

Abstract
The mechanism of LDL receptor (LDLR) degradation mediated by the proprotein convertase subtilisin/kexin type 9 (PCSK9) has been extensively studied; however, many steps within this process remain unclear and still require characterization. Recent studies have shown that PCSK9 lacking its Cys/His-rich domain can still promote LDLR internalization, but the complex does not reach the lysosome suggesting the presence of an additional interaction partner(s). In this study we carried out an unbiased screening approach to identify PCSK9-interacting proteins in the HepG2 cells' secretome using co-immunoprecipitation combined with mass spectrometry analyses. Several interacting proteins were identified, including glypican-3 (GPC3), phospholipid transfer protein, matrilin-3, tissue factor pathway inhibitor, fibrinogen-like 1, and plasminogen activator inhibitor-1. We then validated these interactions by co-immunoprecipitation and Western blotting. Furthermore, functional validation was examined by silencing each candidate protein in HepG2 cells using short hairpin RNAs to determine their effect on LDL uptake and LDLR levels. Only GPC3 and phospholipid transfer protein silencing in HepG2 cells significantly increased LDL uptake in these cells and displayed higher total LDLR protein levels compared with control cells. Moreover, our study provides the first evidence that GPC3 can modulate the PCSK9 extracellular activity as a competitive binding partner to the LDLR in HepG2 cells.
Keywords: lentivirus, low density lipoprotein (LDL), mass spectrometry (MS), proprotein convertase subtilisin/kexin type 9 (PCSK9), short hairpin RNA (shRNA), HepG2 cells, LDL receptor (LDLR), glypican-3, knockdown, secretome
Introduction
Hypercholesterolemia, characterized by elevated plasma levels of low density lipoprotein (LDL)-cholesterol (LDL-C),3 is the major risk factor for atherosclerosis and cardiovascular diseases. LDL particles, which carry most of body cholesterol, are eliminated from the circulation mainly via their binding to the liver hepatocyte cell-surface LDL receptors (LDLR). This interaction occurs between the apolipoprotein B-100 (apoB-100) on LDL particles and the LDLR. Mutations in both LDLR or ApoB genes are associated with familial hypercholesterolemia (FH), an autosomal dominant genetic disorder (1). In 2003, the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene (2) was the third locus validated to be associated with FH (1) and the last member of the proprotein convertase family identified (2).
PCSK9 is mostly expressed and secreted into the plasma from the liver and is also found at lower levels in small intestine and kidneys (2, 3). It is synthesized in the endoplasmic reticulum (ER) as a zymogen pro-PCSK9 (75 kDa) where it must undergo an autocatalytic cleavage of its prodomain (13 kDa) to exit this compartment and be secreted in its mature form (62 kDa). Mature PCSK9 remains non-covalently associated with its cleaved prodomain (2, 4), and this tight complex is secreted as a catalytically inactive serine protease (5). The role of PCSK9 in cholesterol metabolism derived from its ability to bind the EGF-A domain of the LDLR (6) either intracellularly or extracellularly (7), and the PCSK9-LDLR complex is then sorted to the endosome/lysosome pathway for degradation (8). This action reduces LDLR protein levels at the cell surface and thus decreases the elimination of circulating LDL-C. Therefore, PCSK9 promotes LDLR degradation, and PCSK9 inactivation in mice has shown beneficial effects in lowering total cholesterol levels by as much as 42% in the PCSK9 knock-out mouse model (3, 9) or up to 27% in liver-specific PCSK9 knock-out mice (3). Inactivating monoclonal antibodies are now prescribed in lipid clinics worldwide as it was shown that reduction of plasma PCSK9 levels result in ∼60% lowering of plasma LDL-C (10).
The cause and effect of PCSK9 on LDL-C levels has been well described, but the underlying mechanism(s) by which PCSK9 directs cell-surface LDLR to late endosome/lysosome degradation lacks details. Previous studies demonstrated that the deletion (11) and/or substitution (12, 13) of LDLR's cytosolic tail does not prevent PCSK9's ability to mediate LDLR degradation. Moreover, the autosomal recessive hypercholesterolemia adaptor protein, responsible for LDLR internalization through the clathrin-dependent mechanism, is also not absolutely necessary for LDLR degradation mediated by PCSK9 (14). It was also hypothesized that the membrane-bound PCSK9-LDLR complex is first shed into a soluble form upon entry into acidic endosomes via a cathepsin-like cysteine protease inhibited by E64, which may facilitate its efficacious degradation in lysosomes (15). Taken together, we can infer the presence of additional membrane-bound or secreted cellular proteins involved in this mechanism or the presence of another internalization pathway requiring binding partners that remain unidentified.
Additionally, deletion of PCSK9's Cys/His-rich domain (CHRD) does not affect its capacity to bind and internalize LDLR, but its presence is required to direct the complex to lysosomes and disrupt its recycling (5, 16–20). It was proposed that in mildly acidic conditions, the positively charged His-rich PCSK9 CHRD is required to keep the PCSK9 bound to the LDLR ligand-binding domain to reroute the complex to lysosomes (21). However, it is also possible that other proteins could interact with the PCSK9 CHRD and act as a co-receptor for LDLR trafficking. Several studies have demonstrated that such interaction occurs extracellularly with the extrahepatic protein annexin A2 (AnxA2) acting as an endogenous inhibitor of PCSK9 via its ability to bind the CHRD and thus prevent its interaction with the LDLR (22–24).
Although many studies have focused on the identification of new proteins involved in the PCSK9-LDLR degradation pathway, such as AnxA2, LRP1, APLP2, apoB-100, sec24a, GRP94, etc. (13, 22–27), several questions need to be addressed, including the mechanism leading the cell-surface PCSK9-LDLR complex to lysosomal degradation. Herein, we sought to identify novel extracellular interaction partners that could participate and regulate the PCSK9-LDLR complex formation and hence LDLR degradation.
To identify these binding partners we performed co-immunoprecipitation (co-IP) of PCSK9 combined with a mass spectrometry (MS) analysis on the HepG2 cells' secretome. Our studies identified the novel intracellular and extracellular GPC3 interaction with PCSK9 in human hepatocellular carcinoma (HCC) cell lines HepG2 and Huh7. More importantly, we demonstrated that this interaction reduces the PCSK9 extracellular activity on LDLR degradation.
Results
Proteomic Analysis from PCSK9 Immunoprecipitation
To probe for new interaction partners of PCSK9, we performed an immunoprecipitation of PCSK9 (PCSK9-IP) on conditioned media produced from HepG2 cells stably overexpressing V5-tagged PCSK9 (HepG2 pIR/hPCSK9-V5) (27) followed by UHPLC-MS/MS analysis (see under “Experimental Procedures”). This stable cell line expresses ∼8-fold more PCSK9 mRNA in comparison with a control cell line stably transfected with an empty vector pIR (HepG2 pIR) (Fig. 1A). Western blotting on total cell lysate and conditioned media confirms the expression and secretion of hPCSK9-V5 from HepG2 pIR/hPCSK9-V5 cells (Fig. 1B). As predicted, the zymogen pro-hPCSK9-V5 form is observed at ∼75 kDa and the autocatalytically processed mature form (hPCSK9-V5) at ∼65 kDa in the cell lysate, whereas the mature form is only seen in the conditioned media (Fig. 1B), as reported previously (2, 4). Before proceeding to mass spectrometry analysis, we first validated the efficacy of PCSK9-IP on HepG2 pIR/hPCSK9-V5 conditioned media to pull down potential binding partners. A 6.4-fold enrichment ratio was obtained on Western blotting using anti-V5 for PCSK9-IP compared with normal mouse IgG antibody (Fig. 2, left panel), whereas the V5-tagged PCSK9 was not detectable in the conditioned media of control empty vector HepG2 pIR cells as expected (Fig. 2, right panel). Based on the PCSK9 enrichment observed by Western blotting, we prepared four independent immunoprecipitations for proteomic analysis by UHPLC-MS/MS. A total of 42 secreted proteins were identified across all samples (Fig. 3) with enrichment ratios ranging from 1.5- to 7.3-fold, including a 3.0-fold enrichment ratio for PCSK9 (Fig. 3). Twenty one proteins were identified exclusively (>10-fold) in PCSK9-IP using anti-V5 and were not detected in the control IP (Fig. 3). From the list of potential candidates, we narrowed our focus on secreted proteins linked to cholesterol metabolism. Also, we sought to determine whether these proteins were described as potential or validated interactors of an LDLR family member or whether they have been associated with LDL-C levels based on the literature. Six proteins, including GPC3 (7.3-fold), PLTP (>10-fold), MATN3 (6.5-fold), TFPI (5.0-fold), FGL1 (2.5-fold), and PAI-1 (>10-fold), were thus selected.
FIGURE 1.

Stable overexpression of hPCSK9-V5 in HepG2 cells. A, PCSK9 mRNA expression levels from QPCR analysis on RNA extracts from HepG2 cells stably expressing an empty construct pIR and pIR/hPCSK9-V5. Results are the average of four independent experiments relative to HepG2 control pIR with the standard deviation as error bars. Results are normalized using human actin housekeeping gene. *, p < 0.05. B, Western blot (WB) of total cell lysate (25 μg of protein) and conditioned cell media from stable HepG2 empty pIR and pIR/hPCSK9-V5 cells were resolved on SDS-PAGE, blotted onto a nitrocellulose membrane, and incubated with primary mouse anti-V5.
FIGURE 2.

PCSK9 immunoprecipitation from HepG2 cell media. Concentrated conditioned media from HepG2 empty pIR and pIR/hPCSK9-V5 cells (100 μg of protein) were incubated and precipitated overnight with protein G magnetic beads cross-linked with either mouse normal IgG or mouse anti-V5. Immunoprecipitates were washed and separated on SDS-PAGE and probed by Western blotting (WB) with anti-V5. Input lane corresponds to 10% of the total amount of proteins used for each immunoprecipitation (IP).
FIGURE 3.

Mass spectrometry analysis of immunoprecipitated hPCSK9-V5 from HepG2 cells' secretome. List of secreted proteins identified by MS analysis from HepG2 cell secretome stably overexpressing V5-tagged hPCSK9. Unused ProtScore (protein confidence) across all samples was used to calculate the enrichment fold ratio from 1- to 10-fold (ranked as PCSK9-IP anti-V5/PCSK9-IP normal anti-IgG).
Validation of Protein-Protein Interaction by Co-immunoprecipitation
To validate the results obtained by mass spectrometry for the six selected proteins, we co-immunoprecipitated V5-tagged PCSK9 from HepG2 pIR/hPCSK9-V5 conditioned media (Fig. 4A) and assessed the presence of each selected protein by individual Western blottings. All six selected proteins, GPC3, PLTP, MATN3, TFPI, FGL1, and PAI-1, were successfully pulled down from PCSK9-IP. The enrichment ratios (V5/IgG) obtained were >10-fold for GPC3 and 5.8-fold for PLTP (Fig. 4A). We obtained a 1.4-fold enrichment ratio, 3.2- and 2.7-fold for TFPI, FGL1, and PAI-1, respectively (Fig. 4A). However, we could not confirm the specific pulldown of MATN3 from the PCSK9 co-immunoprecipitation because the background signal of MATN3 was equivalent in the negative control to the V5-IP sample (Fig. 4A). We further validated the protein-protein interaction between PCSK9 and selected these proteins by performing the immunoprecipitation of their endogenous levels to confirm the pulldown of PCSK9 from HepG2 pIR/hPCSK9-V5 cell media (Fig. 4B). PCSK9 was increased by ∼4-fold from co-IP of endogenous GPC3, which confirmed GPC3-PCSK9 interaction extracellularly in HepG2 cells. However, we could not demonstrate the reverse immunoprecipitation for PLTP (Fig. 4B), MATN3, TFPI, FGL1, and PAI-1 in HepG2 cells based on their endogenous levels because PCSK9 could not be detected in their co-IP. These results could be explained by the low performance of the antibodies to immunoprecipitate endogenous levels of the proteins. We therefore concentrated mainly on GPC3. To confirm that GPC3-PCSK9 interaction was not because of non-specific binding to the V5 tag on hPCSK9, we performed the same co-immunoprecipitation of endogenously secreted GPC3 from HepG2 cells transiently overexpressing hPCSK9 (no V5 tag). As expected, GPC3-IP pulled down hPCSK9 by >10-fold suggesting that GPC3-PCSK9 interaction is specific (Fig. 4C). Then, we investigated whether this extracellular GPC3-PCSK9 interaction occurred with PCSK9 CHRD or with the catalytic domain/prodomain of PCSK9. We applied the same approach as described in Fig. 4C using conditioned media from HepG2 cells transiently transfected with hPCSK9ΔCHRD-V5 or CHRD-V5. The expression and secretion of both constructs (∼30–35 kDa) were confirmed by Western blotting (Fig. 5, 1st and 4th lanes), as described previously (19, 24). Only hPCSK9ΔCHRD-V5 co-immunoprecipitated with GPC3 by a 4.0-fold enrichment ratio compared with normal IgG (Fig. 5, 2nd and 3rd lanes). GPC3-IP did not pulldown CHRD-V5 suggesting that PCSK9 CHRD is not implicated in the interaction with extracellular GPC3, which occurs with either the prodomain or the catalytic domain of PCSK9 or both.
FIGURE 4.
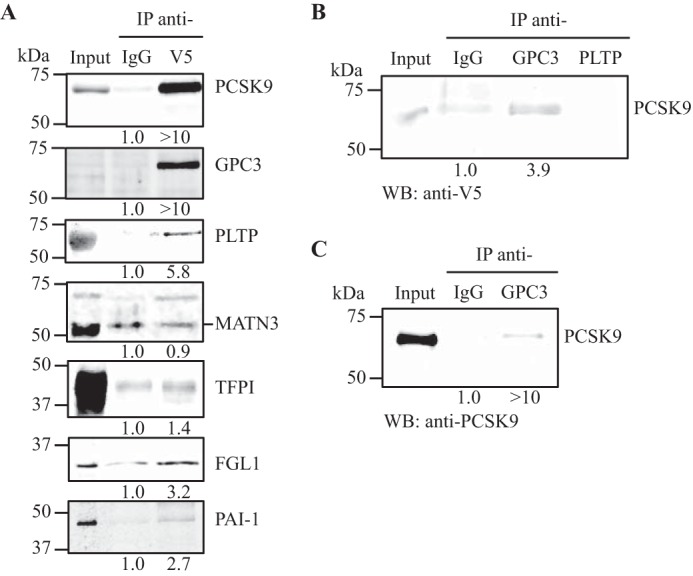
Co-immunoprecipitation of PCSK9 with six selected binding partners. A, PCSK9-V5 was immunoprecipitated (IP) as described in Fig. 2 and analyzed by Western blotting (WB) with anti-V5 as a positive control, anti-GPC3, anti-PLTP, anti-MATN3, anti-TFPI, anti-FGL1, and anti-PAI-1. B, GPC3 and PLTP were immunoprecipitated from HepG2 pIR/hPCSK9-V5 cell-conditioned media (100 μg of protein) using anti-GPC3 mouse monoclonal antibody or anti-PLTP, and the presence of PCSK9 in the immunoprecipitates was assessed with a V5 antibody. C, GPC3 was immunoprecipitated from conditioned media of HepG2 cells overexpressing hPCSK9 (no V5 tag), analyzed, and probed by Western blotting for PCSK9 using rabbit anti-PCSK9. Input lane corresponds to 10% of the total amount of conditioned media used for each immunoprecipitation. Results are representative of three independent experiments.
FIGURE 5.
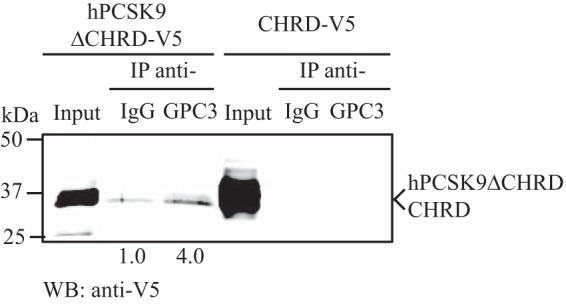
Extracellular GPC3 interaction with PCSK9ΔCHRD. Conditioned media from HepG2 cells overexpressing hPCSK9ΔCHRD-V5 (1st to 3rd lanes) or CHRD-V5 (4th to 6th lanes) were collected to perform GPC3 immunoprecipitation (IP) as described in Figs. 2 and 4. Immunoprecipitates were resolved on SDS-PAGE and probed by Western blotting (WB) with a V5 antibody. Results are representative of three independent experiments.
Intracellular GPC3 Binds to PCSK9-LDLR Complex in Huh7 and HepG2 Cells
Because our objective was to identify novel partners involved in the regulation of the PCSK9-LDLR complex, we investigated further whether intracellular GPC3 was involved in this binding in the Huh7 HCC cell line. Thus, we co-expressed HA-tagged GPC3, V5-tagged PCSK9 WT, and V5-tagged LDLR in Huh7 cells (Fig. 6) and performed the co-IP of HA-GPC3 on total cell lysates to assess its possible interaction with PCSK9 and/or LDLR by Western blotting. Co-expression of GPC3 with PCSK9 resulted in the pulldown of pro-PCSK9 (Fig. 6C, 5th lane), whereas co-expression with PCSK9 and the LDLR led to the pulldown of immature LDLR and pro-PCSK9 in Huh7 cells (Fig. 6C, 8th lane). Because the precursor form of LDLR (95 kDa or non O-glycosylated) is the only form to be pulled down with pro-PCSK9, we suggest that unprocessed GPC3 can bind the PCSK9-LDLR complex very early in the ER (8) before the maturation of PCSK9 and the PC cleavage of GPC3. As expected (28), the co-expression of PCSK9 WT and LDLR resulted in a reduction of 90% of total LDLR protein levels in Huh7 cells (Fig. 6A, 6th lane). No significant difference in total LDLR protein levels were observed when co-expressing GPC3 with LDLR and PCSK9 (Fig. 6A, 8th lane). We repeated this experiment in HepG2 cells which gave very similar results (data not shown). Surprisingly, co-expression of GPC3 and PCSK9 WT in Huh7 and HepG2 cells showed a reduction of total GPC3 levels (GPC3 and secreted GPC3 N-terminal) by 50 and 80% in Huh7 (Fig. 6B, 5th lane) and HepG2 cells, respectively. The same extent of reduction was observed when co-expressed with LDLR in both cell lines (Fig. 6, B and D, 8th lane). Together, these data provide evidence that PCSK9 could be involved in the degradation of GPC3 in an LDLR-independent mechanism. Altogether, our data show that secreted GPC3 interacts with mature PCSK9 in the media of HepG2 cells and with pro-PCSK9 and immature LDLR intracellularly in Huh7 cells.
FIGURE 6.
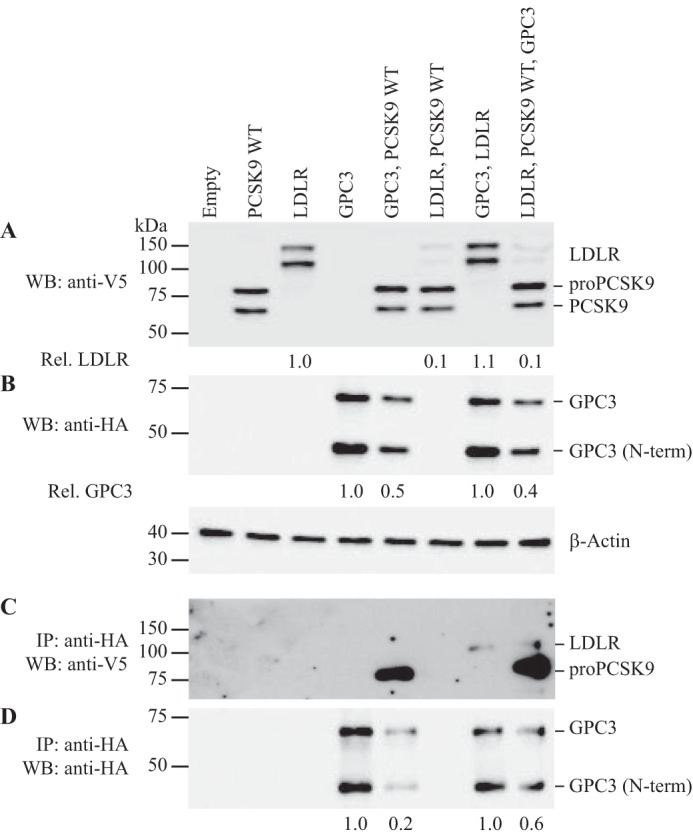
Co-immunoprecipitation of intracellular GPC3 with PCSK9 and LDLR in Huh7 cells. Western blotting (WB) of cell lysates (25 μg) from Huh7 cells overexpressing PCSK9 WT-V5, LDLR-V5, or GPC3-HA. Intracellular protein expression levels from each co-expression conditions were confirmed from 1st to 8th lanes using an anti-V5 (A) and anti-HA (B). GPC3 was immunoprecipitated (IP: anti-HA), and the co-immunoprecipitation of PCSK9 and LDLR was confirmed using an anti-V5 (C, 5th, 7th, and 8th lanes). GPC3 protein levels from the pulldown was analyzed by Western blotting using an anti-HA in each cell lysate (D). Cell lysate β-actin levels were used for normalization. Results are representative of three independent experiments.
Down-regulation of GPC3 and PLTP Exhibits LDL Uptake Phenotype
The extent of the results from mass spectrometry analysis and co-IP experiments is based on HepG2 or Huh7 cells overexpressing PCSK9. We therefore decided to examine the functional significance of interactions with PCSK9. Hence, we used a second approach to study the impact of the six binding candidates on LDL uptake in HepG2 cells by down-regulating their respective expression levels. We have previously assessed the role of AnxA2 and PCSK9 in LDL-cholesterol uptake in Huh7 and HepG2 cells by generating stable knockdown using lentivirus delivery of shRNAs (24). Herein, we generated the stable mRNA knockdown of GPC3, PLTP, MATN3, TFPI, FGL1, and PAI-1 in HepG2 cells using the same strategy in comparison with a stable HepG2 cell line expressing a nonspecific shRNA non-target (shNT) as a control (Table 1). QPCR results confirmed the mRNA reduction, relative to HepG2 shNT cells, of GPC3, PLTP, and MATN3 by 98 ± 1% (means ± S.D.), 93 ± 1, and 85 ± 6%, respectively, and that the mRNA expression of TFPI, FGL1, and PAI-1 were reduced by 92 ± 6, 82 ± 2, and 79 ± 8%, respectively (Fig. 7).
TABLE 1.
List of shRNA TRC numbers for each targeted gene in HepG2 cells
| Genes | TRC no. | TRC version |
|---|---|---|
| GPC3 | TRCN0000315861 | 2 |
| PLTP | TRCN0000150129 | 1 |
| MATN3 | TRCN0000083478 | 1 |
| FGL1 | TRCN0000157414 | 1 |
| PAI1 | TRCN0000331004 | 2 |
| TFPI | TRCN0000073585 | 1 |
FIGURE 7.
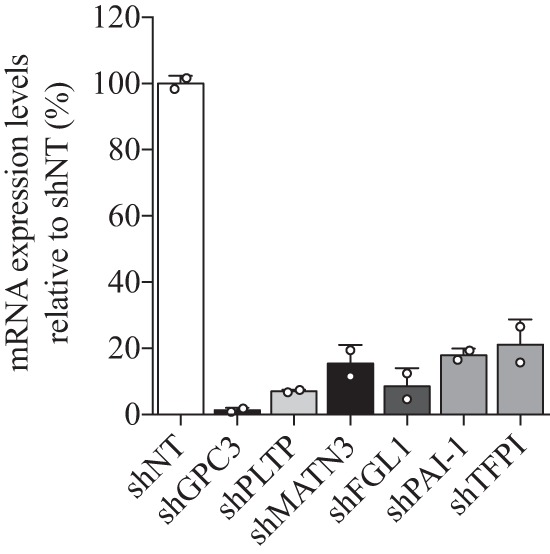
mRNA expression levels from stable knockdown HepG2 cells. QPCR analysis on RNA extracts from HepG2 cells expressing a nonspecific shRNA control (shNon-target) or an shRNA targeting GPC3, PLTP, MATN3, FGL1, PAI1, and TFPI. Results are the average of two independent experiments relative to HepG2 control shNon-target with the standard deviation as error bars. Results are normalized using the human actin housekeeping gene.
To evaluate the function of these proteins on LDL cholesterol uptake in HepG2 cells, we used each stable cell line in a cell-based assay measuring fluorescent 1,1′-dioctadecyl-3,3,3′-3′-tetramethylindocarbocyanine perchlorate (DiI)-labeled LDL (DiI-LDL) uptake. The incorporation of DiI-LDL in stable GPC3 knockdown (shGPC3) cell line was increased by 98 ± 15% (mean ± S.D.) compared with the stable HepG2 shNT cells (Fig. 8). The same effect was observed in reduction of PLTP (shPLTP) mRNA with a 67 ± 16% increase in DiI-LDL uptake, although no significant changes in DiI-LDL uptake were observed in stable MATN3, TFPI, FGL1, and PAI-1 knockdown cell lines (Fig. 8). Thus, the down-regulation of GPC3 and PLTP has a significant impact on basal DiI-LDL uptake in the HepG2 cell model. In contrast, stable knockdown of MATN3, TFPI, FGL1, and PAI-1 did not show any significant difference in DiI-LDL uptake. It is possible that these proteins interact with PCSK9 but that this interaction does not significantly modify LDL-C uptake or the degradation of LDLR mediated by PCSK9.
FIGURE 8.
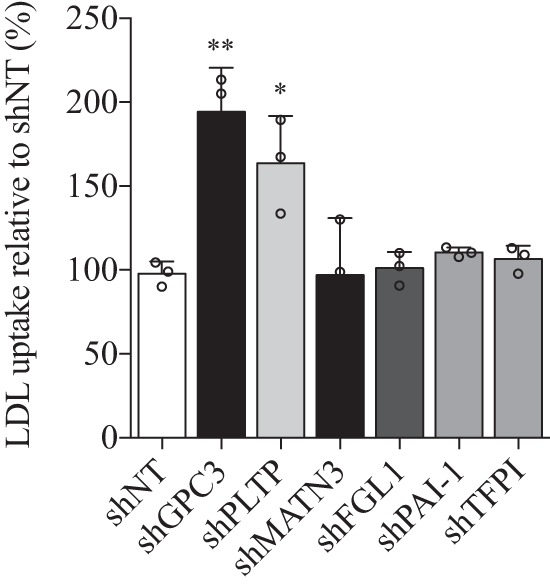
Differences in basal DiI-LDL uptake in stable knockdown HepG2 cells. Each stable knockdown cell line was grown for 24 h followed by serum deprivation for 24 h and incubated with DiI-LDL for 4 h. Raw fluorescence units were normalized by cell numbers per well using CyQUANT® fluorescence units in relation to control shNon-target HepG2 cells. Results are the average of three independent experiments with the standard deviation as error bars. *, p < 0.05; **, p < 0.01.
LDLR mRNA and Protein Levels upon Stable GPC3 and PLTP Knockdown Cell Lines
We further investigated whether higher DiI-LDL uptake observed in the absence of GPC3 or PLTP could be explained by differences in mRNA expression levels or protein levels. QPCR analysis from GPC3 knockdown in HepG2 cells showed no significant increase of LDLR mRNA levels (Fig. 9A), although PLTP knockdown resulted in an increase of LDLR mRNA by only 27% (Fig. 9A), which could explain in part the higher DiI-LDL uptake. However, Western blotting analysis confirmed the significant increase in total LDLR levels by 1.8- and 1.9-fold in GPC3 and PLTP knockdown cell lines, respectively (Fig. 9B), supporting the functional increase in DiI-LDL uptake (Fig. 8).
FIGURE 9.
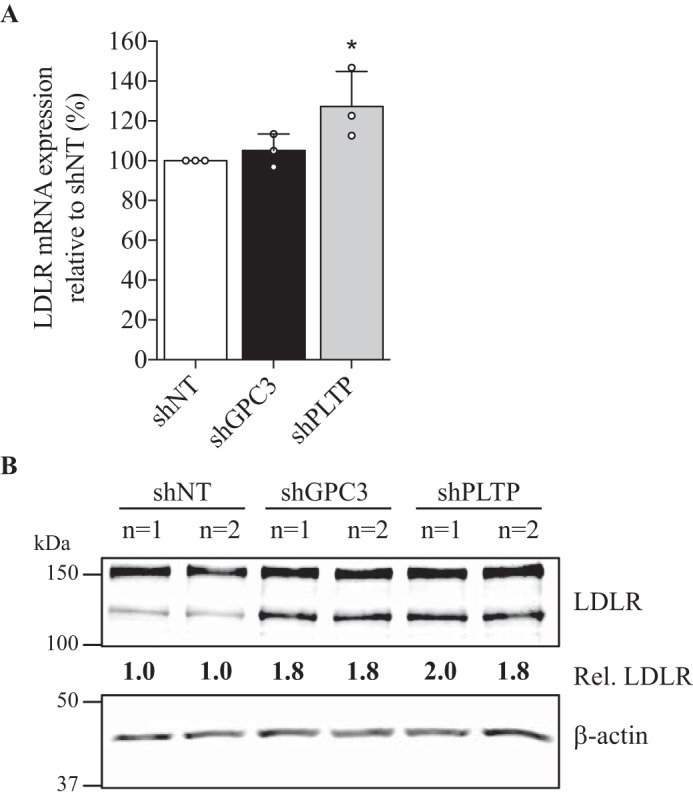
LDLR mRNA expression and total protein levels in stable GPC3 and PLTP knockdown HepG2 cells. A, QPCR analysis of LDLR mRNA in HepG2 shGPC3 and shPLTP stable cell lines relative to shNT cells. Results are shown as three independent experiments with the standard deviation as error bars. Results are normalized using human actin housekeeping gene (*, p < 0.05). B, total cell lysates (25 μg) from stable HepG2 shNT, shGPC3, and shPLTP cells were resolved on SDS-PAGE and subjected to Western blotting using primary goat anti-LDLR and anti-β-actin. Duplicates for each condition are shown, and results are representative of three independent experiments. Protein levels relative to shNT cells were normalized on β-actin levels.
Exogenous hPCSK9 Activity on DiI-LDL Uptake and LDLR Levels in GPC3 and PLTP Knockdown Cell Lines
Because the knockdown of GPC3 and PLTP in HepG2 cells showed higher DiI-LDL uptake (Fig. 8) and LDLR protein levels (Fig. 9B) without major changes in its mRNA levels (Fig. 9A), we hypothesized that these proteins could be implicated in the regulation of PCSK9 extracellular activity. Such direct interaction between extracellular GPC3 or PLTP with PCSK9, demonstrated by co-IP experiments in HepG2 cells (Fig. 4), could have a direct impact on PCSK9 extracellular activity to reduce LDLR levels and DiI-LDL uptake. To verify our premise, we first analyzed DiI-LDL uptake in the presence of exogenous hPCSK9 R218S (hPCSK9 RS), where R218S mutation provides a resistance to furin cleavage/inactivation, on HepG2 shGPC3 and shPLTP cells. The expression, purification, and characterization of recombinant hPCSK9 RS mutant was described previously (24). As expected, DiI-LDL incorporation was initially higher by ∼80% in shGPC3 and shPLTP compared with shNT in the absence of hPCSK9 RS (Fig. 10, A and B), in agreement with basal DiI-LDL uptake measurements in Fig. 8. When hPCSK9 RS was added exogenously from 0.001 to 20 μg/ml (Fig. 10, A and B), no significant difference (p value = 0.2) was observed in the effective concentration values of hPCSK9 RS to reduce DiI-LDL uptake by half (EC50) (Fig. 11) on shNT (1.34 ± 0.14 μg/ml) and shPLTP (1.77 ± 0.36 μg/ml) despite the initial higher uptake. Thus, the reduction of PLTP mRNA yields 1.9-fold higher LDLR protein levels without altering PCSK9 extracellular ability to reduce DiI-LDL uptake. Unlike PLTP, GPC3 knockdown revealed a significant (p value <0.05) 2.9-fold lower EC50 for hPCSK9 RS values (0.46 ± 0.11 μg/ml) relative to shNT cells (Fig. 11). Next, we verified total LDLR protein levels by Western blotting analysis after incubating GPC3 and PLTP knockdown cells with 10 μg/ml hPCSK9 RS. Data showed similar results to those obtained in DiI-LDL uptake because LDLR protein levels in shGPC3 were 30% lower relative to shNT cells (Fig. 12A) corroborating with the better hPCSK9 RS efficiency to reduce DiI-LDL uptake in HepG2 cells (Fig. 11). Western blotting analysis confirmed that LDLR protein levels remained unchanged in HepG2 shPLTP versus shNT cells (Fig. 12B) when incubated with hPCSK9 RS. To validate whether GPC3 can modulate PCSK9 extracellular activity, we performed media swap of hPCSK9 WT on Huh7 cells overexpressing GPC3 and/or LDLR and analyzed LDLR levels. Western blotting analysis showed that extracellular hPCSK9 WT is less efficient for reducing total LDLR levels by 1.6-fold (from 50 to 80%) when GPC3 is overexpressed in Huh7 cells (Fig. 13). This agrees with the results obtained in HepG2 cells where PCSK9 is more efficient in the absence of GPC3. Together, these results suggest that cellular GPC3 reduces exogenous PCSK9's ability to degrade LDLR in HepG2 cells. In that context, we can speculate that GPC3 could compete with the LDLR for binding to PCSK9. Our data also showed that the PLTP effect on DiI-LDL uptake does not come from the modulation of PCSK9 extracellular activity, indicating that this effect could be the result of a yet unknown mechanism.
FIGURE 10.
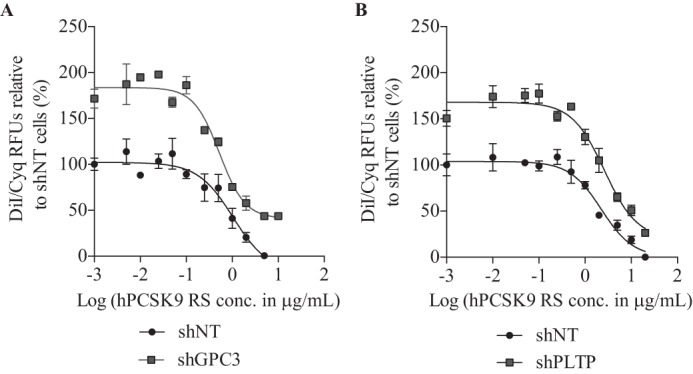
DiI-LDL uptake response curve to hPCSK9 RS in absence of GPC3 and PLTP. Purified human recombinant PCSK9 RS was added to HepG2 cell media at a final concentration of 0.001–20 μg/ml (hPCSK9 RS) followed by the addition of DiI-LDL as described in Fig. 8. DiI-LDL uptake in HepG2 cells shNT versus shGPC3 (A) and shPLTP (B) in the presence of hPCSK9-RS. DiI-LDL incorporation is normalized with CyQUANT® raw fluorescence units (RFU) relative to maximal DiI-LDL incorporation observed in the absence of recombinant hPCSK9 RS. Results are the average of three independent experiments with the standard error of the mean as error bars.
FIGURE 11.
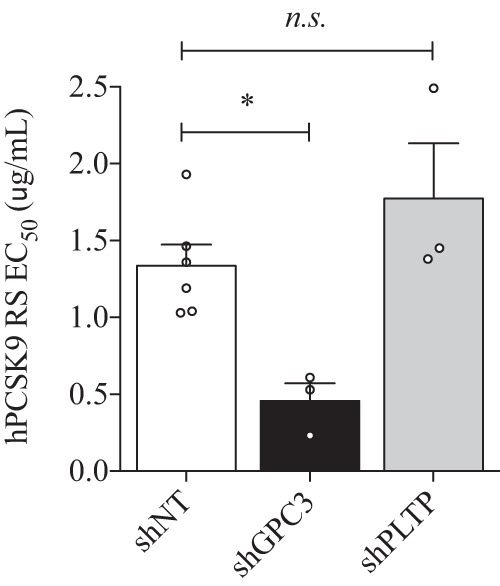
Effective concentration of hPCSK9 RS. Concentrations of hPCSK9 RS necessary to reduce DiI-LDL uptake by 50% in control shNT, shGPC3, and shPLTP are shown in μg/ml. Results are the average of three independent experiments with the standard error of the mean as error bars. *, p < 0.05, and n.s. means non-significant.
FIGURE 12.

GPC3 and PLTP knockdown impact on extracellular hPCSK9 RS activity in HepG2 cells. Total cell lysates (25 μg) from HepG2 shNT and shGPC3 (A) and shPLTP (B) incubated for 6 h with 10 μg/ml hPCSK9 RS (RS) were resolved on SDS-PAGE and analyzed by Western blotting with anti-LDLR and anti-β-actin. Total LDLR protein levels were quantitated, normalized based on β-actin levels, and shown as histograms below a membrane representative of the experiment. LDLR protein levels in each cell line incubated with hPCSK9 RS (RS) are relative to its respective control (Cnt). Results are the average with the standard deviation as error bars from three independent experiments (A) (*, p < 0.05) and two independent experiments (B) (n.s. means non-significant).
FIGURE 13.
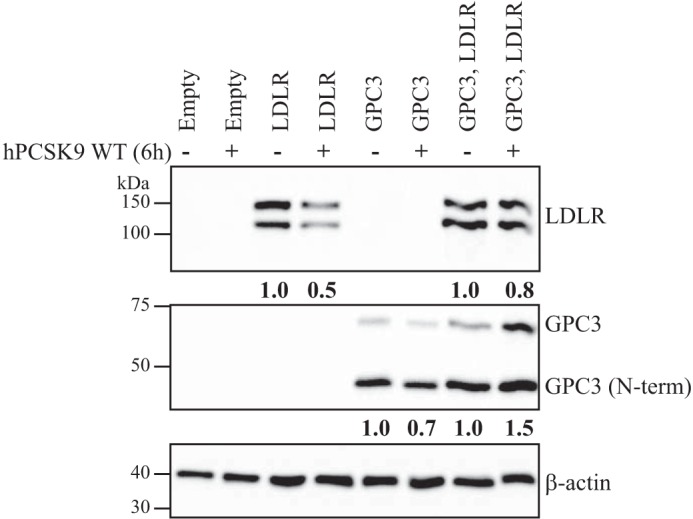
GPC3 impact on extracellular PCSK9 activity in Huh7 cells. Western blotting of total cell lysates (25 μg) from Huh7 cells overexpressing LDLR, GPC3, or both. Huh7 cells were incubated in the presence or in absence of hPCSK9 WT-V5-conditioned media for 6 h. LDLR and GPC3 protein levels were probed, and cell lysate β-actin levels were used for normalization. Quantification is representative of two independent experiments.
Discussion
There is a good case to be made for the presence of additional interaction partners involved in the regulation of LDLR degradation by PCSK9 (5, 10–12, 14, 17, 18). The routing of the PCSK9-LDLR complex from its internalization to its degradation through the lysosomal pathway is still not fully defined. Mass spectrometry analyses have previously been performed to identify novel interaction partners of PCSK9 (22, 29) in different cell lines. Such studies used various mass spectrometry approaches (SILAC-MS, shotgun-MS/MS) to analyze extracts of HEK293 cells (30), subcellular fractions of Huh7 cells (29) stably overexpressing PCSK9, or membrane fractions preparations from mouse livers (13). One study has successfully identified the novel partner AnxA2 by performing immunoprecipitation of PCSK9 in the COS-1 cell lysate (22). It was recently reported that the LDLR does not bind efficiently PCSK9 in the ER because of the presence of a competitive chaperone like-protein GRP94 that prevents such ER interaction (26). Aside from the latter study, no other reports appeared on the identification of strong and stable interaction partners of PCSK9 from the secretome of HepG2 cells by mass spectrometry analysis. Therefore, this study has focused on the identification of novel extracellular interaction partners of PCSK9 from the conditioned media of HepG2 cells. We initially identified 42 secreted proteins (Fig. 3) based on mass spectrometry analysis of PCSK9 immunoprecipitates from the secretome of HepG2 cells. Six proteins were selected from database data linking them to the following: (i) cholesterol homeostasis; (ii) interactions with LDLR family members; or (iii) an association with plasma LDL-C. It has been shown that cell-surface GPC3, TFPI, and PAI-1 can be internalized through an LRP1-dependent mechanism (31–35). TFPI and PLTP have been positively associated with LDL particles in human plasma (36, 37), whereas PLTP has been mostly studied for its important role in HDL metabolism. FGL1 knock-out mice exhibit lower free fatty acid levels and total plasma cholesterol levels (38), and MATN family exhibit epidermal growth factor-like domain, which could possibly interact with PCSK9 such as the one stipulated in an MATN2 EGF-like domain patent (United States patent number 20110150875 A1). Using combined immunoprecipitation and Western blotting analyses (Fig. 4), we could only clearly validate the interaction of endogenous GPC3 with PCSK9.
GPC3 is one of the six members from the glypican family, a subgroup of heparin sulfate proteoglycan family. It is bound to the cell surface of the plasma membrane through a glycosylphosphatidylinositol anchor on its C-terminal domain along with two heparin sulfate side chains (39). In situ hybridization histochemistry revealed that GPC3 mRNA is abundant in many tissues during embryogenesis, including liver primordia (Fig. 14). However, its mRNA expression levels in adult liver are barely detectable (Fig. 14), as reported previously (40–42). Furthermore, GPC3 is overexpressed in 80% of HCCs and correlates with the severity of the tumor (41, 43). The core protein of 70 kDa with the heparan sulfate side chains contains a recognition motif for furin and PC5/6B (44). The cleavage of GPC3 at RQYR↓S359 results in the release of a secreted 40-kDa GPC3 N-terminal domain and a cell membrane-associated fragment of 30 kDa (44). The ∼65-kDa molecular mass of GPC3 observed in the co-IP media with PCSK9 (Fig. 4A) does not correspond to the small N-terminal fragment produced by furin, but it most likely is the shed molecular form of intact GPC3 produced by the C-terminal cleavage of its glycosylphosphatidylinositol anchor by endogenous lipases (45, 46). GPC3 binds the growth factor Wnt presenting it to its receptor frizzled. Therefore, GPC3 acts as a co-receptor at the cell membrane and promotes the downstream Wnt-frizzled signaling pathway (44, 47–49). However, other studies have shown that GPC3 can also negatively regulate another pathway by competing for the binding of hedgehog growth factor to its receptor Ptc (50).
FIGURE 14.
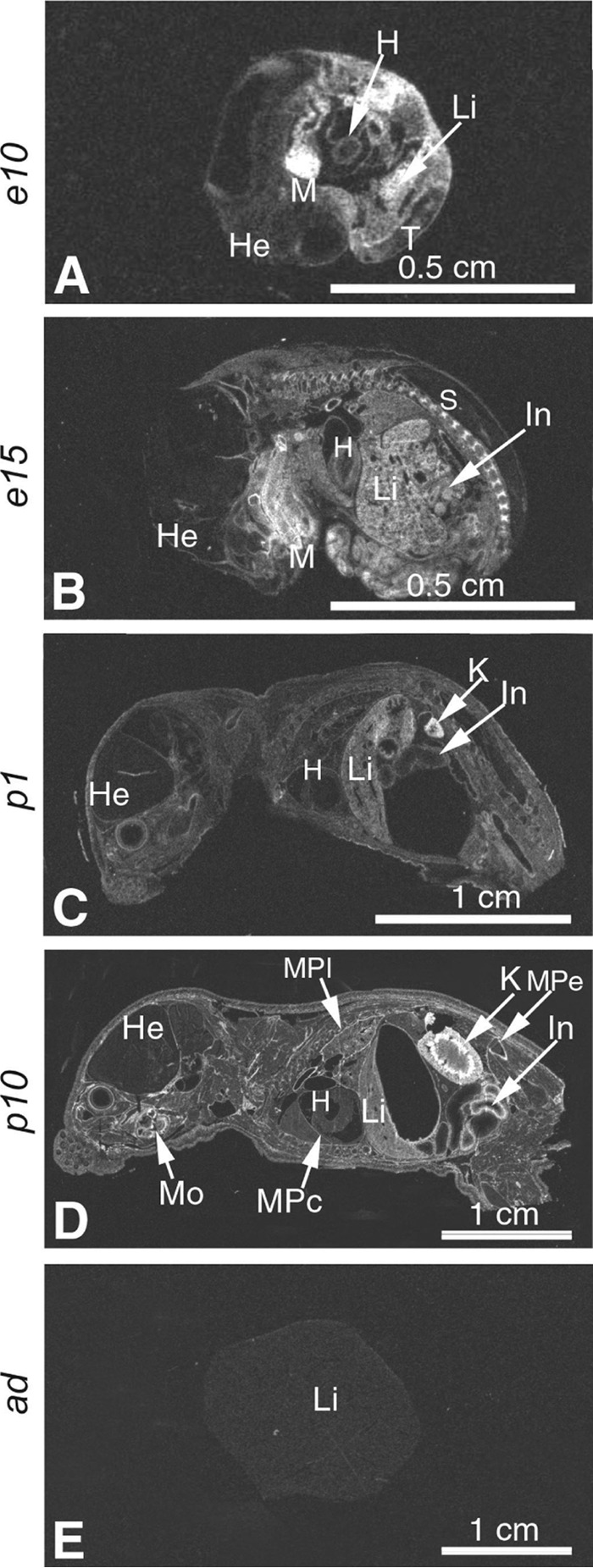
In situ hybridization evidence for developmentally regulated GPC3 expression in the mouse. A and B, mouse embryos at mid-gestation stage. X-ray film autoradiography showing mRNA labeling seen as bright under dark field illumination. High level of GPC3 expression is evident in the mandible mesenchyme. Down-regulated GPC3 expression is evident in the liver from high levels in e10 to moderate in e15 embryos. C and D, postnatal GPC3 expression in the kidney and intestine, up-regulated from low levels in p1 to moderate and high in p10; increasing levels of GPC3 expression in the membranes of the pleura, pericardium, and periosteum. Moderate level of GPC3 expression is noted in the liver of a newborn mouse (in C) and low level in postnatal p10 (in D). E, undetectable GPC3 expression in the adult mouse liver. Abbreviations used are as follows: H, heart; He, head; K, kidney; In, intestine; Li, liver; MPc, membranes of the pericardium; MPe, membranes of the periosteum; MPl, membranes of the pleura; Mo, molar with the membranes. Magnifications: A and B, ×8; C, ×4; D and E, ×2.
Overexpression of GPC3 with PCSK9 and LDLR in Huh7 cells allowed us to demonstrate the interaction of GPC3 with pro-PCSK9 and immature LDLR intracellularly (Fig. 6C), thus likely in the ER as this is the only place where pro-PCSK9 is found (2). These observations suggest that GPC3 can interact with the PCSK9-LDLR complex very early in the ER. The co-IP of GPC3, when co-expressed with PCSK9 in Huh7 cells, did not pull down intracellular mature PCSK9 (Fig. 6C) most likely because of PCSK9's rapid exit from the ER leading to its secretion. Nassoury et al. (8) have described that only the co-expression of PCSK9 fused to the KDEL ER retention sequence could result in the co-immunoprecipitation of immature LDLR with both pro and mature PCSK9 forms.
Surprisingly, the co-expression of GPC3 with PCSK9 resulted in a decrease in total GPC3 protein levels relative to control in Huh7 cells, suggesting that a putative PCSK9-GPC3 complex could be degraded via an intracellular pathway (Fig. 6B) (7). Because our data showed that overexpression of intracellular membrane-bound GPC3 does not significantly affect the overall intracellular ability of PCSK9 to induce the degradation of the LDLR (Fig. 6A) and that co-expression of LDLR did not change the reduction in total GPC3 protein levels (Fig. 6B), we postulate that PCSK9 can degrade GPC3 in HepG2 cells via an LDLR-independent mechanism. The precise mechanism by which overexpressed PCSK9 can induce the cellular degradation of GPC3 will need further studies, but it was shown that LRP1, an LDLR family member, can mediate the endocytosis and degradation of the hedgehog-GPC3 complex at the cell surface by binding to the heparan sulfate chains of GPC3 (32). Thus, it is possible that a putative PCSK9-GPC3-LRP1 could be formed, especially because PCSK9 has already been shown to interact and enhance the degradation of LRP1 in HepG2 cells in an LDLR-independent fashion (13).
To establish a functional role of identified interacting partners on LDL uptake in HepG2 cells, we chose to perform specific mRNA knockdowns (Fig. 7) on all six identified interacting partners, even though they were not all validated in the immunoprecipitation studies (Fig. 4B). Only the mRNA knockdowns of GPC3 and PLTP showed a significant increase of basal DiI-LDL uptake (Fig. 8).
PLTP knockdown in HepG2 cells increased LDLR mRNA expression and total protein levels (Fig. 9). However, the ability of exogenous hPCSK9 R218S to reduce DiI-LDL uptake (Fig. 11) and protein levels (Fig. 12B) did not change in HepG2 shPLTP versus shNT cells. Taken together, these results clearly demonstrate a role of PLTP on LDLR regulation, but its effect does not involve the modulation of PCSK9 extracellular activity. The PLTP effect on LDLR mRNA results most likely from another indirect pathway because no direct transcriptional function has ever been described from this protein. As our aim was to identify extracellular PCSK9-interacting proteins able to modulate PCSK9 activity toward LDLR degradation, we did not further investigate the role of PLTP on LDLR levels in HepG2 cells. However, it may be interesting to characterize more in detail the cellular function of PLTP on LDLR because studies have focused mostly on its role in HDL metabolism, in the production of apoB-containing lipoproteins, in insulin resistance, or in type 2 diabetes mellitus.
We also observed that LDLR protein levels were higher upon GPC3 knockdown (shGPC3 cells), although LDLR mRNA levels did not change (Fig. 9). This suggests that GPC3 has post-transcriptional effects on LDLR levels. This hypothesis was supported in DiI-LDL uptake experiments in HepG2 shGPC3 cells using exogenous hPCSK9 R218S, which was nearly three times more efficient to reduce DiI-LDL uptake (Fig. 11) and also more efficient to reduce total LDLR protein levels (Fig. 12A). Based upon these results, we propose that the extracellular interaction between PCSK9 and GPC3 competes with PCSK9 binding to the cell-surface LDLR (Fig. 15). This was confirmed in media swap experiments on HepG2 cells co-expressing GPC3 and LDLR combined with Western blotting analyses of total LDLR protein levels (Fig. 13). As expected, overexpression of GPC3 with LDLR reduces the hPCSK9 WT capacity to degrade the LDLR by 1.6-fold compared with overexpression of LDLR alone in Huh7 cells (Fig. 13). We conclude that GPC3 is not only a novel extracellular and intracellular interactor of PCSK9 but also a competitive extracellular binding partner to the LDLR in HepG2 and Huh7 cell models (Fig. 15). Our study also confirmed that this extracellular binding does not involve PCSK9 CHRD similar to the endogenous inhibitor AnxA2 previously described (19, 24). Based on our results, GPC3 interacts with the prodomain and/or catalytic domain of PCSK9 (Fig. 5). It is possible that GPC3 competes with the LDLR to bind the catalytic domain of PCSK9. Further studies will be necessary to validate the precise region or residues implicated in this protein-protein interaction.
FIGURE 15.

PCSK9 and GPC3 interaction model. Illustration of PCSK9 extracellular activity on LDLR degradation in the absence (A) or presence (B) of intracellular and extracellular GPC3 in HepG2 and Huh7 cells. A, cell-surface LDLR interaction with PCSK9 leads to its internalization and its degradation via the endosomal/lysosomal pathway. This action results in lower LDLR at the cell surface with an accumulation of circulating LDL particles. B, early interaction of pro-PCSK9-GPC3 in the ER is illustrated based on co-immunoprecipitation results presented in Fig. 6. The impact of this intracellular interaction on pro-PCSK9 remains to be determined. We also propose that the presence of either cell membrane or secreted/soluble GPC3 can interact with PCSK9 and prevent its binding to cell-surface LDLR. This competitive binding of GPC3 and PCSK9 to the LDLR reduces PCSK9 extracellular activity resulting in higher LDLR levels and higher LDL uptake.
GPC3 is abundantly expressed in mouse (Fig. 14) and human embryonic liver and in human HCC (40). However, its expression levels in adult human (40, 51) and mouse (Fig. 14) liver is very low, whereas PCSK9 is mainly expressed in human and mouse liver throughout embryogenesis and in the adult. Because our data in Huh7 and HepG2 cells demonstrated that GPC3 can reduce PCSK9 activity to degrade cell-surface LDLR, it is highly plausible that GPC3 might have an important physiological role on the regulation of PCSK9's extracellular activity during embryogenesis, in liver regeneration, or in HCC. Our results also suggest that GPC3 would reduce PCSK9 activity on LDLR degradation (and possibly other PCSK9 targets) during embryogenesis and in certain cancers. It is noteworthy that GPC3 is also very abundant in the small intestine and in the kidney (Fig. 14) where PCSK9 is also significantly expressed (2). Because the role of PCSK9 in the small intestine is not yet fully understood, it is difficult to define the impact of GPC3 on PCSK9 activity in this tissue at this time.
In conclusion, our studies successfully identified a novel and functional interaction between GPC3 and PCSK9. Most importantly, we demonstrate that extracellular GPC3 can act as an endogenous competitive binding partner of PCSK9 to the LDLR in HepG2 and Huh7 cells.
Experimental Procedures
Cell Culture, Transfections, and Western Blotting Analysis
HepG2 and Huh7 cell lines were grown in Eagle's minimum essential medium and DMEM, respectively, with 10% FBS (Wisent), and cells were maintained at 37 °C under 5% CO2. At 70% confluence, the cells were transfected with Effectene (Qiagen) with an empty vector pIR or pIR/hPCSK9-V5 and stably selected with geneticin. Transient transfection of hGPC3-HA, hPLTP-V5, hLDLR-V5, hPCSK9-V5, hPCSK9 (no V5 tag), hPCSK9ΔCHRD-V5 (L455X), and CHRD-V5 were performed as described previously (7–9, 19, 24). Twenty four hours post-transfection, cells were washed and incubated in serum-free medium for an additional 24 h prior Western blotting analysis. Medium was recovered, and the cells were lysed in ice-cold radioimmunoprecipitation assay buffer (RIPA: 50 mm Tris-HCl, pH 7.8, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mm NaCl) containing a mixture of protease inhibitors (Roche Applied Science). For media swap experiments, stable HEK293 cells overexpressing hPCSK9-V5 were plated, and 24 h later, the media were replaced with serum-free media for an overnight incubation. Then, serum-free conditioned media were collected, and PCSK9 levels were measured by ELISA (13, 52) and added onto corresponding cell lines for a 6-h incubation preceding cell lysis.
PCSK9 Immunoprecipitation and Sample Preparation for Mass Spectrometry
HepG2 cells overexpressing an empty vector (pIR) or PCSK9-V5 were plated at 4 × 106 cells in a 100-mm dish and grown for 24 h with 10% FBS. Cells were washed five times with PBS and incubated for 24 h with serum-free media. Conditioned media were collected and concentrated 15-fold on an Amicon 3-kDa cutoff (Millipore). Immunoprecipitation of PCSK9-V5 was carried out overnight at 4 °C on 100 μg of conditioned media supplemented with protease inhibitors using 5 μl of protein G magnetic beads (ReSyn Biosciences) cross-linked with 2.5 μg of mouse anti-V5 (R960–25, Thermo Fisher Scientific). Beads were washed four times with PBS/Tween 0.1% (PBST) to remove unbound proteins. Samples were heated with 25 μl of reducing Laemmli buffer, resolved on SDS-PAGE, and blotted onto nitrocellulose membranes (GE Healthcare). Following 1 h of incubation in 5% BSA in TBS/Tween 0.1% (TBST), the membranes were incubated with mouse anti-V5 overnight at 4 °C. After incubation, membranes were washed four times in TBST and incubated with anti-mouse IRDye 800 (LI-COR) and exposed to near-infrared fluorescence imaging (LI-COR Odyssey). PCSK9 immunoprecipitations were prepared differently for mass spectrometry analysis. After overnight incubation at 4 °C and four washes with PBST, three additional washes with 50 mm Tris-HCl, pH 7.5, were performed and followed with trypsin/Lys-C digestion (Promega) at 37 °C overnight directly on beads in a final volume of 100 μl of 50 mm Tris-HCl, pH 7.5. Supernatant was collected, and trypsin/Lys-C was inactivated using formic acid. Sample pH was neutralized, reduced with 10 mm DTT final concentration for 15 min at 95 °C, and alkylated at 15 mm final concentration for 30 min at room temperature. Then, samples were cleaned on solid reversed phase extraction column (Phenomenex) and eluted with a solution of 75% acetonitrile with 2% formic acid. Finally, samples were dried, resuspended in 5% acetonitrile with 3% DMSO, and analyzed on micro flow UHPLC-MS/MS system (ABSciex TripleTOF 5600, PhenoSwitch Bioscience). Each sample was analyzed under data-independent acquisition mode, and protein identification score was assigned using ProteinPilot software. False-positive proteins were removed, and unused ProtScore (protein confidence) across all samples was used to calculate the enrichment fold ratio (ranked as PCSK9-IP anti-V5/PCSK9-IP normal anti-IgG) and to identify binding candidates.
Co-IP
Co-IP experiments were performed as described earlier using mouse anti-V5 on HepG2 pIR/hPCSK9-V5 conditioned media. PCSK9 immunoprecipitates were probed by Western blotting with primary antibodies mouse anti-GPC3 (MAB2119, R&D Systems), rabbit anti-PLTP (AB85303, Abcam), rabbit anti-MATN3 (NBP1-77022, Novus Biologicals), goat anti-TFPI (AF2974, R&D Systems), goat anti-FGL1 (AF1614, R&D Systems), and rabbit anti-PAI-1 (11909, Cell Signaling Technology). GPC3 and PLTP primary antibodies were used to co-immunoprecipitate endogenous levels on the same conditioned media, and PCSK9-V5 was detected with anti-V5 antibody. Co-IP of GPC3-HA in Huh7 and HepG2 cells were performed using anti-HA and probed for LDLR and PCSK9 using a V5-HRP-conjugated antibody by Western blotting. Additionally, endogenous GPC3 was immunoprecipitated from HepG2 cells transiently overexpressing hPCSK9 (no V5 tag), hPCSK9ΔCHRD-V5 (L455X), or CHRD-V5 using the mouse anti-GPC3. The immunoprecipitates were resolved on SDS-PAGE and probed for PCSK9 by Western blotting using a rabbit anti-PCSK9 (24).
Lentivirus Production
HEK293 cells were used to produce lentiviral particles as described previously (24). The lentiviral vector pLKO.1/puro (Sigma Mission shRNA) containing shRNA specific to hGPC3, hPLTP, hMATN3, hTFPI, hFGL1, and hPAI-1 mRNAs (Table 1) was used in co-transfection with the packaging plasmids pLp1, pLp2, and VSVG (Invitrogen). Lentiviral titer were determined by transducing HT-1080 cells for upcoming infection in HepG2 cells as described before (24).
Stable shRNA Knockdown in HepG2
HepG2 cells were plated, 1 day prior to infection, at 200,000 cells/well in a 6-well plate in complete (10% FBS) Eagle's minimum essential medium. Lentiviral infection was performed as described previously (24) using a multiplicity of infection of 3 in the presence of 4 μg/μl Polybrene over a period of 24 h. Puromycin (2 μg/ml) was used for stable selection 48 h post-infection. RNA isolation was performed using the RNeasy kit (Qiagen), and RT-QPCR was carried out with RNA samples (1 μg) using SyBrGreen and QPCR system Mx3005p (Stratagene). The primers sequences used for QPCR analysis are listed in Table 2. Results are normalized to human actin and are presented as an average ± S.D. of triplicates relative to the control shNT.
TABLE 2.
List of forward and reverse primers for RT-qPCR analysis
| Genes | Forward primer (5′–3′) | Reverse primer (5′–3′) | Product size |
|---|---|---|---|
| bp | |||
| PCSK9 | ATCCACGCTTCCTGCTGC | CACGGTCACCTGCTCCTG | 86 |
| GPC3 | TCATGCAAGGCTGTATGGCA | TGCCAATCTGTAAGTCTAGCCC | 267 |
| PLTP | GGATGGTGTATGTGGCCTTC | CTGCAGCATGGTCTTCAGAG | 473 |
| MATN3 | TAACACCCACGGATGTGAGC | ACACTTGTCACGGACTGAACA | 247 |
| FGL1 | AACCTGAGCTGGGTCTCTGA | CCATTGTCAGAGCGGTGGTA | 103 |
| PAI1 | ACAACCCCACAGGAACAGTC | GATGAAGGCGTCTTTCCCCA | 73 |
| TFPI | AGACAGCAGCGACTTTAGGC | AGCAGATCAAGAAACTGGCGA | 118 |
| LDLR | AGCCGTAAGGACACAGCACAC | GGAAGACGAGGAGCACGATGG | 203 |
| ACTB | GCCTCGCCTTTGCCGATCCG | ACATGCCGGAGCCGTTGTCG | 99 |
DiI-LDL Uptake Cell-based Assay
The experiment was conducted as described previously (24) with HepG2 cells. Purified recombinant hPCSK9 RS was added to the medium at a final concentration of 0.001–20 μg/ml for 4 h followed by the addition of 2 mg/ml DiI-LDL (BT-904, Alfa Aesar) for 4 h at 37 °C. Cells were washed twice with PBS, and DiI-LDL raw fluorescence units were measured with Molecular Geminix XS at excitation/emission of 520/575 nm. Each value is relative to the maximal DiI-LDL incorporation value in the control.
Author Contributions
K. L. conducted most of the experiments, analyzed the results, and wrote the manuscript. R. E. performed the experiment on the co-immunoprecipitation of PCSK9-GPC3-LDLR (Fig. 6) and the effect of GPC3 overexpression on PCSK9 extracellular activity in Huh7 cells (Fig. 13). R. Desjardins helped in the production of stable knockdown cell lines. N. G. S. and R. Day conceived and designed the project and wrote the paper with K. L. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Jessica Fortin-Mimeault for help with cell line preparation and Jadwiga Marcinkiewicz for the in situ hybridization analyses (Fig. 14).
This work was supported in part by Fondation Leducq Grant 13 CVD 03 (to R. D. and N. G. S.), by Canada Research Chair 126684 (to N. G. S.), and in part by a Pfizer ASPIRE CV IIR Grant (to N. G. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- LDL-C
- low density lipoprotein-cholesterol
- CHRD
- Cys/His-rich domain
- GPC3
- glypican-3
- PLTP
- phospholipid transfer protein
- LDLR
- LDL receptor
- FH
- familial hypercholesterolemia
- ER
- endoplasmic reticulum
- AnxA2
- annexin A2
- co-IP
- co-immunoprecipitation
- TFPI
- tissue factor pathway inhibitor
- UHPLC-MS/MS
- micro-flow high performance liquid chromatography coupled with tandem mass spectrometry
- shNT
- shRNA non-target
- DiI-LDL
- 1,1′-dioctadecyl-3,3,3′-3′-tetramethylindocarbocyanine perchlorate-labeled LDL
- shGPC3
- stable GPC3 knockdown
- shPLTP
- stable PLTP knockdown
- hPCSK9 RS
- hPCSK9 R218S
- IP
- immunoprecipitation
- QPCR
- quantitative PCR
- HCC
- human hepatocellular carcinoma.
References
- 1. Abifadel M., Varret M., Rabès J.-P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., et al. (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 2. Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., and Chretien M. (2003) The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., Seidah N. G., and Prat A. (2008) Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 48, 646–654 [DOI] [PubMed] [Google Scholar]
- 4. Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M.-C., Hamelin J., Varret M., Allard D., Trillard M., Abifadel M., Tebon A., Attie A. D., Rader D. J., et al. (2004) NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279, 48865–48875 [DOI] [PubMed] [Google Scholar]
- 5. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., et al. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 6. Kwon H. J., Lagace T. A., McNutt M. C., Horton J. D., and Deisenhofer J. (2008) Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. U.S.A. 105, 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Poirier S., Mayer G., Poupon V., McPherson P. S., Desjardins R., Ly K., Asselin M.-C., Day R., Duclos F. J., Witmer M., Parker R., Prat A., and Seidah N. G. (2009) Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J. Biol. Chem. 284, 28856–28864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nassoury N., Blasiole D. A., Tebon Oler A., Benjannet S., Hamelin J., Poupon V., McPherson P. S., Attie A. D., Prat A., and Seidah N. G. (2007) The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 8, 718–732 [DOI] [PubMed] [Google Scholar]
- 9. Rashid S., Curtis D. E., Garuti R., Anderson N. N., Bashmakov Y., Ho Y. K., Hammer R. E., Moon Y. A., and Horton J. D. (2005) Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc. Natl. Acad. Sci. U.S.A. 102, 5374–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seidah N. G., Awan Z., Chrétien M., and Mbikay M. (2014) PCSK9: a key modulator of cardiovascular health. Circ. Res. 114, 1022–1036 [DOI] [PubMed] [Google Scholar]
- 11. Strøm T. B., Holla Ø. L., Tveten K., Cameron J., Berge K. E., and Leren T. P. (2010) Disrupted recycling of the low density lipoprotein receptor by PCSK9 is not mediated by residues of the cytoplasmic domain. Mol. Genet. Metab. 101, 76–80 [DOI] [PubMed] [Google Scholar]
- 12. Holla Ø. L, Strøm T. B., Cameron J., Berge K. E., and Leren T. P. (2010) A chimeric LDL receptor containing the cytoplasmic domain of the transferrin receptor is degraded by PCSK9. Mol. Genet. Metab. 99, 149–156 [DOI] [PubMed] [Google Scholar]
- 13. Canuel M., Sun X., Asselin M.-C., Paramithiotis E., Prat A., and Seidah N. G. (2013) Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS ONE 8, e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Norman D., Sun X. M., Bourbon M., Knight B. L., Naoumova R. P., and Soutar A. K. (1999) Characterization of a novel cellular defect in patients with phenotypic homozygous familial hypercholesterolemia. J. Clin. Invest. 104, 619–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leren T. P. (2014) Sorting an LDL receptor with bound PCSK9 to intracellular degradation. Atherosclerosis 237, 76–81 [DOI] [PubMed] [Google Scholar]
- 16. Zhang D.-W., Garuti R., Tang W.-J., Cohen J. C., and Hobbs H. H. (2008) Structural requirements for PCSK9-mediated degradation of the low density lipoprotein receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ni Y. G., Condra J. H., Orsatti L., Shen X., Di Marco S., Pandit S., Bottomley M. J., Ruggeri L., Cummings R. T., Cubbon R. M., Santoro J. C., Ehrhardt A., Lewis D., Fisher T. S., Ha S., et al. (2010) A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J. Biol. Chem. 285, 12882–12891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holla Ø. L, Cameron J., Tveten K., Strøm T. B., Berge K. E., Laerdahl J. K., and Leren T. P. (2011) Role of the C-terminal domain of PCSK9 in degradation of the LDL receptors. J. Lipid Res. 52, 1787–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saavedra Y. G., Day R., and Seidah N. G. (2012) The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 protein is needed for the extracellular low density lipoprotein receptor (LDLR) degradation pathway. J. Biol. Chem. 287, 43492–43501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weider E., Susan-Resiga D., Essalmani R., Hamelin J., Asselin M.-C., Ashraf Y., Wycoff K. L., Zhang J., Prat A., and Seidah N. G. (2016) Proprotein convertase subtilisin/Kexin type 9 (PCSK9) single domain antibodies are potent inhibitors of LDL receptor degradation. J. Biol. Chem. 291, 16659–16671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tveten K., Holla Ø. L., Cameron J., Strøm T. B., Berge K. E., Laerdahl J. K., and Leren T. P. (2012) Interaction between the ligand-binding domain of the LDL receptor and the C-terminal domain of PCSK9 is required for PCSK9 to remain bound to the LDL receptor during endosomal acidification. Hum. Mol. Genet. 21, 1402–1409 [DOI] [PubMed] [Google Scholar]
- 22. Mayer G., Poirier S., and Seidah N. G. (2008) Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J. Biol. Chem. 283, 31791–31801 [DOI] [PubMed] [Google Scholar]
- 23. Seidah N. G., Poirier S., Denis M., Parker R., Miao B., Mapelli C., Prat A., Wassef H., Davignon J., Hajjar K. A., and Mayer G. (2012) Annexin A2 is a natural extrahepatic inhibitor of the PCSK9-induced LDL receptor degradation. PLoS ONE 7, e41865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ly K., Saavedra Y. G., Canuel M., Routhier S., Desjardins R., Hamelin J., Mayne J., Lazure C., Seidah N. G., and Day R. (2014) Annexin A2 reduces PCSK9 protein levels via a translational mechanism and interacts with the M1 and M2 domains of PCSK9. J. Biol. Chem. 289, 17732–17746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun H., Samarghandi A., Zhang N., Yao Z., Xiong M., and Teng B.-B. (2012) Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low density lipoprotein receptor. Arterioscler. Thromb. Vasc. Biol. 32, 1585–1595 [DOI] [PubMed] [Google Scholar]
- 26. Poirier S., Mamarbachi M., Chen W.-T., Lee A. S., and Mayer G. (2015) GRP94 regulates circulating cholesterol levels through blockade of PCSK9-induced LDLR degradation. Cell Rep. 13, 2064–2071 [DOI] [PubMed] [Google Scholar]
- 27. Butkinaree C., Canuel M., Essalmani R., Poirier S., Benjannet S., Asselin M.-C., Roubtsova A., Hamelin J., Marcinkiewicz J., Chamberland A., Guillemot J., Mayer G., Sisodia S. S., Jacob Y., Prat A., and Seidah N. G. (2015) Amyloid precursor-like protein 2 and sortilin do not regulate the PCSK9 convertase-mediated low density lipoprotein receptor degradation but interact with each other. J. Biol. Chem. 290, 18609–18620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poirier S., Mayer G., Benjannet S., Bergeron E., Marcinkiewicz J., Nassoury N., Mayer H., Nimpf J., Prat A., and Seidah N. G. (2008) The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 283, 2363–2372 [DOI] [PubMed] [Google Scholar]
- 29. Denis N., Palmer-Smith H., Elisma F., Busuttil A., Wright T. G., Bou Khalil M., Prat A., Seidah N. G., Chrétien M., Mayne J., and Figeys D. (2011) Quantitative proteomic analysis of PCSK9 gain of function in human hepatic HuH7 cells. J. Proteome Res. 10, 2011–2026 [DOI] [PubMed] [Google Scholar]
- 30. Xu W., Liu L., and Hornby D. (2012) c-IAP1 binds and processes PCSK9 protein: linking the c-IAP1 in a TNF-α pathway to PCSK9-mediated LDLR degradation pathway. Molecules 17, 12086–12101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cortese K., Sahores M., Madsen C. D., Tacchetti C., and Blasi F. (2008) Clathrin and LRP-1-independent constitutive endocytosis and recycling of uPAR. PLoS ONE 3, e3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Capurro M. I., Shi W., and Filmus J. (2012) LRP1 mediates Hedgehog-induced endocytosis of the GPC3-Hedgehog complex. J. Cell Sci. 125, 3380–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hamik A., Setiadi H., Bu G., McEver R. P., and Morrissey J. H. (1999) Down-regulation of monocyte tissue factor mediated by tissue factor pathway inhibitor and the low density lipoprotein receptor-related protein. J. Biol. Chem. 274, 4962–4969 [DOI] [PubMed] [Google Scholar]
- 34. Llorente-Cortés V., Otero-Viñas M., Camino-López S., Llampayas O., and Badimon L. (2004) Aggregated low density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells. Circulation 110, 452–459 [DOI] [PubMed] [Google Scholar]
- 35. Strickland D. K., Au D. T., Cunfer P., and Muratoglu S. C. (2014) Low-density lipoprotein receptor-related protein-1: role in the regulation of vascular integrity. Arterioscler. Thromb. Vasc. Biol. 34, 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dashty M., Motazacker M. M., Levels J., de Vries M., Mahmoudi M., Peppelenbosch M. P., and Rezaee F. (2014) Proteome of human plasma very low density lipoprotein and low density lipoprotein exhibits a link with coagulation and lipid metabolism. Thromb. Haemost. 111, 518–530 [DOI] [PubMed] [Google Scholar]
- 37. Murdoch S. J., Carr M. C., Kennedy H., Brunzell J. D., and Albers J. J. (2002) Selective and independent associations of phospholipid transfer protein and hepatic lipase with the LDL subfraction distribution. J. Lipid Res. 43, 1256–1263 [PubMed] [Google Scholar]
- 38. Demchev V., Malana G., Vangala D., Stoll J., Desai A., Kang H. W., Li Y., Nayeb-Hashemi H., Niepel M., Cohen D. E., and Ukomadu C. (2013) Targeted deletion of fibrinogen like protein 1 reveals a novel role in energy substrate utilization. PLoS ONE 8, e58084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Filmus J., and Selleck S. B. (2001) Glypicans: proteoglycans with a surprise. J. Clin. Invest. 108, 497–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsu H. C., Cheng W., and Lai P. L. (1997) Cloning and expression of a developmentally regulated transcript MXR7 in hepatocellular carcinoma: biological significance and temporospatial distribution. Cancer Res. 57, 5179–5184 [PubMed] [Google Scholar]
- 41. Capurro M., Wanless I. R., Sherman M., Deboer G., Shi W., Miyoshi E., and Filmus J. (2003) Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 125, 89–97 [DOI] [PubMed] [Google Scholar]
- 42. Fu S.-J., Qi C.-Y., Xiao W.-K., Li S.-Q., Peng B.-G., and Liang L.-J. (2013) Glypican-3 is a potential prognostic biomarker for hepatocellular carcinoma after curative resection. Surgery 154, 536–544 [DOI] [PubMed] [Google Scholar]
- 43. Filmus J., and Capurro M. (2013) Glypican-3: a marker and a therapeutic target in hepatocellular carcinoma. FEBS J. 280, 2471–2476 [DOI] [PubMed] [Google Scholar]
- 44. De Cat B., Muyldermans S.-Y., Coomans C., Degeest G., Vanderschueren B., Creemers J., Biemar F., Peers B., and David G. (2003) Processing by proprotein convertases is required for glypican-3 modulation of cell survival, Wnt signaling, and gastrulation movements. J. Cell Biol. 163, 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Haruyama Y., and Kataoka H. (2016) Glypican-3 is a prognostic factor and an immunotherapeutic target in hepatocellular carcinoma. World J. Gastroenterol. 22, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Traister A., Shi W., and Filmus J. (2008) Mammalian notum induces the release of glypicans and other GPI-anchored proteins from the cell surface. Biochem. J. 410, 503–511 [DOI] [PubMed] [Google Scholar]
- 47. Capurro M. I., Xiang Y.-Y., Lobe C., and Filmus J. (2005) Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. 65, 6245–6254 [DOI] [PubMed] [Google Scholar]
- 48. Gao W., and Ho M. (2011) The role of glypican-3 in regulating Wnt in hepatocellular carcinomas. Cancer Rep. 1, 14–19 [PMC free article] [PubMed] [Google Scholar]
- 49. Capurro M., Martin T., Shi W., and Filmus J. (2014) Glypican-3 binds to Frizzled and plays a direct role in the stimulation of canonical Wnt signaling. J. Cell Sci. 127, 1565–1575 [DOI] [PubMed] [Google Scholar]
- 50. Capurro M. I., Xu P., Shi W., Li F., Jia A., and Filmus J. (2008) Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev. Cell 14, 700–711 [DOI] [PubMed] [Google Scholar]
- 51. Iglesias B. V., Centeno G., Pascuccelli H., Ward F., Peters M. G., Filmus J., Puricelli L., and de Kier Joffé E. B. (2008) Expression pattern of glypican-3 (GPC3) during human embryonic and fetal development. Histol. Histopathol. 23, 1333–1340 [DOI] [PubMed] [Google Scholar]
- 52. Benjannet S., Hamelin J., Chrétien M., and Seidah N. G. (2012) Loss- and gain-of-function PCSK9 variants: cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J. Biol. Chem. 287, 33745–33755 [DOI] [PMC free article] [PubMed] [Google Scholar]