQC spectrum of the TMEM5 product (anomeric region) without 13C-decoupling (shown in “red”). The NMR spectra were collected at 298 K. The final digital resolution in the 1H dimension was set to 4.9 Hz/point for the 13C-decoupled 1H-13C HSQC spectrum (black) and to 1.2 Hz/point for 13C-coupled 1H-13C HSQC spectrum (red).
Effect of Mutations Found in α-Dystroglycanopathy Patients on TMEM5 Activity
Several mutations in the TMEM5 gene have been identified in α-dystroglycanopathy patients (16, 18). We examined the effects of two missense mutations, Y339C and R340L, on TMEM5 activity. The Tyr339 and Arg340 residues are located in the exostosin domain (putative catalytic domain) of TMEM5 (16). We prepared soluble forms of these TMEM5 mutants, which we immunoprecipitated using the same method as for the wild-type protein (Fig. 2A). These mutants were incubated with the FKRP product and UDP-[14C]Xyl and subjected to HPLC. Although the expression levels of the two mutant proteins were slightly reduced compared with the wild-type protein, determined by Western blotting analysis (Fig. 2A), the incorporation of radioactivity into the FKRP product was completely absent in both mutants (Fig. 2F). The results indicated that these missense mutations disrupted TMEM5 xylosyltransferase activity.
Inhibition of GlcA/Xyl Repeat Biosynthesis in TMEM5-CRISPR Cells
We also generated TMEM5-deficient HAP1 cells using the CRISPR/Cas9 system and examined whether the Xylβ1-4Rbo5P structure was disrupted. To this end, we expressed recombinant DG/Fc fusion protein (DGFc) in the TMEM5-deficient cells and then analyzed the sugar chain structure by MS, as previously reported (13). We confirmed the disruption of the functional glycosylation of α-DG by Western blotting analysis using the IIH6 antibody, which recognizes the (-3GlcAβ1-3Xylα1-) repeat synthesized by LARGE (Fig. 6A). Rescue experiments confirmed that the abnormal glycosylation of α-DG was caused by the disruption of TMEM5. When DGFc was expressed in normal HAP1 cells, glycopeptides modified with tandem Rbo5P structures, an additional GlcAβ1-4Xyl, and 3GlcAβ1-3Xylα1 units were mainly eluted from an ion exchange column with 0.5 m ammonium acetate (13) (Fig. 6D). In the case of the TMEM5-deficient HAP1 cells, several signals were assigned to glycopeptides containing tandem Rbo5P structure; however, no signals were assigned to glycopeptides containing the additional Xyl moiety (Fig. 6, B and C). These data confirmed that TMEM5 synthesizes the Xylβ1-4Rbo5P linkage in the α-DG sugar chain and that the absence of this structure results in the lack of functional glycan.
FIGURE 6.

Abnormal glycosylation in TMEM5-deficient cells. A, the enrichment of α-DG with wheat germ agglutinin-agarose was analyzed by Western blotting with antibodies against the α-DG core protein (core) and glycosylated α-DG (IIH6). Normal, parent HAP1 cells; TMEM-KO, TMEM5-deficient cells; KO rescue, TMEM5-deficient cells transfected with V5-TMEM5. V5-TMEM5 expression was confirmed by an anti-V5 antibody. B–D, MS of DGFc expressed in TMEM5-KO HAP1 cells (B and C) or normal HAP1 cells (D). Glycopeptides were recovered from 0.25 m (B) and 0.5 m (C and D) ammonium acetate fractions via ion exchange chromatography. The ions containing core M3 are indicated with their estimated compositions and summarized in the table. Peaks labeled with “a” and “b” in panel D indicate glycopeptides modified with GlcA/Xyl unit(s). The graphical annotations show the structure abbreviations in the table. DGFc peptide contains five Thr residues. pep, peptide; H, Hex; N, HexNAc; P, phosphate. Note that there were variations in the Hex/HexNAc modification patterns on the glycopeptides.
The TMEM5 Product Acts as an Acceptor Substrate for B4GAT1
Previously, the formation of a GlcA/Xyl unit was reported to serve as a primer for the (-3GlcAβ1-3Xylα1-) repeat, with the GlcA residue being transferred by B4GAT1 (Fig. 1) (11, 12). Because our data indicate that TMEM5 forms the Xylβ1-4Rbo5P linkage, we examined whether the TMEM5 product served as an acceptor for B4GAT1 or LARGE. Immunoprecipitated soluble B4GAT1 (sB4GAT1) and soluble LARGE (sLARGE) migrated at the appropriate molecular weights on SDS-PAGE gels, and the expression levels of these two proteins were comparable in Western blots (Fig. 7A). The TMEM5 product and UDP-[14C]GlcA were incubated with sB4GAT1 or sLARGE and subjected to HPLC (Fig. 7B). Radioactivity was incorporated into the TMEM5 product only in the reaction with sB4GAT1, but not in the reaction with sLARGE (Fig. 7C). The sLARGE activity was confirmed using p-nitrophenyl-α-Xyl as an acceptor substrate (Fig. 7, D and E). This result verified that the Xyl residue transferred by TMEM5 acts as an acceptor for B4GAT1.
FIGURE 7.

The TMEM5 product is a substrate for B4GAT1. A, immunoprecipitation of the Myc-tagged proteins. Soluble Myc-tagged proteins in the culture supernatant were immunoprecipitated with anti-Myc-agarose (rabbit polyclonal) and subjected to Western blotting with an anti-Myc antibody (goat polyclonal). The migration positions of the molecular weight standards are shown on the left. B–E, after a 12-h reaction at 37 °C, the substrate and product were separated by HPLC, and the UV absorbance was detected at 215 nm (B and D); the [14C]GlcA-labeled product was detected via liquid scintillation counting (C and E).
Discussion
In the present study, we determined that the TMEM5 enzyme is a UDP-d-xylose:ribitol-5-phosphate β1,4-xylosyltransferase; our data offer the last piece of the biosynthetic pathway of functional α-DG glycan.
TMEM5 is one of the gene products responsible for α-dystroglycanopathy (16, 17) and was thought to be a candidate enzyme involved in the synthesis of the LARGE-independent GlcA/Xyl unit (16). Recent work revealed TMEM5 as a potential xylosyltransferase that was responsible for transferring Xyl to Rbo5P (15). The observation that CDP-Rbo functioned as an acceptor substrate suggested that TMEM5 requires a phosphodiester-linked Rbo (15). However, there was no direct evidence showing that the Xyl was transferred to Rbo5P in the core M3 glycan. Therefore, we sought to elucidate the role of TMEM5 in α-DG glycan biosynthesis (Fig. 1).
We first prepared a glycopeptide substrate that contains the full glycan structure of the FKRP product (13) to examine whether TMEM5 transfers Xyl to Rbo5P of the FKRP product. As shown in Fig. 2, C and D, TMEM5 transferred Xyl to the FKRP product but not to the FKTN product. Next, we determined the position, anomeric configuration, and glycosidic linkage of the transferred Xyl residue in the TMEM5 product by NMR (Figs. 3–5) and found that TMEM5 attaches β-Xyl to the C4 position of the second Rbo′ in the tandem Rbo5P structure. The C4 position of the second Rbo′ is chemically close to the phosphodiester linkage connecting the first Rbo and second Rbo′. Experimental evidence showed that TMEM5 did not add a significant amount of Xyl to the mono-Rbo found in the FKTN product. Thus, TMEM5 likely requires a tandem Rbo5P structure, at least in part, for efficient Xyl addition. Based on these results, we concluded that TMEM5 is a UDP-d-xylose:ribitol-5-phosphate β1,4-xylosyltransferase, and the complete structure of core M3 is proposed (Fig. 1).
TMEM5 is similar to exostosin (EXT1), a glycosyltransferase that participates in heparan sulfate proteoglycan synthesis (16). TMEM5 is a type II membrane protein, and the extracellular domain (amino acids 31–443) contains an exostosin domain (amino acids 213–353), which is a putative catalytic domain. Several missense frameshift mutations in the TMEM5 gene have been detected in α-dystroglycanopathy patients, and these mutations are located around the exostosin domain (16, 18). We found that two missense mutations, Y339C and R340L, abolished TMEM5 activity (Fig. 2F). This result suggests that these mutations disrupt the function of the catalytic domain and abolish the generation of the (-3GlcAβ1-3Xylα1-) repeat. The absence of the (-3GlcAβ1-3Xylα1-) repeat in TMEM5-deficient HAP1 cells suggests the lack of an alternative pathway to compensate for TMEM5 function in core M3 glycan processing.
As shown Fig. 7, we confirmed that the TMEM5 product served as an acceptor substrate for B4GAT1. B4GAT1 has been reported to prefer β-linked Xyl over α-linked Xyl (12). This observation is consistent with our finding that the Xyl is linked to the second Rbo′ on the TMEM5 product via a β-linkage. Although B4GAT1 clearly recognizes at least β1,4-linked Xyl, further experiments are needed to characterize the substrate specificity of B4GAT1, such as determining the importance of the linkage and necessity of Rbo5P. On the other hand, LARGE did not transfer GlcA to the β1,4-linked Xyl residue on the TMEM5 product. This result is consistent with the finding that the glucuronosyltransferase activity of LARGE is specific to α-linked Xyl and not to β-linked Xyl (9). Therefore, formation of the GlcAβ1-4 linkage by B4GAT1 is required for LARGE action. The (-3GlcAβ1-3Xylα1-) repeat generated by LARGE is also known to require an additional GlcA/Xyl unit. Our data revealed this unit to be GlcAβ1-4Xylβ1-4Rbo5P. Interestingly, core M3 uses two types of GlcA/Xyl units, GlcAβ1-3Xylα1-3 and GlcAβ1-4Xylβ1-4, to form functional glycans. Furthermore, the (-3GlcAβ1-3Xylα1-) repeat, which is synthesized by LARGE, is composed of α-Xyl. The different Xyl linkages produced by TMEM5 and LARGE might be important for the step-by-step construction of O-mannosyl glycan.
Here, we have identified the enzymatic function of TMEM5, which produces a Xylβ1-4Rbo5P. We believe that TMEM5 synthesizes this structure on the core M3 of α-DG but may possibly act on other glycans and/or other proteins. The (-3GlcAβ1-3Xylα1-) repeat generated by LARGE resembles glycosaminoglycan chains (heparan sulfate, chondroitin sulfate, etc.), which consist of repeating disaccharides are synthesized by copolymerases with dual glycosyltransferase activities. It is important to survey other proteins for TMEM5 activity to elucidate the function of the (-3GlcAβ1-3Xylα1-) repeat and laminin binding.
Experimental Procedures
Plasmid Construction
The cDNA encoding human TMEM5 was cloned from HEK293 cells by RT-PCR using primers TMEM5F (TAGGGATCCGCCGCCACCATGGAGGCCGGGCCGCCGGGCAGCGCCAG) and TMEM5R (AGATCTAGATGCTATCAGAAGCTGACCAATGAGTCCAG). The PCR product was inserted into BamHI and XbaI sites of the pEF1V5/His vector. To generate sTMEM5, the cDNA encoding Arg31 to the C-terminal stop codon was amplified using primers recTMEM5 (TATGAATTCACGCCGCCGCCAGGCGCCGGCCGGGT) and recTMEM5R (AGACTCGAGTTAACTTTTATTATTCATTAAAAATGAGCTTTCTAA). The PCR product encoding sTMEM5 was digested with EcoRI/XbaI and inserted into the EcoRI and XbaI sites of ss2-His/Myc-pcDNA3.1.
Missense mutations (16) were generated by site-directed mutagenesis using a two-sided splicing method involving overlapping extension. The ss2-His/Myc-TMEM5-pcDNA3.1 plasmid was used as the template. The following primer pairs were used for the first PCR: p.Y339C (Y3339C.F (CGGAGTAAACACAGAATGCTGTCGAATCTATGAGGCTTGCTC) and BGHpA (CTAGAAGGCACAGTCGAGGC)) and (T7 (TAATACGACTCACTATAGG), and Y3339C.R (GAGCAAGCCTCATAGATTCGACAGCATTCTGTGTTTACTCCG)), and p.R340L (R340L.F (GAGTAAACACAGAATGCTATCTTATCTATGAGGCTTGCTCCTATG) and BGHpA) and (T7 and R340L.R (CATAGGAGCAAGCCTCATAGATAAGATAGCATTCTGTGTTTACTC)). The PCR products for each mutant were mixed, extended, and amplified using the T7 and BGHpA primer pair. The second PCR products were digested with EcoRI/XbaI and then inserted into the EcoRI and XbaI sites of ss2-His/Myc-pcDNA3.1.
The cDNA encoding human B4GAT1 was cloned by RT-PCR from human brain total RNA (Clontech) using primers iGnT1f (CCGGAATTCGCCGCCACCATGCAGATGTCCTACGCCATCCGGTGC) and iGnT1r (GCCTCTAGAGCAGCGTCGGGGAGAGTTGGGGTACTTG). The PCR product was inserted into the EcoRI and XbaI sites of the pcDNA3.1 Myc/His vector. The sB4GAT1 was generated by PCR amplification of the cDNA encoding Gly35 to the C-terminal stop using primers recB3GNT1F (TATGAATTCAGGACTGCACGGGCAGGAGGAGCAAGACCAA) and recB3GNT1R (AGACTCGAGTCAGCAGCGTCGGGGAGAGTTGGGGTACTT). The PCR product encoding sB4GNT1 was digested with EcoRI/XhoI and inserted into the EcoRI and XbaI sites of ss1-His/Myc-pcDNA3.1. The cDNA encoding human LARGE and the sLARGE expression vector were described previously (13).
Preparation and Analysis of the Cells with CRISPR/Cas9 Genome Editing
The CRISPR/Cas9 targeting sequence for TMEM5 was GAAGAATGGAATCCTTGGGA. The oligonucleotide was inserted into the Cas9 Smart Nuclease All-in-One vector (System Biosciences, Mountain View, CA) with an additional 8-base sequence at the 5′ end. The vector was transfected into HAP1 cells (Haplogen, Vienna, Austria). IIH6-negative cells were sorted by fluorescence-activated cell sorting (FACS; MoFlo, Beckman Coulter, Brea, CA). HAP1 cells were cultured in Iscove's modified Dulbecco's medium (IMDM, Wako Pure Chemical Industries) supplemented with 10% FBS and penicillin/streptomycin. Each cell clone was verified for IIH6 reactivity by Western blotting and DNA sequencing. The mutation comprised a 1-bp insertion that introduced a frameshift. For the rescue experiments, TMEM5-V5 was expressed in TMEM5-deficient HAP1 cells. Normal, TMEM5-deficient, and TMEM5-rescued HAP1 cells were solubilized with 1% Triton X-100 in Tris-buffered saline (TBS; 50 mm Tris-HCl, pH 7.4, and 150 mm NaCl) containing a protease inhibitor mixture (Nacalai Tesque, Kyoto, Japan). Protein concentrations were determined using Lowry's method, and then equal amounts of protein were incubated with wheat germ agglutinin beads (Vector Laboratories) or anti-V5 beads (Medical and Biological Laboratories, Nagoya, Japan) to prepare DG or TMEM5, respectively. The bound materials were eluted with Laemmli sample buffer and analyzed by Western blotting. The following antibodies were used for Western blotting: mouse monoclonal IIH6 against glycosylated α-DG (Merck Millipore, Darmstadt, Germany), a rat monoclonal antibody against the α-DG core protein (3D7) (19), and a mouse monoclonal antibody against the V5 tag (Nacalai Tesque). Preparation and MS analysis of DGFc were performed as described previously (13).
Preparation of Enzyme Sources
The expression plasmids were transfected into HEK293T cells with Lipofectamine 3000 (Life Technologies Japan, Tokyo, Japan). Recombinant proteins were immunoprecipitated from 1 ml of culture supernatant using 10 μl of anti-c-Myc antibody-agarose (rabbit polyclonal, Sigma). The protein-bound agaroses were used as the enzyme sources. Samples (2.5 μl of agarose) were separated by SDS-PAGE (10% gel), and the proteins were transferred to PVDF membranes. The membranes were blocked with 5% skim milk in 137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, and 0.05% Tween 20 (PBS-T), incubated with anti-c-Myc polyclonal antibody (A-14-goat, Santa Cruz Biotechnology, Dallas, TX) in 5% skim milk PBS-T, and treated with horseradish peroxidase-conjugated anti-goat IgG (GE Healthcare, Buckinghamshire, England) in 5% skim milk PBS-T. Proteins that bound to the antibody were visualized with an ECL kit (GE Healthcare).
Preparation of the Substrates for the Glycosyltransferase Assay
A series of glycopeptides were used as native-like substrates for the glycosyltransferase assay (Fig. 1) and synthesized from the mannosyl peptide (Ac-AT(Man)PAPVAAIGPK-NH2; Man-peptide) using GTDC2, B3GALNT2, SGK196, fukutin, and FKRP as described previously (13). Briefly, the Man-peptide was generated by solid-phase synthesis using N-(9-fluorenyl)methyoxycarbonyl (Fmoc) chemistry (13). The peptide sequence corresponds to the amino acid sequence of mouse α-DG (Ala314-Pro324) and replaces the Thr residues at the potential O-GlcNAc glycosylation sites with Ala. The recombinant sGTDC2, sB3GALNT2, sSGK196, sfukutin, sFKRP, and sTMEM5 proteins were expressed in HEK293T cells and immunoprecipitated from the culture supernatants with the anti-c-Myc antibody-agarose (Sigma). The protein-bound agaroses were used as the enzyme sources. The enzyme reactions were performed in a 400-μl reaction volume at 37 °C for 16 h using the following buffer conditions: sGTDC2, sB3GALNT2, sfukutin, sFKRP, and sTMEM5, 100 mm MES (pH 6.5), 2.5–5 mm donor substrate, 0.2–2 mm acceptor peptide, 10 mm MnCl2, 10 mm MgCl2, 0.5% Triton X-100, and 200 μl of enzyme-bound agarose; sSGK196, 100 mm sodium acetate (pH 5.5), 5 mm ATP, 1 mm sB3GALNT2 product, 0.5% Triton X-100, and 200 μl of sSGK196-bound agarose. Each donor substrate was obtained as follows: UDP-GlcNAc and UDP-GalNAc were purchased from TOYOBO (Osaka, Japan), ATP was purchased from Sigma, CDP-Rbo was synthesized as described previously (13), and UDP-Xyl was obtained from the Complex Carbohydrate Research Center at the University of Georgia. Each product was separated by reverse-phase HPLC with a Mightysil RP-18GP Aqua column (10 × 250 mm) (KANTO CHEMICAL, Tokyo, Japan). Solvent A was 0.1% TFA in distilled water, and solvent B was 0.1% TFA in acetonitrile. Peptides were eluted at a flow rate of 3 ml/min using a linear gradient of 0–40% solvent B. Peptide separation was monitored by determining the absorbance at 214 nm. Each product was used as an acceptor substrate. The TMEM5 product was analyzed by MS and NMR.
Glycosyltransferase Assay
sTMEM5, the sTMEM5 mutants, sB4GAT1, or sLARGE were expressed and immunoprecipitated with the anti-c-Myc antibody-agarose conjugate. The protein-bound agaroses were used as the enzyme sources. The enzymatic reactions were performed in a 20-μl reaction volume containing 100 mm MES (pH 6.5), 1 mm 14C-labeled donor substrate (75,000 dpm/nmol), 0.1 mm acceptor peptide, 10 mm MnCl2, 10 mm MgCl2, 0.5% Triton X-100, and 5 μl of the enzyme-bound agarose at 37 °C for 1–12 h. p-Nitrophenyl-α-Xyl (1 mm, Sigma) was also used as an acceptor for sLARGE activity. Donor substrates were purchased from the following sources: UDP-[14C]Xyl, PerkinElmer Life Sciences, Waltham, MA; UDP-[14C]GlcA ([14C]glucuronyl), American Radiolabeled Chemicals, Saint Louis, MO; and UDP-GlcA (TOYOBO). Each product was separated with a Mightysil RP-18GP Aqua column (5 × 250 mm) (KANTO CHEMICAL). Solvent A was 0.1% TFA in distilled water, and solvent B was 0.1% TFA in acetonitrile. Peptides were eluted at a flow rate of 1 ml/min using a linear gradient of 0–40% solvent B. Peptide separation was monitored by determining the absorbance at 214 nm, and the radioactivity of each fraction (1 ml) was measured with a liquid scintillation counter.
Mass Spectrometry
Matrix-assisted laser desorption/ionization (MALDI) time-of-flight (TOF) MS was used to elucidate the glycopeptide structures. MALDI linear TOF measurements were carried out on a Voyager DE Pro MALDI TOF mass spectrometer equipped with a nitrogen pulsed laser (337 nm) (Applied Biosystems, Foster City, CA). Typically, 0.1–1 pmol of the glycopeptide samples was dissolved in a 1-μl solution of 10 mg/ml of 2,5-dihydroxybenzoic acid in 0.1% TFA and 30% acetonitrile on a MALDI sample target and dried. The measurements were carried out in positive ion mode.
NMR Analysis
NMR spectra were recorded with 500 and 600 MHz NMR spectrometers (BrukerBioSpin) equipped with a 5-mm TXI cryogenic probe and a BBO probe, respectively. The probe temperature was set to 25 or 6 °C. The sample was dissolved in D2O (99.99 atom % D), and the sample concentration was 0.25 mm for the TMEM5 substrate and 0.33 mm for the TMEM5 product. 1H chemical shifts were reported relative to the external standard 4,4-dimethyl-4-silapentane-1-sulfonic acid. The 13C and 31P chemical shifts were calibrated using an indirect reference based on the X/1H resonance ratios of 0.251449530 (13C/1H) and 0.404808636 (31P/1H). NMR signals were assigned by a series of one- and two-dimensional measurements, including one-dimensional 1H, one-dimensional 31P, one-dimensional selective nuclear Overhauser effect spectroscopy (NOESY), two-dimensional 1H-13C HSQC, two-dimensional 1H-13C HSQC-total correlation spectroscopy (TOCSY), and two-dimensional 1H-31P HMBC spectra. The 3JHH and 1JCH coupling constants of the Xyl anomeric signal were obtained from one-dimensional 1H and two-dimensional 13C-coupled 1H-13C HSQC spectra, respectively. To estimate the coupling constants, the final digital resolution in the 1H-dimension was set to 4.9 Hz/point for the 13C-decoupled HSQC spectrum and 1.2 Hz/point for the 13C-coupled HSQC spectrum. The NMR data were processed with XWIN-NMR (version 3.5) or TopSpin (version 2.1), and the spectra were displayed using XWIN-PLOT (version 3.5).
Author Contributions
H. M., Y. Y., M. K., and K. K. designed the project, performed the experiments, analyzed and interpreted the data, and wrote the manuscript. H. M. and K. A.-M. identified the enzyme activities and generated glycopeptides for NMR. M. K. and K. K. constructed the plasmids and generated the CRISPR cells. Y. Y. performed the NMR analysis. M. T. performed the MS analysis. H. K. and M. M. generated essential reagents. Y. W., T. T., and T. E. designed the project, edited the manuscript, and jointly supervised the research. H. M. and T. E. are responsible for all reagents and data. All authors discussed the data and the manuscript.
This work was supported by the National Center of Neurology and Psychiatry (NCNP) Intramural Research Grant 26-8 (to T. T. and T. E.), the Japan Agency for Medical Research and Development (AMED) Grants 16gm0810010h0201 (to H. M.), 16gm0810010h0001 (to M. K.), and 15dk0310041h0002 (to T. T.), the Japan Society for the Promotion of Science Grants 26253057 (to T. T.), 16K08262 (to T. E.), 23390081 (to Y. W.), 15H04352 and 24687017 (to M. K.), 26670499 and 16H05353 (to K. K.), and 24619014 (to M. T.), the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) Grants 26110712 (to M. K.) and 26110727 (to H. M.), and the Mizutani Foundation for Glycoscience Grant 150171 (to H. M.). The authors declare that they have no conflicts of interest with the contents of this article.
- α-DG
- α-dystroglycan
- B3GALNT2
- β-1,3-N-acetylgalactosaminyltransferase 2
- B4GAT1
- β-1,4-glucuronyltransferase 1
- core M1
- GlcNAcβ1-2Man
- core M2
- GlcNAcβ1-2 (GlcNAcβ1-6)Man
- core M3
- GalNAcβ1-3GlcNAcβ1-4Man
- CRISPR
- clustered regularly interspaced short palindromic repeats
- Cas9
- CRISPR-associated protein 9
- FKTN
- fukutin
- FKRP
- fukutin-related protein
- GlcA
- glucuronic acid
- GlcNAc
- N-acetylglucosamine
- GTDC2
- protein O-mannose β-1,4-N-acetylglucosaminyltransferase 2
- HexNAc
- N-acetylhexosamine
- HMBC
- heteronuclear multiple-bond correlation
- HSQC
- heteronuclear single quantum correlation
- LARGE
- like-acetylglucosaminyltransferase
- POMT
- protein O-mannosyltransferase
- Rbo
- ribitol
- Rbo5P
- ribitol 5-phosphate
- SGK196
- protein kinase-like protein sugen kinase 196
- TMEM5
- transmembrane protein 5
- DGFc
- DG/Fc fusion protein.
References
- 1. Endo T. (2015) Glycobiology of α-dystroglycan and muscular dystrophy. J. Biochem. 157, 1–12 [DOI] [PubMed] [Google Scholar]
- 2. Manya H., and Endo T. (2015) O-Mannosyl glycan and muscular dystrophy. in Sugar Chains: Decoding the Functions of Glycans (Suzuki T., Ohtsubo K., and Taniguchi N., eds) pp. 235–258, Springer Japan, Tokyo [Google Scholar]
- 3. Chiba A., Matsumura K., Yamada H., Inazu T., Shimizu T., Kusunoki S., Kanazawa I., Kobata A., and Endo T. (1997) Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve α-dystroglycan: the role of a novel O-mannosyl-type oligosaccharide in the binding of α-dystroglycan with laminin. J. Biol. Chem. 272, 2156–2162 [DOI] [PubMed] [Google Scholar]
- 4. Inamori K., Endo T., Ide Y., Fujii S., Gu J., Honke K., and Taniguchi N. (2003) Molecular cloning and characterization of human GnT-IX, a novel β1,6-N-acetylglucosaminyltransferase that is specifically expressed in the brain. J. Biol. Chem. 278, 43102–43109 [DOI] [PubMed] [Google Scholar]
- 5. Kaneko M., Alvarez-Manilla G., Kamar M., Lee I., Lee J. K., Troupe K., Zhang Wj., Osawa M., and Pierce M. (2003) A novel β(1,6)-N-acetylglucosaminyltransferase V (GnT-VB)(1). FEBS Lett. 554, 515–519 [DOI] [PubMed] [Google Scholar]
- 6. Inamori K., Endo T., Gu J., Matsuo I., Ito Y., Fujii S., Iwasaki H., Narimatsu H., Miyoshi E., Honke K., and Taniguchi N. (2004) N-Acetylglucosaminyltransferase IX acts on the GlcNAc β1,2-Man α1-Ser/Thr moiety, forming a 2,6-branched structure in brain O-mannosyl glycan. J. Biol. Chem. 279, 2337–2340 [DOI] [PubMed] [Google Scholar]
- 7. Yoshida-Moriguchi T., Yu L., Stalnaker S. H., Davis S., Kunz S., Madson M., Oldstone M. B., Schachter H., Wells L., and Campbell K. P. (2010) O-Mannosyl phosphorylation of α-dystroglycan is required for laminin binding. Science 327, 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Manya H., Chiba A., Yoshida A., Wang X., Chiba Y., Jigami Y., Margolis R. U., and Endo T. (2004) Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc. Natl. Acad. Sci. U.S.A. 101, 500–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Inamori K., Yoshida-Moriguchi T., Hara Y., Anderson M. E., Yu L., and Campbell K. P. (2012) Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoshida-Moriguchi T., Willer T., Anderson M. E., Venzke D., Whyte T., Muntoni F., Lee H., Nelson S. F., Yu L., and Campbell K. P. (2013) SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 341, 896–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Praissman J. L., Live D. H., Wang S., Ramiah A., Chinoy Z. S., Boons G. J., Moremen K. W., and Wells L. (2014) B4GAT1 is the priming enzyme for the LARGE-dependent functional glycosylation of α-dystroglycan. elife 3, e03943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Willer T., Inamori K., Venzke D., Harvey C., Morgensen G., Hara Y., Beltran Valero de Bernabe D., Yu L., Wright K. M., and Campbell K. P. (2014) The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated α-dystroglycan functional glycosylation. elife 3, e03941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanagawa M., Kobayashi K., Tajiri M., Manya H., Kuga A., Yamaguchi Y., Akasaka-Manya K., Furukawa J., Mizuno M., Kawakami H., Shinohara Y., Wada Y., Endo T., and Toda T. (2016) Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Rep. 14, 2209–2223 [DOI] [PubMed] [Google Scholar]
- 14. Gerin I., Ury B., Breloy I., Bouchet-Seraphin C., Bolsée J., Halbout M., Graff J., Vertommen D., Muccioli G. G., Seta N., Cuisset J. M., Dabaj I., Quijano-Roy S., Grahn A., Van Schaftingen E., and Bommer G. T. (2016) ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto α-dystroglycan. Nat. Commun. 7, 11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Praissman J. L., Willer T., Sheikh M. O., Toi A., Chitayat D., Lin Y. Y., Lee H., Stalnaker S. H., Wang S., Prabhakar P. K., Nelson S. F., Stemple D. L., Moore S. A., Moremen K. W., Campbell K. P., and Wells L. (2016) The functional O-mannose glycan on α-dystroglycan contains a phospho-ribitol primed for matriglycan addition. elife 5, e14473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vuillaumier-Barrot S., Bouchet-Séraphin C., Chelbi M., Devisme L., Quentin S., Gazal S., Laquerrière A., Fallet-Bianco C., Loget P., Odent S., Carles D., Bazin A., Aziza J., Clemenson A., Guimiot F., et al. (2012) Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am. J. Hum. Genet. 91, 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jae L. T., Raaben M., Riemersma M., van Beusekom E., Blomen V. A., Velds A., Kerkhoven R. M., Carette J. E., Topaloglu H., Meinecke P., Wessels M. W., Lefeber D. J., Whelan S. P., van Bokhoven H., and Brummelkamp T. R. (2013) Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science 340, 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Astrea G., Pezzini I., Picillo E., Pasquariello R., Moro F., Ergoli M., D'Ambrosio P., D'Amico A., Politano L., and Santorelli F. M. (2016) TMEM5-associated dystroglycanopathy presenting with CMD and mild limb-girdle muscle involvement. Neuromuscul. Disord. 26, 459–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ohtsuka Y., Kanagawa M., Yu C. C., Ito C., Chiyo T., Kobayashi K., Okada T., Takeda S., and Toda T. (2015) Fukutin is prerequisite to ameliorate muscular dystrophic phenotype by myofiber-selective LARGE expression. Sci. Rep. 5, 8316. [DOI] [PMC free article] [PubMed] [Google Scholar]