

VSMC phenotype plays a role in growth of blood vessels during embryogenesis and in pathogenesis of vascular diseases. A contractile phenotype of VSMC is critical in normal physiological function of blood vessels. The processes that control VSMC phenotype are still not clearly known. This study defines a novel role for G6PD in regulating the contractile gene program in VSMCs.
Keywords: epigenetics, metabolism, pentose phosphate pathway, RNA-seq, NADPH redox, protein kinase G, vascular smooth muscle phenotype
Abstract
Homeostatic control of vascular smooth muscle cell (VSMC) differentiation is critical for contractile activity and regulation of blood flow. Recently, we reported that precontracted blood vessels are relaxed and the phenotype of VSMC is regulated from a synthetic to contractile state by glucose-6-phosphate dehydrogenase (G6PD) inhibition. In the current study, we investigated whether the increase in the expression of VSMC contractile proteins by inhibition and knockdown of G6PD is mediated through a protein kinase G (PKG)-dependent pathway and whether it regulates blood pressure. We found that the expression of VSMC-restricted contractile proteins, myocardin (MYOCD), and miR-1 and miR-143 are increased by G6PD inhibition or knockdown. Importantly, RNA-sequence analysis of aortic tissue from G6PD-deficient mice revealed uniform increases in VSMC-restricted genes, particularly those regulated by the MYOCD-serum response factor (SRF) switch. Conversely, expression of Krüppel-like factor 4 (KLF4) is decreased by G6PD inhibition. Interestingly, the G6PD inhibition-induced expression of miR-1 and contractile proteins was blocked by Rp-β-phenyl-1,N2-etheno-8-bromo-guanosine-3′,5′-cyclic monophosphorothioate, a PKG inhibitor. On the other hand, MYOCD and miR-143 levels are increased by G6PD inhibition through a PKG-independent manner. Furthermore, blood pressure was lower in the G6PD-deficient compared with wild-type mice. Therefore, our results suggest that the expression of VSMC contractile proteins induced by G6PD inhibition occurs via PKG1α-dependent and -independent pathways.
NEW & NOTEWORTHY
VSMC phenotype plays a role in growth of blood vessels during embryogenesis and in pathogenesis of vascular diseases. A contractile phenotype of VSMC is critical in normal physiological function of blood vessels. The processes that control VSMC phenotype are still not clearly known. This study defines a novel role for G6PD in regulating the contractile gene program in VSMCs.
glucose-6-phosphate dehydrogenase (G6PD) generates nicotinamide adenine dinucleotide phosphate reduced (NADPH), a key cofactor for various redox-sensitive enzymes like NADPH oxidases, glutathione/thioredoxin reductases, and other reductive and anabolic reactions in the cell (21). We have shown that G6PD is involved in regulation of coronary and pulmonary artery contraction and relaxation (2), and pulmonary artery SMC phenotype (13). Adult VSMC may exhibit a contractile “differentiated” or synthetic “dedifferentiated” phenotype (1, 8, 9, 24). These phenotypes are not necessarily stable, irreversible or mutually exclusive (24, 36). Contractile phenotype of VSMC is required for proper functionality of blood vessels, i.e., to contract and relax in response to sympathetic stimuli or to nitric oxide, respectively. It is well known that VSMCs switch from a contractile to synthetic phenotype in response to injury or chemical stimuli. This phenotypic switching contributes to vascular remodeling associated with medial hypertrophy, acute neointimal formation following iatrogenic interventions, and atherosclerosis (1, 8, 9).
Expression of VSMC contractile genes is regulated by a network of transcription factors and cofactors such as: myocardin (MYOCD), Krüppel-like factor 4 (KLF4), and ELK1. MYOCD is a master regulator of smooth muscle contractile gene expression (10, 44). MYOCD forms a ternary complex with serum response factor (SRF) over CArG boxes to transactivate the contractile program of gene expression in VSMC (31, 44). KLF4 negatively regulates expression of MYOCD (43) and together with SRF and ELK1, mediates a mechanism driving expression of synthetic genes in VSMC (1, 3, 27, 34, 37, 44, 45). Thus, carefully orchestrated levels of these transcription factors regulate the phenotypic plasticity of VSMC.
Protein kinase G (PKG) has been known to decrease the transcriptional activity of ELK1 by sumoylation (14). MicroRNAs (miR) are also involved in fine-tuning the expression of KLF4 and ELK1. For example, miR-143 downregulates ELK1 (15, 24, 25) and miR-145 and miR-1 reduces KLF4 (15, 24, 25, 46). Since G6PD inhibition increases PKG activity and H2O2 that stimulates miR production (2, 11), we postulated that PKG and miR contribute to increase G6PD inhibition-induced expression of VSMC marker/contractile phenotype proteins. We tested this hypothesis in arteries and VSMC isolated from bovine and rat aorta that express G6PD, as well as aorta of G6PD-deficient mice.
MATERIALS AND METHODS
Coronary arteries (LAD) were isolated from bovine heart purchased from a slaughterhouse. Aorta used in this study were isolated from rats and from wild-type and G6PD-deficient mice. Hemizygous (XbY) G6PD-deficient male and female mice and wild-type (XY) control mice (WT) age 12–16 wk were studied. Animals were genotyped as described previously (39). Normally, G6PD-deficient mice do not exhibit any abnormal cardiovascular phenotype and live up to 2–2.5 yr. Experimental protocols using animals had approval from the New York Medical College (A3362-01) and University of South Alabama (11036) Institutional Animal Care and Use Committees. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Cell culture.
Rat aortic smooth muscle cells (RASMCs) were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (Life Technologies, Grand Island, NY) and 1% antibiotics (Gibco) at 37°C and 5% CO2. Cells were cultured for a span of 48 h and then used for further experiments.
MicroRNA expression by unsupervised whole genome quantitative microarray and QPCR.
MicroRNA (miR) levels were determined by global miR profiling using LNA-enhanced microarrays by Exiqon A/S, Denmark. Additionally, miR analysis was performed by quantitative RT-PCR (qRT-PCR) as previously described (11). Briefly, total RNA was extracted from aorta using Qiagen miRNEasy kit. Input RNA quality and concentration were measured on NanoDrop and cDNA was prepared using miR-specific TaqMan miR assays (Applied Biosystems, Foster City, CA). Quantitative PCR was performed in triplicate using TaqMan Universal PCR Master mix. Standard curves were made for each miR using synthetic miR oligonucleotides (IDT, Coralville, IA) with the following sequence: Rno-miR-145: GUCCAGUUUUCCCAGGAAUCCCU; Rno-miR-1: UGGAAUGUAAAGAAGUGUGUAU; Rno-miR-143: UGAGAUGAAGCACUGUAGCUC; and Rno-miR-133a: UUUGGUCCCCUUCAACCAGCUG.
Ct values from standard curves were then used to quantify relative expression of specific miR using the 2−ddCt formula.
RNA-Seq analysis.
Total RNA was extracted from aorta of wild-type and G6PD-deficient mice using Qiagen miRNEasy kit. Input RNA quality and concentration were measured on NanoDrop. Following bioanalyzer quality control confirmation, RNA-seq was performed (triplicate samples for each genotype) on total RNA by stranded ribo-depletion with RiboZero using the Illumina HiSeq 2500 platform at the University of Rochester Genomics Research Center (http://www.urmc.rochester.edu/fgc/) as described previously (5). Single-end sequencing was done at a depth of 10 million reads per replicate (n = 3). Quantitative analysis, including the statistical analysis of differentially expressed genes, was done with Cufflinks 2.0.2 and Cuffdiff2 (http://cufflinks.cbcb.umd.edu). The Benjamini-Hochberg method was applied for multiple test correction (FDR < 0.05). Data output files such as Volcano plots were generated with the R package, ggplot (http://ggplot2.org/). Gene ontology (GO) analysis was done using DAVID (6, 17).
Western blot analysis.
Proteins were extracted from snap frozen coronary arteries, aorta and RASMCs in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Nonidet P-40, 100 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 200 mM pepstatin. Following resolution in a 10% PAGE gel, proteins were transferred to nitrocellulose membrane and probed using smooth muscle myosin heavy chain (MYH11: Biomedical Technologies; BT562; 1:1,000); SM22α (TAGLN: Abcam; ab14106; 1:1,000); Calponin (CNN1: Sigma; C2687; 1:1,000); KLF4 (Cell Signaling; 4038p; 1:1,000); glyceraldehyde-3-phosphate dehydrogenase (GAPDH: Cell Signaling; 2118S; 1:1,000); and actin (ACTB; Sigma; A2228; 1:1,000) antibodies.
cGMP measurement.
cGMP levels were estimated by ELISA assay kit from Cayman Chemicals according to manufacturer's instructions.
Immunoprecipitation and liquid chromatography-tandem mass spectrometry.
One milligram of protein extract was precleared with agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). Pull down was done with anti-G6PD antibody (Sigma-Aldrich, St Louis, MO) and agarose beads overnight at 4°C. The beads-antibody-protein complex was washed and resuspended in 2x SDS buffer and run on SDS-PA gel. The protein was eluted and analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS; Applied Biomics, Hayward, CA). NanoLC fractionation and matrix-assisted laser desorption ionization-time of flight/time of flight (MALDI-TOF/TOF) were followed by a standard search of the National Center for Biotechnology Information and SwissProt databases using MASCOT.
Blood pressure measurement.
Adult mice were anesthetized by 4% isoflurane and 2% isoflurane was used to maintain anesthesia for the entire duration of the surgery and data acquisition. Body temperature of the animal during the surgery was maintained using a heating pad. About 3 cm2 area of skin over the ventral neck region was exposed to locate right common carotid artery. 1.4F Millar Micro-Tip pressure catheter was carefully inserted into the right common carotid artery and systolic blood pressure was measured by MVPS-Ultra system and ADInstruments Power Lab system.
Statistical analysis.
All miR analysis and Western blot data were analyzed using Student's t-tests. Values of P < 0.05 were considered significant. Values are presented as means ± SE.
RESULTS
VSMC contractile protein expression is increased by G6PD inhibition and deficiency.
To test the hypothesis that expression of VSMC contractile proteins is increased by G6PD inhibition, bovine coronary arteries (BCA) were isolated and treated with a competitive G6PD inhibitor, 6-aminonicotinamide (6AN, 1 mM), for 12 h and contractile protein expression was measured by immunoblotting. The expression of smooth muscle-myosin heavy chain (MYH11), calponin (CNN1), and SM22α (TAGLN) was increased by G6PD inhibition (Fig. 1A). A similar increase in the expression of contractile proteins was observed when the arteries were treated with another G6PD inhibitor, dehydroepiandrosterone (data not shown). To gain insight into the spectrum of mRNA changes that occur genomewide in vessels lacking G6pd, we performed RNA-seq experiments on aortic samples stripped of endothelium and adventitia in G6PD-deficient vs. wild-type controls. Essentially all genes associated with the VSMC contractile phenotype were upregulated > 1.5 log2 fold in samples of aorta lacking G6PD vs. control [GEO Submission (GSE80972) (NCBI tracking system no. 17863684); Table 1]. Included among these were the SRF-MYOCD-dependent Lmod1 (35), Cnn1 (32), Kcnmb1 (28), and Actg2 (41) genes (Fig. 1B). Gene Ontology analysis of the 987 top (>1.5 log2fold_change) upregulated genes revealed G6PD-deficiency increased genes involved in translation, actin filament based processes, cell adhesion, and various cytoskeletal structures and the 178 top (>1.5 log2fold_change) downregulated genes showed GO terms associated with homeostatic processes, lipid transport, and lipid localization (Table 2). We also analyzed transcription factor binding site enrichment using oPOSSUM (26). This analysis disclosed the SRF-binding CArG box as the most enriched TFBS in genes upregulated with G6PD deficiency (Table 3). Western blot confirmed that the MYH11 expression was higher in the aorta of G6PD-deficient compared with wild-type mice (Fig. 1C). Together, these results establish an important role for G6PD in antagonizing the VSMC contractile gene program in the aorta.
Fig. 1.

VSMC contractile protein expression is increased by G6PD inhibition and deficiency. Isolated coronary arteries were treated with G6PD inhibitor 6-aminonicotinamide (6AN; 1 mM; n = 5) for 12 h. A: expression of contractile proteins SM22α (TAGLN), calponin (CNN1) and smooth muscle myosin heavy chain (MYH11) were measured by immunoblotting. Proteins were normalized to glyceraldehyde-6-phosphate dehydrogenase (GAPDH). B: Volcano plot demonstrates several genes are up- and downregulated in aorta of G6PD-deficient mice (n = 3). C: a representative immunoblot of MYH11 expression in G6PD-deficient mice aorta is shown (n = 3). Green circles: significant (P < 0.05); yellow circles: significant, but unreliable replicate data; red circles: significant (P < 0.05), but inadequate Log2_FoldChange; orange circles: not significant but considerable Log2_FoldChange; gray Circles: not significant.
Table 1.
Upregulated (log2_fold change) gene associated with SMC Contractile Gene Program
| Gene | log2 |
|---|---|
| Actg2 | 3.56588 |
| Casq1 | 2.933 |
| Myl9 | 2.83493 |
| Tagln | 2.5053 |
| Cnn1 | 2.43311 |
| Acta2 | 2.42846 |
| Myh11 | 2.39866 |
| Myl6 | 2.37437 |
| Kcnmb1 | 2.30992 |
| Dstn | 2.28147 |
| Lmod1 | 2.25203 |
| Mylk | 2.19593 |
| Csrp1 | 2.10506 |
| Csrp2 | 2.01419 |
| Tpm2 | 2.00505 |
| Cspg4 | 1.99633 |
| Itga8 | 1.98484 |
| Tspan2 | 1.98444 |
Table 2.
Top GO terms for genes upregulated or downregulated with G6PD deficiency
| Biological Process | Count | P-Value | Fold Enrichment | FDR |
|---|---|---|---|---|
| GO terms for genes upregulated with G6PD deficiency | ||||
| Translation | 60 | 2.59E-21 | 4.2 | 4.51E-18 |
| Actin filament-based process | 22 | 3.60E-05 | 2.8 | 0.062635 |
| Regulation of cytoskeleton organization | 15 | 1.17E-04 | 3.4 | 0.203316 |
| GO terms for genes downregulated with G6PD deficiency | ||||
| Homeostatic process | 7 | 0.001 | 5.3 | 2.1 |
| Lipid transport | 4 | 0.002 | 14.7 | 3.1 |
| Lipid localization | 4 | 0.003 | 13.7 | 3.9 |
Table 3.
Transcription factor binding site enrichment using oPOSSUM
| TF | TF Class | TF Supergroup | IC | Background Gene Hits | Background Gene Non-hits | Target Gene Hits | Target Gene Non-hits | Background TFBS Hits | Background TFBS Rate | Target TFBS Hits | Target TFBS Rate | Z-Score | Fisher Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SRF | MADS | Vertebrate | 17.965 | 713 | 14437 | 57 | 542 | 793 | 0.0004 | 77 | 0.0009 | 23.83 | 1.09E-06 |
| RELA | REL | Vertebrate | 14.757 | 4846 | 10304 | 222 | 377 | 8017 | 0.0035 | 419 | 0.004 | 9.486 | 5.57E-03 |
| REL | REL | Vertebrate | 10.515 | 7798 | 7352 | 354 | 245 | 18677 | 0.0081 | 921 | 0.0089 | 8.491 | 1.40E-04 |
| NFκB | REL | Vertebrate | 13.345 | 5960 | 9190 | 278 | 321 | 11447 | 0.005 | 570 | 0.0055 | 7.405 | 3.32E-04 |
| NHLH1 | bHLH | Vertebrate | 14.132 | 4534 | 10616 | 199 | 400 | 7563 | 0.0039 | 379 | 0.0044 | 7.039 | 4.75E-02 |
| Dl_2 | REL | Insect | 14.123 | 4779 | 10371 | 225 | 374 | 7910 | 0.0034 | 396 | 0.0038 | 6.504 | 1.26E-03 |
| ELF5 | ETS | Vertebrate | 8.693 | 12185 | 2965 | 495 | 104 | 73478 | 0.0287 | 3428 | 0.0297 | 5.829 | 9.80E-02 |
| TAL1-TCF3 | bHLH | Vertebrate | 14.07 | 5459 | 9691 | 249 | 350 | 10058 | 0.0052 | 487 | 0.0056 | 5.35 | 3.46E-03 |
| SQUA | MADS | Plant | 12.389 | 7349 | 7801 | 318 | 281 | 21167 | 0.0129 | 998 | 0.0134 | 5.146 | 1.55E-02 |
| TP53 | P53 | Vertebrate | 26.239 | 21 | 15129 | 2 | 597 | 21 | 0 | 2 | 0 | 4.716 | 2.17E-01 |
Myocardin expression is increased by G6PD inhibition and knockdown.
MYOCD plays a key role in upregulating the expression of VSMC contractile proteins (10, 45). RNAseq analysis showed that Myocd gene expression was higher in G6PD-deficient compared with wild-type mice aorta (Fig. 2A). Furthermore, MYOCD mRNA expression was increased in RASMC by G6PD inhibition (Fig. 2B). However, SRF expression was not modulated by G6PD inhibition with 6AN or dehydroepiandrosterone (data not shown).
Fig. 2.
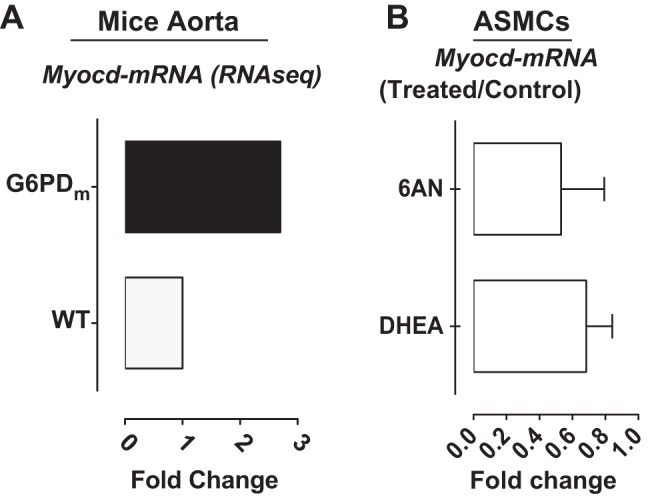
Myocardin expression is increased by G6PD inhibition and knockdown. A: myocardin gene expression is higher in G6PD-deficient compared with wild-type mice aorta (n = 3 analyzed in triplicates). B: myocardin message is increased in RASMC treated with 6-aminonicotinamide (6AN; 1 mM; n = 5).
Expression of VSMC contractile proteins induced by G6PD inhibition is PKG-dependent.
Previously, we have shown that in pulmonary arteries G6PD forms a complex with PKG1α, increases PKG activity, and promotes TAGLN expression (13). We also found that G6PD forms a complex with phosphodiesterase 5 (PDE5; Table 4). This indicated that cGMP-dependent activation of PKG mediated the expression of contractile proteins elicited by G6PD inhibition. Therefore, we measured cGMP levels in isolated bovine arteries. G6PD inhibition with 6AN for 12 h decreased (P < 0.05) basal as well as sildenafil (10 μM)-induced cGMP levels compared with control (Fig. 3A). Similar decrease was obtained by DHEA treatment. Since G6PD inhibition decreased cGMP, we speculated that PKG activity increased presumably following upregulation of PKG1α expression in VSMCs by G6PD inhibition. Application of 6AN (Fig. 3, B and C) to the arteries for 12 h indeed increased PKG1α expression compared with untreated control and total PKG activity, measured as phosphorylation of VASP. ELK1 together with KLF4 represses the expression of myocardin-induced SMC genes (44, 47) and PKG1α decreases the activity of ELK1 by sumoylation (14). To determine whether the increased expression of VSMC contractile proteins elicited by G6PD inhibition is mediated through PKG signaling pathway, we applied Rp-β-phenyl-1,N2-etheno-8-bromo-guanosine-3′,5′-cyclic monophosphorothioate (Rp-cGMPs, 100 nM), a PKG inhibitor, along with G6PD inhibitors to arteries. As demonstrated (Fig. 3D), G6PD inhibition-induced expression of TAGLN, and CNN1 and MYH11 was suppressed by Rp-cGMPs.
Table 4.
LC-MS/MS results showing G6PD forms a complex with phosphodiesterase 5 and endothelial nitric oxide synthase in endothelium-intact coronary artery
| Top Ranked Protein Name | Accession No. | Protein MW | Protein PI | Peptide Count | Total Ion Score | Total Ion CI, % |
|---|---|---|---|---|---|---|
| cGMP-specific 3′,5′-cyclic phosphodiesterase | gi 359066284 | 20,895 | 5.8 | 3 | 54 | 100 |
| Nitric oxide synthase, ECNOS | gi 252390 | 132,901 | 7.0 | 3 | 47 | 99 |
MW, molecular weight.
Fig. 3.

Total protein kinase G activity and the expression of VSMC contractile proteins are increased in isolated coronary arteries by treatment with G6PD inhibitor. A: cGMP levels are decreased by G6PD inhibitor, 6-aminonicotinamide (6AN; 1 mM; n = 5), under basal conditions and after inhibiting phosphodiesterase-5 (PDE5) with sildenafil (Sil; 10 μM). Protein kinase G-1α (PKG-1α) expression (B) and PKG activity measured as phosphorylation of vasodilator-stimulated phosphoprotein (VASP) (C) is increased by G6PD inhibition (n = 5). D: SM22α (TAGLN), calponin (CNN1) and smooth muscle myosin heavy chain (MYH11) expression were measured after inhibiting PKG with Rp-β-phenyl-1,N2-etheno-8-bromo-guanosine-3′,5′-cyclic monophosphorothioate (cGMPs; 100 nM) in the absence and presence of G6PD inhibitor, 6AN (1 mM). N = 5 in each group. *P < 0.05 vs. control, #P < 0.05 vs. 6AN.
miR-1 and miR-143 are increased and KLF4 expression is decreased by G6PD inhibition and knockdown.
MYOCD_v3, is the predominant MYOCD isoform expressed in the vascular wall (16, 22), and promotes miR-145 and miR-143 expression and differentiation of VSMC (15). In contrast to miR-1, miR-133a is highly expressed in adult VSMCs and it suppresses proliferation and promotes differentiation of VSMCs (42). Therefore, our next goal was to determine whether expression of miRs, which play a role in regulating contractile protein expression, is regulated by the G6PD-PKG pathway.
We used rat and mouse aorta to study the effect of G6PD-PKG signaling on miR expression profile because sequence of rat and mouse miR is known and miR assay kits are commercially available. First, we determined miR profile in the aorta by unbiased whole genome quantitative miR microarray. A total of 242 miRs were detected in rat aorta treated with and without 6AN and DHEA. We found 93 and 135 miRs were increased by DHEA and 6AN treatment, respectively, compared with untreated control. In contrast, 147 and 107 miRs were decreased by DHEA and 6AN treatment, respectively, compared with untreated control. Strikingly, miRs (such as miR-1-3p, miR-133a-3p, -133b-3p, -143-3p, 143-5p, 145-3p, and 145-5p) that upregulate VSMC contractile proteins in a myocardin-dependent manner were increased (>1.5-fold) and miRs (for example: miR-206 and -21) that promote cell proliferation and migration were decreased (>1.5-fold) by DHEA and 6AN compared with untreated controls.
Next, we measured the expression levels of miR-143-3p, -145-3p, -133a-3p, and -1-3p in rat aorta by qRT-PCR (Fig. 4A). Inhibition of G6PD with DHEA and 6AN increased miR-143-3p and miR-1-3p, but not miR-145-3p or miR-133a-3p (Fig. 4, B and C). Rp-cGMPs, a PKG inhibitor, prevented DHEA- and 6AN-induced increase of miR-1-3p, but not miR-143-3p, expression (Fig. 4, D and E). Furthermore, miR-143-3p levels were higher in the aorta from G6PD-deficient compared wild-type mice (Fig. 4F).
Fig. 4.

miR-1 and miR-143 levels are increased and KLF4 expression is suppressed by G6PD inhibition and knockdown. A: expression of various miRs measured in aorta by quantitative RT-PCR (n = 5). miR-143 and miR-145 (B), and miR-1 and miR-133 (C) expression were measured in aorta treated with or without G6PD inhibitors, 6-aminonicotinamide (6AN; 1 mM) and dehydroepiandrosterone (DHEA; 100 μM). D and E: inhibiting PKG activity with Rp-β-phenyl-1,N2-etheno-8-bromo-guanosine-3′,5′-cyclic monophosphorothioate (cGMPs) suppressed expression of miR-1, but not miR-143, increased by G6PD inhibition. F: expression of miR-143 is increased in the aorta of G6PD-deficient compared with wild-type mice. G: KLF4 expression is decreased in VSMCs by DHEA (100 μM; n = 5). *P < 0.05 vs. cGMPs.
Since KLF4 gene is the target of miR-1, we determined KLF4 levels in aorta treated with and without 6AN and DHEA. KLF4 expression was decreased by application of G6PD inhibitors to the arteries for 12 h (Fig. 4G).
Blood pressure is lower in G6PD-deficient compared with wild-type mice.
The systolic, diastolic and mean blood pressure were lower by 20 mmHg (P < 0.05; Fig. 5) in the G6PD-deficient compared with wild-type mice.
Fig. 5.

Blood pressure is lower in G6PD-deficient mice. Systolic (A), diastolic (B), and mean blood pressure (C) determined by catheterization of right carotid artery is lower in the G6PD-deficient compared with wild-type mice (n = 5). Arterial elastance (stiffness) (D) is not different in both G6PD-deficient and wild-type group. *P < 0.05 vs. wild type.
DISCUSSION
A fundamental characteristic of VSMCs is an oscillation between contractile and synthetic states in normal health and disease. The contractile phenotype defines VSMC that express VSMC-restricted contractile and cytoskeletal proteins such as MYH11, ACTA2, ACTG2, CALD1, CNN1, smooth muscle tropomyosins, TAGLN and smoothelin-B isoform (1). Smooth muscle cells that have a higher tendency to proliferate, migrate and secrete a variety of cytokines, chemokines, growth factors and extracellular matrix are generally classified as synthetic/proliferative (36). Previously, we have shown that the contractility of the blood vessel is reduced and the fate of VSMC phenotype is regulated from a synthetic to contractile state by G6PD inhibition (2, 13, 20). In this study, we demonstrate that MYOCD- and PKG-dependent pathways mediated G6PD inhibition-induced expression of VSMC restricted proteins.
G6PD is responsible for the generation of NADPH, a key cofactor in the cell (21), and its activity changes with alteration in glucose metabolism. Our studies suggest that G6PD activity is increased in the vessel wall of pigs and rats with metabolic syndrome (23, 40) and rats with pulmonary hypertension (12, 38). However, the relationship between VSMC metabolism and functional phenotype remains largely unknown. Our current results demonstrated that the expression of MYOCD mRNA as well as VSMC contractile proteins is increased in intact arteries and cultured RASMC by inhibition and knockdown of G6PD. The expression of VSMC contractile genes is upregulated primarily by SRF-MYOCD (10, 45) and by miR-143/145 and miR-1/133a (1, 15, 24, 47). Since miR-1 and miR-143 posttranscriptionally downregulate the expression of KLF4 and ELK1 (46), we suggest that G6PD inhibition-induced increase of miR-1 and miR-143, and decrease of KLF4 contributed to increased expression of contractile genes in VSMC. Long and short noncoding RNAs are implicated to regulate SMC phenotype (4, 5, 33). While miR-145 and miR-143 are cotranscribed only miR-143 was upregulated by G6PD inhibition and deficiency. This indicates that these miRs may be differentially regulated posttranscriptionally by G6PD. Since we found some of the noncoding RNAs were significantly up- or downregulated in aorta of G6PD-deficient mice (Gm genes in Fig. 1), a plausibility of noncoding RNAs in G6PD-mediated differential regulation of miR145 and miR43 expression cannot be ruled out.
RNA-seq results suggested that in samples of aorta lacking G6PD vs. wild-type control mice essentially all genes associated with the VSMC contractile phenotype program including, but not limited to, the SRF-MYOCD-dependent Lmod1 (35), Cnn1 (32), Kcnmb1 (28), and Actg2 (41) genes were upregulated. In contrast, the genes in synthetic phenotype category such as 1) adhesion and secretory proteins, including COL1 and -3, CALD1, VCL and TGM2; and 2) synthetic protein markers, including OPN and VCAN, were neither up- nor downregulated with G6PD deficiency. While 987 genes were upregulated, only 178 genes were downregulated in aorta of G6PD-deficient compared with wild-type mice. KEGG pathway and GO analysis of the 987 top upregulated genes showed G6PD-deficiency increased genes involved in ribosome, translation, vascular smooth muscle contraction, actin filament based processes, cell adhesion, and various cytoskeletal structures. In contrast, the 178 top downregulated genes showed GO terms associated with homeostatic processes, fatty acid metabolic process, lipid transport and lipid localization. TFBS enrichment analysis using oPOSSUM (26) disclosed the SRF-binding CArG box as the most enriched TFBS in genes upregulated with G6PD deficiency. In contrast, PPARG-RXRA was the most enriched TFBS in genes downregulated with G6PD deficiency. Western blot confirmed that the MYH11, TAGLN, and CNN1 expression was higher in the aorta of G6PD-deficient compared with wild-type mice. Altogether, these results established an important role for G6PD in antagonizing the VSMC contractile gene program in the aorta.
Previously, we showed that G6PD inhibition increases activity of PKG (13). In this study, we found that PKG1α expression and total PKG activity is increased by application of G6PD inhibitors to bovine coronary artery. Therefore, to determine whether the expression of contractile proteins was a consequence of increased PKG1α expression or activity, we applied Rp-cGMPs, a competitive PKG inhibitor (7), along with G6PD inhibitors to the coronary arteries. Rp-cGMPs blocked the increase of VSMC contractile proteins and miR-1 expression induced by G6PD inhibition. These observations suggest that increased PKG1α activity by G6PD inhibition upregulated miR-1, which is known to decrease KLF4 expression in VSMCs (15), and are consistent with the other studies that suggest ectopic expression of PKG1α in VSMCs upregulates the expression of contractile protein genes by downregulating ELK1 (14). While MYOCD expression decreases in ovine fetal pulmonary artery smooth muscle cells by knockdown of PKG1α (49), we reported that the expression of PKG1α in PKG-deficient cells enhances MYOCD-induced Tagln promoter activity without increasing Myocd gene expression (14). Consistently, our results suggest that Rp-cGMPs decreased the expression of MYOCD-target genes but not MYOCD, which was elicited by G6PD-inhibition or knockdown. Since KLF4 represses Myocd gene (27), we suggest that decrease of KLF4 expression may be one of the causes for G6PD inhibition-mediated increase in MYOCD expression. Transcription factors such as MEF2, FOXO4, and TEAD1-2 positively regulate Myocd gene expression during early cardiovascular development (16). Intriguingly, transcriptional activity of FOXO4 is increased by elevated H2O2 (18). We have shown that MYOCD expression is induced by H2O2 in aorta of Goto-Kakizaki rats and in aortic smooth muscle cells (11). Since G6PD inhibition can potentially reduce the clearance of intracellular H2O2, a role for H2O2 in G6PD inhibition-mediated upregulation of Myocd gene expression cannot be ruled out. Thus these observations suggest that an increased expression of MYOCD by G6PD inhibition is, at least partly, mediated through PKG-independent signaling pathways.
To further determine the consequence of elevated PKG and MYOCD-target genes by G6PD deficiency on vascular function, we measured blood pressure by catheterization of right carotid artery. The blood pressure was significantly lower in G6PD-deficient than wild-type mice. Along these lines, others have reported that G6PD-deficiency prevents ANG II-induced hypertension (30). Conversely, inhibition of Na+-K+-ATPase by various agents including ouabain stimulates G6PD activity and elevated G6PD activity is associated with essential hypertension in humans (29). Therefore, it is reasonable to suggest that G6PD plays a potential role in regulating blood pressure and reduction of G6PD expression and activity is beneficial in reducing hypertension.
Although G6PD inhibition and deficiency increased MYOCD/SRF-dependent VSMC-restricted genes and contractile proteins that promote a contractile phenotype, G6PD inhibition and deficiency paradoxically opposes a contractile state and relaxes blood vessels precontracted with KCl and U46619 (19). G6PD inhibition antagonizes the depolarization of the membrane potential- and agonist-induced Ca2+ influx through the L-type Ca2+ channels and increases of cytosolic Ca2+, and facilitates Ca2+ uptake by sarco(endo)plasmic reticulum Ca2+ ATPase (2). Furthermore, PKG-dependent phosphorylation of MYPT1, which increases MYPT1 activity, reduces Ca2+ sensitivity to the myofilaments (48). Therefore, we suggest that G6PD inhibition and deficiency lowers blood pressure by reducing the intracellular Ca2+ and decreasing Ca2+ sensitivity to the myofilaments via a PKG-mediated increase of MYPT1 activity. These, results taken together with our current findings suggest that G6PD inhibition and knockdown has two independent actions on vascular smooth muscle function: 1) it relaxes the arteries and 2) increases expression of vascular smooth muscle cell-restricted genes and contractile protein. Increased VSMC-restricted proteins could be a compensatory response of the VSMC to an acute and potent vasodilation/relaxation evoked by G6PD inhibition/knockdown.
In summary, the cellular pool of MYOCD-dependent genes and KLF4 that determine the VSMC contractile phenotype is increased and decreased, respectively, by inhibition or knockdown of G6PD. Furthermore, we demonstrate for the first time that PKG1α is involved in G6PD-mediated regulation of miR-1 and VSMC contractile protein expression. Taken together, our findings suggest (Fig. 6) that SRF/MYOCD-dependent contractile proteins upregulated through PKG-dependent and PKG-independent pathways that increase miR-1 and MYOCD/miR-143 expression, respectively, regulate vascular function.
Fig. 6.
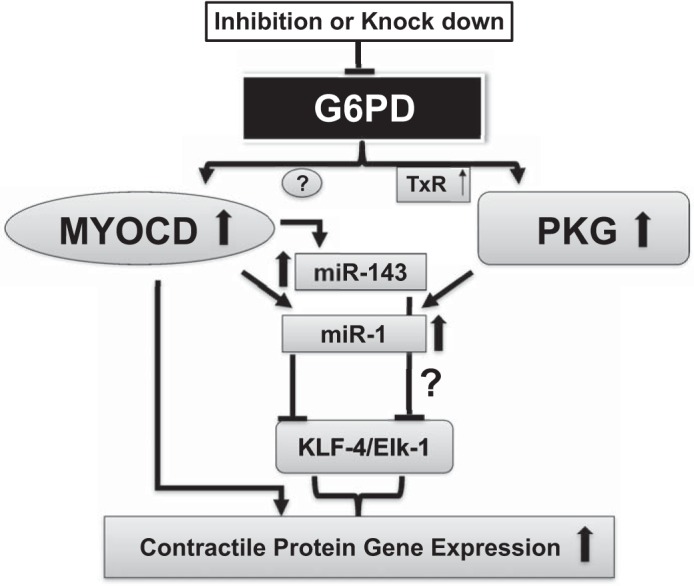
Schematic representation showing the possible mechanisms by which G6PD inhibition increases contractile protein expression. G6PD inhibition increases MYOCD expression, which in turn increases the transcription of VSMC contractile proteins. G6PD inhibition also increases PKG activity via thioredoxin reductase (TrX)-mediated thiol oxidation, which promotes the expression of miR-1 and miR-143 leading to increase of VSMC contractile protein expression.
GRANTS
This study was support by National Heart, Lung, and Blood Institute Grants HL-085352 (S. A. Gupte) and HL-112793 and HL-117907 (J. M. Miano).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.C., S.R.J., and V.D. performed experiments; S.C., S.R.J., V.D., J.M.M., and S.A.G. analyzed data; S.C., S.R.J., V.D., and A.A.I. prepared figures; S.C., S.R.J., V.D., and S.A.G. drafted manuscript; S.C., S.R.J., A.A.I., T.M.L., R.G., J.M.M., and S.A.G. edited and revised manuscript; S.C., S.R.J., V.D., A.A.I., T.M.L., R.G., J.M.M., and S.A.G. approved final version of manuscript; T.M.L., R.G., J.M.M., and S.A.G. conception and design of research; T.M.L., R.G., J.M.M., and S.A.G. interpreted results of experiments.
ACKNOWLEDGMENTS
Present address of S. Chettimada: Cancer Immunology & Virology, Dana Farber Cancer Institute, Harvard Medical School, Boston, MA.
REFERENCES
- 1.Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 74: 13–40, 2012. [DOI] [PubMed] [Google Scholar]
- 2.Ata H, Rawat DK, Lincoln T, Gupte SA. Mechanism of glucose-6-phosphate dehydrogenase-mediated regulation of coronary artery contractility. Am J Physiol Heart Circ Physiol 300: H2054–H2063, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Autieri MV. Kruppel-like factor 4: transcriptional regulator of proliferation, or inflammation, or differentiation, or all three? Circ Res 102: 1455–1457, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballantyne MD, Pinel K, Dakin R, Vesey AT, Diver L, Mackenzie R, Garcia R, Welsh P, Sattar N, Hamilton G, Joshi N, Dweck MR, Miano JM, McBride MW, Newby DE, McDonald RA, Baker AH. Smooth muscle enriched long non-coding RNA (SMILR) regulates cell proliferation. Circulation 133: 2050–2065, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell RD, Long X, Lin M, Bergmann JH, Nanda V, Cowan SL, Zhou Q, Han Y, Spector DL, Zheng D, Miano JM. Identification and initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler Thromb Vasc Biol 34: 1249–1259, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benson CC, Zhou Q, Long X, Miano JM. Identifying functional single nucleotide polymorphisms in the human CArGome. Physiol Genomics 43: 1038–1048, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butt E, Eigenthaler M, Genieser HG. (Rp)-8-pCPT-cGMPS, a novel cGMP-dependent protein kinase inhibitor. Eur J Pharmacol 269: 265–268, 1994. [DOI] [PubMed] [Google Scholar]
- 8.Campbell GR, Campbell JH. Smooth muscle phenotypic changes in arterial wall homeostasis: implications for the pathogenesis of atherosclerosis. Exp Mol Pathol 42: 139–162, 1985. [DOI] [PubMed] [Google Scholar]
- 9.Campbell JH, Campbell GR. Smooth muscle phenotypic modulation—a personal experience. Arterioscler Thromb Vasc Biol 32: 1784–1789, 2012. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol 34: 1345–1356, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Chettimada S, Ata H, Rawat DK, Gulati S, Kahn AG, Edwards JG, Gupte SA. Contractile protein expression is up-regulated by reactive oxygen species in aorta of Goto-Kakizaki rat. Am J Physiol Heart Circ Physiol 306: H214–H224, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chettimada S, Joshi SR, Alzoubi A, Gebb SA, McMurtry IF, Gupte R, Gupte SA. Glucose-6-phosphate dehydrogenase plays a critical role in hypoxia-induced CD133+ progenitor cells self-renewal and stimulates their accumulation in the lungs of pulmonary hypertensive rats. Am J Physiol Lung Cell Mol Physiol 307: L545–L556, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chettimada S, Rawat DK, Dey N, Kobelja R, Simms Z, Wolin MS, Lincoln TM, Gupte SA. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol 303: L64–L74, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi C, Sellak H, Brown FM, Lincoln TM. cGMP-dependent protein kinase and the regulation of vascular smooth muscle cell gene expression: possible involvement of Elk-1 sumoylation. Am J Physiol Heart Circ Physiol 299: H1660–H1670, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460: 705–710, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Creemers EE, Sutherland LB, McAnally J, Richardson JA, Olson EN. Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development 133: 4245–4256, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: P3, 2003. [PubMed] [Google Scholar]
- 18.Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, Burgering BM. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J 23: 4802–4812, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupte RS, Ata H, Rawat D, Abe M, Taylor MS, Ochi R, Gupte SA. Glucose-6-phosphate dehydrogenase is a regulator of vascular smooth muscle contraction. Antioxid Redox Signal 14: 543–558, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupte RS, Rawat DK, Chettimada S, Cioffi DL, Wolin MS, Gerthoffer WT, McMurtry IF, Gupte SA. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem 285: 19561–19571, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupte SA, Wolin MS. Relationships between vascular oxygen sensing mechanisms and hypertensive disease processes. Hypertension 60: 269–275, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imamura M, Long X, Nanda V, Miano JM. Expression and functional activity of four myocardin isoforms. Gene 464: 1–10, 2010. [DOI] [PubMed] [Google Scholar]
- 23.Joshi SR, Alloossh M, Gupte SA. Glucose-6-phosphate dehydrogenase is involved in regulation of arterial smooth muscle phenotype in metabolic syndrome. FASEB J 28: 1076–1075., 2014. [Google Scholar]
- 24.Joshi SR, Comer BS, McLendon JM, Gerthoffer WT. MicroRNA regulation of smooth muscle phenotype. Mol Cell Pharmacol 4: 1–16, 2012. [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi SR, McLendon JM, Comer BS, Gerthoffer WT. MicroRNAs-control of essential genes: implications for pulmonary vascular disease. Pulmon Circ 1: 357–364, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon AT, Arenillas DJ, Worsley Hunt R, Wasserman WW. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 2: 987–1002, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem 280: 9719–9727, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Long X, Tharp DL, Georger MA, Slivano OJ, Lee MY, Wamhoff BR, Bowles DK, Miano JM. The smooth muscle cell-restricted KCNMB1 ion channel subunit is a direct transcriptional target of serum response factor and myocardin. J Biol Chem 284: 33671–33682, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacGregor GA, Fenton S, Alaghband-Zadeh J, Markandu N, Roulston JE, de Wardener HE. Evidence for a raised concentration of a circulating sodium transport inhibitor in essential hypertension. Br Med J 283: 1355–1357, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsui R, Xu S, Maitland KA, Hayes A, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin II. Circulation 112: 257–263, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Miano JM. Myocardin in biology and disease. J Biomed Res 29: 3–19, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miano JM, Carlson MJ, Spencer JA, Misra RP. Serum response factor-dependent regulation of the smooth muscle calponin gene. J Biol Chem 275: 9814–9822, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Miano JM, Long X. The short and long of noncoding sequences in the control of vascular cell phenotypes. Cell Mol Life Sci 72: 3457–3488, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol 292: C70–C81, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Nanda V, Miano JM. Leiomodin 1, a new serum response factor-dependent target gene expressed preferentially in differentiated smooth muscle cells. J Biol Chem 287: 2459–2467, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev 20: 1545–1556, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Rawat DK, Alzoubi A, Gupte R, Chettimada S, Watanabe M, Kahn AG, Okada T, McMurtry IF, Gupte SA. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 64: 1266–1274, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Rawat DK, Hecker P, Watanabe M, Chettimada S, Levy RJ, Okada T, Edwards JG, Gupte SA. Glucose-6-phosphate dehydrogenase and NADPH redox regulates cardiac myocyte L-type calcium channel activity and myocardial contractile function. PLoS One 7: e45365, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serpillon S, Floyd BC, Gupte RS, George S, Kozicky M, Neito V, Recchia F, Stanley W, Wolin MS, Gupte SA. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart Circ Physiol 297: H153–H162, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Q, Taurin S, Sethakorn N, Long X, Imamura M, Wang DZ, Zimmer WE, Dulin NO, Miano JM. Myocardin-dependent activation of the CArG box-rich smooth muscle gamma-actin gene: preferential utilization of a single CArG element through functional association with the NKX3.1 homeodomain protein. J Biol Chem 284: 32582–32590, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res 109: 880–893, 2011. [DOI] [PubMed] [Google Scholar]
- 43.Turner EC, Huang CL, Govindarajan K, Caplice NM. Identification of a Klf4-dependent upstream repressor region mediating transcriptional regulation of the myocardin gene in human smooth muscle cells. Biochim Biophys Acta 1829: 1191–1201, 2013. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428: 185–189, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci USA 100: 7129–7134, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie C, Huang H, Sun X, Guo Y, Hamblin M, Ritchie RP, Garcia-Barrio MT, Zhang J, Chen YE. MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem Cells Dev 20: 205–210, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida T, Gan Q, Owens GK. Kruppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol 295: C1175–C1182, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuen S, Ogut O, Brozovich FV. MYPT1 protein isoforms are differentially phosphorylated by protein kinase G. J Biol Chem 286: 37274–37279, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou W, Negash S, Liu J, Raj JU. Modulation of pulmonary vascular smooth muscle cell phenotype in hypoxia: role of cGMP-dependent protein kinase and myocardin. Am J Physiol Lung Cell Mol Physiol 296: L780–L789, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]