

In prediabetes induced by chronic high-fat diet and a low single dose of streptozotocin in rats, mild diastolic dysfunction and ventricular hypertrophy are observed. Elevated cardiac lipid accumulation, subsarcolemmal mitochondrial reactive oxygen species production, and early changes in cardiac mitophagy may be responsible for cardiac effects of prediabetes.
Keywords: obesity, type 2 diabetes, high-fat diet, reactive oxygen species, diabetic cardiomyopathy
Abstract
Although incidence and prevalence of prediabetes are increasing, little is known about its cardiac effects. Therefore, our aim was to investigate the effect of prediabetes on cardiac function and to characterize parameters and pathways associated with deteriorated cardiac performance. Long-Evans rats were fed with either control or high-fat chow for 21 wk and treated with a single low dose (20 mg/kg) of streptozotocin at week 4. High-fat and streptozotocin treatment induced prediabetes as characterized by slightly elevated fasting blood glucose, impaired glucose and insulin tolerance, increased visceral adipose tissue and plasma leptin levels, as well as sensory neuropathy. In prediabetic animals, a mild diastolic dysfunction was observed, the number of myocardial lipid droplets increased, and left ventricular mass and wall thickness were elevated; however, no molecular sign of fibrosis or cardiac hypertrophy was shown. In prediabetes, production of reactive oxygen species was elevated in subsarcolemmal mitochondria. Expression of mitofusin-2 was increased, while the phosphorylation of phospholamban and expression of Bcl-2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3, a marker of mitophagy) decreased. However, expression of other markers of cardiac auto- and mitophagy, mitochondrial dynamics, inflammation, heat shock proteins, Ca2+/calmodulin-dependent protein kinase II, mammalian target of rapamycin, or apoptotic pathways were unchanged in prediabetes. This is the first comprehensive analysis of cardiac effects of prediabetes indicating that mild diastolic dysfunction and cardiac hypertrophy are multifactorial phenomena that are associated with early changes in mitophagy, cardiac lipid accumulation, and elevated oxidative stress and that prediabetes-induced oxidative stress originates from the subsarcolemmal mitochondria.
NEW & NOTEWORTHY
In prediabetes induced by chronic high-fat diet and a low single dose of streptozotocin in rats, mild diastolic dysfunction and ventricular hypertrophy are observed. Elevated cardiac lipid accumulation, subsarcolemmal mitochondrial reactive oxygen species production, and early changes in cardiac mitophagy may be responsible for cardiac effects of prediabetes.
type 2 diabetes mellitus is a common civilization disease with a growing prevalence worldwide (2, 68, 81). It is well established that type 2 diabetes mellitus is a risk factor of cardiovascular diseases, such as heart failure and myocardial infarction, contributing to their increased morbidity and mortality (6, 68). However, before the development of overt diabetes, a period of prediabetic state (i.e., impaired glucose and insulin tolerance, insulin and leptin resistance, oscillations of normo- and hyperglycemic states, mild to moderate obesity) occurs (54), which may also promote cardiovascular complications (21, 30, 45). Although cardiac pathophysiological alterations are relatively well characterized in fully developed diabetes (i.e., diabetic cardiomyopathy), information about prediabetes is quite limited. It has been reported that prediabetes induces mild diastolic dysfunction in OLETF rats, which is a genetic model for spontaneous long-term hyperglycemia (51); however, cardiac consequences of prediabetes and their molecular mechanism are unknown in nongenetic prediabetic settings.
Contractile dysfunction in diabetic cardiomyopathy has been attributed to numerous factors and pathways [i.e., increased oxidative stress or activated apoptosis (13, 78), which could be connected to an impaired mitochondrial function (24), autophagy (41, 78), or to an imbalance in the calcium homeostasis (60)]. Although these pathways are well studied in diabetes, their role in prediabetes has not been uncovered. Furthermore, because mitochondrial function is heavily influenced by mitochondrial dynamics including mitochondrial biogenesis, fusion, fission, and autophagy-mitophagy and because these processes have been linked to the development of diabetic cardiomyopathy (13, 32, 41, 78), we hypothesized that altered mitochondrial dynamics might be involved in the mechanism of deteriorated cardiac functions in prediabetes. Moreover, development of diabetes leads to systemic sensory neuropathy that has been shown to result in diastolic dysfunction in the rat heart (7, 86).
Therefore, here we aimed to systematically characterize the cardiac effect of prediabetes on functional, morphological, and molecular levels in a nongenetic rodent model.
MATERIALS AND METHODS
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996) and was approved by the animal ethics committee of the Semmelweis University, Budapest, Hungary (registration numbers: XIV-I-001/450-6/2012). Chemicals were purchased from Sigma (St. Louis, MO) unless otherwise noted.
Animal model and experimental design.
Male Long-Evans rats of 5–7 wk of age were purchased from Charles River Laboratories (Wilmington, MA). Animals were housed in a room maintained in a 12-h:12-h light/dark cycle and constant temperature of 21°C. Animals were allowed access to food and water ad libitum. After 1 wk of acclimatization, rats were divided into two groups: control (CON; n = 20) and prediabetic group (PRED; n = 20) (Fig. 1). The control group was fed control chow, whereas the prediabetic group was fed a chow supplemented with 40% lard as a high-fat diet. Body weights were measured weekly. Blood was taken, and fasting blood glucose levels were measured from the saphenous vein every second week with a blood glucose monitoring system (Accu-Check; Roche, Basel, Switzerland). To facilitate the development of prediabetes and to avoid hypoinsulinemia, animals on high-fat diet received 20 mg/kg streptozotocin (STZ; Santa Cruz Biotechnology, Dallas, TX) intraperitoneally (i.p.) at the fourth week of the diet according to Mansor et al. (50), whereas the control group was treated with the same volume of ice-cold citrate buffer as vehicle. At the 20th wk, oral glucose tolerance test (OGTT) was performed in overnight fasted rats with per os administration of 1.5 g/kg glucose and measurements of plasma glucose levels at 15, 30, 60, and 120 min. Insulin tolerance test (ITT) was also performed at the 20th wk in overnight fasted rats. Insulin (0.5 IU/kg, Humulin R; Ely Lilly, Utrecht, The Netherlands) was injected i.p., and plasma glucose levels were checked at 15, 30, 45, 60, 90, and 120 min. At week 21 of the diet, animals were anesthetized with pentobarbital (60 mg/kg, i.p.; Euthasol; Produlab Pharma, Raamsdonksveer, The Netherlands). Echocardiography and cardiac catheterization were performed, and then hearts were excised, shortly perfused with oxygenated Krebs-Henseleit buffer in Langendorff mode as described earlier, and weighed. Epididymal and interscapular brown fat tissue, which are the markers of adiposity (9, 34), were isolated, and their weights were measured. Blood and tissue samples were collected and stored at −80°C.
Fig. 1.

Experimental protocol. Long-Evans rats were fed with either control (CON) diet for 21 wk, or with high-fat diet and treated with 20 mg/kg streptozotocin (STZ) at week 4 (PRED) to induce prediabetes. Body weights were measured weekly, and blood samples were taken from the saphenous vein every second week. Sensory neuropathy was measured at week 15. Oral glucose tolerance test (OGTT), insulin tolerance test (ITT), and computer tomography (CT) were performed at week 20. Echocardiography, hemodynamic analysis, and parameters of mitochondrial function were measured at week 21 of diet. Tissue sampling was performed after terminal procedures.
Assessment of sensory neuropathy.
To test whether sensory neuropathy develops in prediabetes, plantar Von Frey test was performed. At week 15 of the diet, rats were placed in a plastic cage with a wire mesh bottom to allow full access to the paws. After 5–10-min acclimation time, mechanical hind paw withdrawal thresholds were measured by a dynamic plantar aesthesiometer (UGO-Basile, Monvalle, Italy) as previously described (56).
Evaluation of body fat content.
At week 20 of the diet, computed tomography (CT) measurements were performed on NanoSPECT/CT PLUS (Mediso, Budapest, Hungary). The semicircular CT scanning was acquired with a 55-kV tube voltage, 500-ms exposure time, 1:4 binning, and 360 projections in 18 min, 7 s. During the acquisitions, rats were placed in a prone position in a dedicated rat bed and were anesthetized with 2% isoflurane in oxygen. Temperature of the animals was kept at 37.2 ± 0.3°C during imaging. In the reconstruction, 0.24-mm in-plane resolution and slice thickness were set, and Butterworth filter was applied (volume size: 76.8 × 76.8 × 190 mm). Images were further analyzed with VivoQuant (inviCRO, Boston, MA) dedicated image analysis software products by placing appropriate volumes of interest (VOI) on the whole body fat of animals. The aim of segmentation was to separate the fat from other tissues. The connected threshold method helped to choose the adequate attenuated pixels for fat tissue analysis, and then the isolated points were detected by erode 4 voxel and dilate 4 voxel steps. After the measurements, animals recovered from anesthesia.
Cardiac function by echocardiography.
Before euthanasia, to measure cardiac function, echocardiography was performed as previously described (42, 64). Briefly, anesthetized animals were placed on a controlled heating pad, and the core temperature, measured via rectal probe, was maintained at 37°C. Transthoracic echocardiography was performed with animals in the supine position by one investigator blinded to the experimental groups. Two-dimensional and M-mode echocardiographic images of long and short (midpapillary muscle level) axis were recorded, using a 13-MHz linear transducer (GE 12L-RS; GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom), connected to an echocardiographic imaging unit (Vivid i, GE Healthcare). The digital images were analyzed by a blinded investigator using an image analysis software (EchoPac, GE Healthcare). On two-dimensional recordings of the short axis at the midpapillary level, left ventricular (LV) anterior (LVAWT) and posterior wall thickness (LVPWT) in diastole (index: d) and systole (index: s) as well as LV end-diastolic (LVEDD) and end-systolic diameter (LVESD) were measured. In addition, end-diastolic and end-systolic LV areas were planimetered from two-dimensional recordings of short and long axis. End-systole was defined as the time point of minimal LV dimensions and end-diastole as the time point of maximal dimensions. All values were averaged over three consecutive cycles. The following parameters were derived from these measurements (65). Fractional shortening (FS) was calculated as [(LVEDD − LVESD)/LVEDD] × 100. LV mass was calculated according to the following formula: [LVmass = (LVEDD + AWTd + PWTd)3 − LVEDD × 3]1.04 × 0.8 + 0.14.
Hemodynamic measurements, LV pressure-volume analysis.
After echocardiographic measurements, hemodynamic measurement was performed as previously described (62, 63). Briefly, rats were tracheotomized, intubated, and ventilated, while core temperature was maintained at 37°C. A median laparotomy was performed. A polyethylene catheter was inserted into the left external jugular vein. A 2-Fr microtip pressure-conductance catheter (SPR-838; Millar Instruments, Houston, TX) was inserted into the right carotid artery and advanced into the ascending aorta. After stabilization for 5 min, mean arterial blood pressure (MAP) was recorded. After that, the catheter was advanced into the LV under pressure control. After stabilization for 5 min, signals were continuously recorded at a sampling rate of 1,000/s using a pressure-volume (P-V) conductance system (MPVS-Ultra, Millar Instruments) connected to the PowerLab 16/30 data acquisition system (AD Instruments, Colorado Springs, CO), stored, and displayed on a personal computer by the LabChart5 Software System (AD Instruments). After positioning the catheter, we registered baseline P-V loops. With the use of a special P-V analysis program (PVAN, Millar Instruments), LV end-systolic pressure, LV end-diastolic pressure, the maximal slope of LV systolic pressure increment (dP/dtmax) and diastolic pressure decrement, time constant of LV pressure decay (τ; according to the Glantz method), ejection fraction (EF), stroke work (SW), and LV maximal power were computed and calculated. Stroke volume and cardiac output (CO) were calculated and corrected according to in vitro and in vivo volume calibrations using the PVAN software. Total peripheral resistance (TPR) was calculated by the following equation: TPR = MAP/CO. In addition to the above parameters, P-V loops recorded at different preloads can be used to derive other useful systolic function indexes that are less influenced by loading conditions and cardiac mass (37, 58). Therefore, LV P-V relations were measured by transiently compressing the inferior vena cava (reducing preload) under the diaphragm with a cotton-tipped applicator. The slope of the LV end-systolic P-V relationship (according to the parabolic curvilinear model), preload recruitable stroke work, and the slope of the dP/dtmax-end-diastolic volume (EDV) relationship (dP/dtmax-EDV) were calculated as load-independent indexes of LV contractility. The slope of the LV end-diastolic P-V relationship (EDPVR) was calculated as a reliable index of LV stiffness (37). At the end of each experiment, 100 μl of hypertonic saline was injected intravenously, and, from the shift of P-V relations, parallel conductance volume was calculated by the software and used for the correction of the cardiac mass volume. The volume calibration of the conductance system was performed as previously described (37).
Adipokine array from rat plasma.
Adipokine array was performed from 1 ml rat plasma according to manufacturer's instructions (Proteome Profiler Rat Adipokine Array Kit; R&D Systems, Abingdon, United Kingdom).
Biochemical measurements.
Serum cholesterol, high-density lipoprotein (HDL), and triglyceride levels were measured by colorimetric assays (Diagnosticum, Budapest, Hungary) as previously described (19). Plasma leptin (Invitrogen, Camarillo, CA), tissue inhibitor of matrix metalloprotease (TIMP) metallopeptidase inhibitor 1 (TIMP-1; R&D System, Minneapolis, MN), and angiotensin-II (Phoenix Pharmaceuticals, Karlsruhe, Germany) were measured by ELISA according to manufacturer's instructions. Urea, glutamate oxaloacetate transaminase, glutamate pyruvate transaminase, low-density lipoprotein, C-reactive protein (CRP), cholesterol, uric acid, and creatinine were measured by automated clinical laboratory assays (Diagnosticum).
Histology.
Heart, liver, and pancreas samples were fixed in 4% neutral-buffered formalin. After 24 h, samples were washed with PBS and stored in 70% ethanol in PBS until embedded in paraffin. Samples were stained with hematoxylin-eosin (HE) and Masson's trichrome (MA) staining. LV samples were analyzed to examine histopathological differences and evaluate cardiomyocyte hypertrophy and fibrosis. The level of fibrosis was measured on MA-stained LV sections, and transverse transnuclear width (cardiomyocyte diameter) was assessed on longitudinally oriented cardiomyocytes on HE-stained LV sections by a Zeiss microscope (Carl Zeiss, Jena, Germany). Digital images were acquired using an imaging software (QCapture Pro 6.0; QImaging, Surrey, British Columbia, Canada) at ×200 magnification. Quantification of cardiomyocyte diameter and fibrosis was performed with ImageJ Software (v1.48; NIH, Bethesda, MD). Liver samples were evaluated for hepatic steatosis/fibrosis and scored as previously described (40).
Nitrotyrosine immunostaining of LV samples.
After embedding and cutting 5-μm-thick sections, heat-induced antigen epitope retrieval was performed (95°C, 10 min, in citrate buffer with a pH of 6.0). Sections were stained with rabbit polyclonal anti-nitrotyrosine antibody (5 μg/ml; Cayman Chemical, Ann Arbor, MI) by using the ABC kit of Vector Laboratories (Burlingame, CA) according to the manufacturer's protocol. Nitrotyrosine-stained sections were counterstained with hematoxylin. Specific staining was visualized, and images were acquired using a BX-41 microscope (Olympus, Tokyo, Japan).
Quantitative RT-PCR.
Total RNA was isolated from LV tissue with Reliaprep RNA Tissue Miniprep kit (Promega, Madison, WI) according to the manufacturer's instructions. cDNA was synthesized using Tetro cDNA Synthesis Kit (Bioline, London, UK) according to the manufacturer's protocol. PCR reaction was performed with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) or TaqMan Universal PCR MasterMix (Thermo Fisher Scientific, Waltham, MA) and 3 nM forward and reverse primers for collagen type 1 and 3 (COL1 and COL3), atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) (Integrated DNA Technologies, Leuven, Belgium), assay mixes for α-myosin heavy chain (α-MHC, assay ID: Rn00691721_g1), β-MHC (assay ID: Rn00568328_m1), TNF-α (assay ID: Rn99999017_m1), and IL-6 (assay ID: Rn01410330_m1, Thermo Fisher Scientific) were used. β2-Microglobulin or GAPDH (assay ID: Rn01775763_g1) were used as reference genes. Quantitative real-time PCR was performed with the StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). Expression levels were calculated using the CT comparative method (2−ΔCT).
Measurement of pancreatic insulin.
Freeze-clamped and pulverized pancreas samples were used to determine pancreatic insulin content. Analysis was performed with insulin (I-125) IRMA Kit (Izotop Kft, Budapest, Hungary) according to the manufacturer's instructions.
Electron microscopy.
LV tissue samples (1 × 1 mm) were placed in modified Kranovsky fixative (2% paraformaldehyde, 2.5% glutaraldehyde, 0.1 M Na-cacodylate buffer, pH 7.4, and 3 mM CaCl2). After being washed in cacodylate buffer, samples were incubated in 1% osmium tetroxide in 0.1 M PBS for 35 min. Samples were then washed in buffer several times for 10 min and dehydrated in an ascending ethanol series, including a step of uranyl acetate (1%) solution in 70% ethanol to increase contrast. Dehydrated blocks were transferred to propylene oxide before being placed into Durcupan resin. Blocks were placed in a thermostat for 48 h at 56°C. From the embedded blocks, 1-μm-thick semithin and serial ultrathin sections (70 nm) were cut with a Leica ultramicrotome and mounted either on mesh or on Collodion-coated (Parlodion; Electron Microscopy Sciences, Fort Washington, PA) single-slot copper grids. Additional contrast was provided to these sections with uranyl acetate and lead citrate solutions, and they were examined with a JEOL1200EX-II electron microscope. Areas of subsarcolemmal (SSM), interfibrillar mitochondria (IFM), and lipid droplets were measured by freehand polygon selection in iTEM Imaging Platform.
Mitochondrial enzyme activity measurements.
Fresh myocardial samples were homogenized in 1/30 weight per volume Chappel-Perry buffer (100 mM KCl, 5 mM MgCl2, 1 mM EDTA, 50 mM Tris, pH 7.5) supplemented with 15 mg/l trypsin inhibitor, 15.5 mg/l benzamidine, 5 mg/l leupeptin, and 7 mg/l pepstatin A. All enzyme activities were measured as duplicates with a photometer (Cary 50 Scan UV-Visible Spectrophotometer; Agilent Technologies, Palo Alto, CA). Before we added substrate or cofactor, the reaction mix was incubated at 30°C for 10 min (except for cytochrome c oxidase). Enzyme activities were expressed relative to citrate synthase activity or total protein levels (measured with bicinchoninic acid assay). The activity of rotenone-sensitive NADH:ubiquinone-oxidoreductase (Complex I) was measured at 340 nm in the presence of 1 mM EDTA, 2.5 mM KCN, 1 μM antimycin A, and 20 μM rotenone after the addition of coenzyme Q and NADH to a final concentration of 60 μM. The activity of NADH:cytochrome c-oxidoreductase (Complex I+III) was measured at 550 nm as the antimycin A- and rotenone-sensitive fraction of total NADH-cytochrome c oxidoreductase in the presence of 0.1 mM EDTA, 3 mM KCN, and 0.1% cytochrome c after adding NADH to a final concentration of 0.2 mM. The activity of succinate:cytochrome c-oxidoreductase (Complex II+III) was measured at 550 nm in the presence of 0.1 mM EDTA, 2.5 mM KCN, 0.1% bovine serum albumin, and 4 mM succinate after the addition of cytochrome c to a final concentration of 0.1%. The activity of succinate-dehydrogenase was measured at 600 nm in the presence of 0.1 mM EDTA, 2.5 mM KCN, 0.1% bovine serum albumin, and 2 mM succinate after the addition of 2,6-dichloroindophenol and phenazine-methosulfate to a final concentration of 34.900 μM and 1.625 mM, respectively. The activity of cytochrome c-oxidase was measured at 550 nm in the presence of 0.08% reduced cytochrome c. The activity of citrate synthase was measured at 412 nm in the presence of 0.1% Triton X-100, 0.1 mM 5,5′-dithiobis (2-nitrobenzoic acid), and 0.1 mM acetyl-coenzyme A after the addition of oxaloacetate to a final concentration of 0.5 mM.
Preparation of isolated mitochondria.
SSM and IFM fractions were isolated according to a protocol described previously (74) with the use of homogenization buffer (buffer A) containing the following (in mM): 100 KCl, 50 MOPS, 5 MgSO4, 1 EGTA, and pH 7.4 Tris·HCl. Isolation buffer (buffer B) contained the following (in mM): 250 sucrose, 10 HEPES, 1 EGTA, and pH 7.4 Tris·HCl. Before the isolation, 1 mM ATP was added freshly to the homogenization buffer. All steps were carried out on ice. After Langendorff perfusion of the heart, LV samples were cut to small species with scissors and washed in buffer A, then homogenized with five strokes of Teflon pistils in a glass potter. The homogenate was centrifuged for 10 min at 800 g, 4°C. For isolation of SSM, the supernatant was centrifuged for 10 min at 8,000 g. This pellet was suspended in buffer A and centrifuged for 10 min at 8,000 g, and the resulting sediment was resuspended in a small volume of buffer A. The pellet of the first centrifugation was used for isolation of IFM fraction and resuspended in buffer B (10 ml/g tissue) and after addition of 8 U/g of bacterial protease incubated for 1 min on ice and then homogenized with five strokes of Teflon pistil in a glass potter and centrifuged for 10 min at 800 g. The supernatant was centrifuged for 10 min at 8,000 g, and the resulting mitochondrial pellet was finally resuspended in buffer A and used for mitochondrial respiration, membrane potential, H2O2 production, and Ca2+ uptake measurements. For Western blots, the resulting SSM and IFM pellets were finally resuspended in a 200 μl volume of Buffer B, which were layered on 30% Percoll solution and ultracentrifuged (Rotor type: Beckman Type 70.1 Ti) for 30 min at 18,700 g at 4°C. After ultracentrifugation, lower rings were collected (100 μl/tube) and filled with 1 ml Buffer B and centrifuged for 10 min at 12,200 g, 4°C. After being washed, pellets were stored at −80°C.
Measurement of mitochondrial respiration.
Protein concentration of SSM and IFM samples was determined by the Biuret method (11). Mitochondrial oxygen consumption was measured by high-resolution respirometry with Oxygraph-2K (Oroboros Instruments, Innsbruck, Austria), a Clark-type O2 electrode, for 40 min. The mitochondrial protein content was 0.1 mg/ml in the measurements. Measuring mitochondrial respiration followed the substrate-uncoupler-inhibitor titration protocol. Mitochondria were energized with 5 mM glutamate and 5 mM malate. Mitochondrial respiration was initiated with 2 mM adenosine diphosphate. Cytochrome c (4 μM), succinate (5 mM), rotenone (1 μM), and carboxyatractyloside (2 μM) were used as indicated. Measurements were performed in an assay medium containing 125 mM KCl, 20 mM HEPES, 100 μM EGTA, 2 mM K2HPO4, 1 mM MgCl2, and 0.025% BSA. Data were digitally recorded using DatLab4 software.
Measurement of mitochondrial membrane potential.
To detect mitochondrial membrane potential, we used the fluorescent, cationic dye, safranine O (2 μM), which can bind to the protein possessing negative charge in the inner mitochondrial membrane depending on the mitochondrial membrane potential. The excitation/emission wavelengths were 495/585 nm, respectively. Fluorescence was recorded at 37°C by Hitachi F-4500 spectrofluorometer (Hitachi High Technologies, Maidenhead, UK). The reaction medium was the following: 125 mM KCl, 20 mM HEPES, 100 μM EGTA, 2 mM K2HPO4, 1 mM MgCl2, and 0.025% BSA.
Detection of H2O2 formation in mitochondria.
H2O2 production of SSM and IFM was assessed by Amplex UltraRed fluorescent dye method (52). Horseradish peroxidase (2.5 U/ml) and Amplex UltraRed reagent (1 μM) and then 0.05 mg/ml mitochondria were added to the incubation medium. H2O2 formation was initiated by the addition of 5 mM glutamate and 5 mM malate or 5 mM succinate, and fluorescence was detected at 37°C with Deltascan fluorescence spectrophotometer (Photon Technology International, Lawrenceville, NJ). The excitation wavelength was 550 nm, and the fluorescence emission was detected at 585 nm. A calibration signal was generated with known quantities of H2O2 at the end of each experiment.
Measurement of Ca2+ uptake in mitochondria.
The free Ca2+ concentration at each added concentration of Ca2+ was calculated and measured. Ca2+ uptake by mitochondria was followed by measuring calcium-green-5N (100 nM) fluorescence at 505 nm excitation and 535 emission wavelengths at 37°C using a Hitachi F-4500 spectrofluorometer (Hitachi High Technologies). The reaction medium was the following: 125 mM KCl, 20 mM HEPES, 100 μM EGTA, 2 mM K2HPO4, 1 mM MgCl2, and 0.025% BSA.
Western blot of LV lysates and isolated mitochondria fractions.
Freeze-clamped LVs were pulverized under liquid nitrogen and homogenized in homogenization buffer containing (in mmol/l) 20 Tris·HCl, 250 sucrose, 1.0 EGTA, and 1.0 dithiothreitol or in radioimmunoprecipitation assay buffer (Cell Signaling Technology, Danvers, MA), supplemented with 1 mM PMSF (Roche, Basel, Switzerland), 0.1 mM sodium fluoride, 200 mM sodium orthovanadate, and complete protease inhibitor cocktail (Roche) with TissueLyser LT (Qiagen, Venlo, The Netherlands) to obtain LV soluble protein fraction or LV whole cell lysate. Previously isolated mitochondrial samples were resuspended in ice-cold 1× cell lysis buffer (Cell Signaling Technology). Concentration of proteins was assessed with Lowry's assay or bicinchoninic acid assay kit (Thermo Fisher Scientific).
For tropomyosin oxidation analysis, tissue samples were homogenized in ice-cold PBS, pH 7.2, containing an antiprotease mixture (Complete, Roche) and 5 mM EDTA. Just before use, the protein samples were stirred under vacuum and bubbled with argon to maximally reduce the oxygen tension. The protein suspension was centrifuged at 12,000 g for 10 min at 4°C. The resulting pellet was resuspended in sample buffer (2% SDS, 5% glycerol, 1% β-mercaptoethanol, 125 mM Tris·HCl, pH 6.8) and denatured by 10 min of boiling. This procedure referred to as reducing condition was compared with the nonreducing condition obtained without the addition of β-mercaptoethanol. To avoid artifacts attributable to the oxidation of thiol groups in vitro, nonreducing conditions were performed in the presence of 1 mM N-ethylmaleimide.
Protein samples were resolved on precast 4–20% Criterion TGX gels (Bio-Rad) or Bis-Tris gels depending on the protein of interest and transferred to nitrocellulose or Immun-Blot PVDF membranes (Bio-Rad). Quality of transfer was verified with Ponceau S staining. Membranes were blocked with 5% nonfat milk (Bio-Rad) or 2–5% BSA (Santa Cruz Biotechnology) in Tris-buffered saline with 0.05% Tween 20 (TBS-T) for 0.5–2.0 h. Membranes were incubated with primary antibodies in 1–5% nonfat milk or BSA in TBS-T: anti-tropomyosin (Tm; 1:250), anti-phospho-phospholamban (PLB-Ser16; 1:5,000), p-PLB (Thr17; 1:5,000), anti-sarco/endoplasmic reticulum Ca2+-ATPase II (SERCA2A; 1:5,000; Badrilla, Leeds, United Kingdom), anti-heat shock protein-60 (HSP-60; 1:500), anti-HSP-70 (1:500), anti-HSP-90 (1:500), anti-B-cell lymphoma 2 (Bcl-2; 1:500), anti-caspase-3 (1:500), anti-Ca2+/calmodulin-dependent protein kinase II (CaMKIIδ; 1:2,000), anti-Parkin (1:5,000, Santa Cruz Biotechnology), anti-Shc (1:1,000), anti-dynamin-related/like protein 1 (DRP1/DLP1; 1:5,000), anti-optic atrophy 1 protein (OPA1; 1:2,500, BD Biosciences, Franklin Lakes, NJ), anti-mitofusin-2 (MFN2; 1:2,500, Abcam, Cambridge, UK), anti-phospho-CaMKIIδ (Thr287; 1:2,000), anti-phospho-HSP-27 (Ser82; 1:1,000), anti-HSP-27 (1:1,000), anti-Bax (1:1,000), anti-sequestosome 1 (SQSTM1/p62; 1:1,000), anti-microtubule-associated protein 1 light chain 3 A/B (LC3 A/B; 1:5,000), anti-Beclin-1 (1:1,000), anti-Bcl-2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3; 1:5,000), anti-phospho-Akt (Ser473; 1:1,000), anti-Akt (1:1,000), anti-phospho-AMP-activated protein kinase α (AMPKα-Thr172; 1:1,000), anti-AMPKα (1:1,000), anti-phospho-ribosomal S6 (Ser235/236; 1:1,000), anti-ribosomal S6 (1:1,000), anti-phospho-glycogen synthase kinase-3β (GSK3β-Ser9; 1:1,000), anti-GSK3β (1:1,000), and anti-GAPDH (1:5,000) as loading control (Cell Signaling Technology). For isolated mitochondria, the following primary antibodies were used in 5% nonfat milk in TBS-T: anti-OPA1 (1:2,500) from BD Bioscience, anti-SQSTM1/p62 (1:1,000), anti-LC3 A/B (1:1,000), and anti-cytochrome c oxidase subunit 4 (COX4; 1:5,000) as loading control from Cell Signaling Technology. After three washes with TBS-T, horseradish peroxidase-conjugated secondary antibody was added for 2 h at room temperature (1:5,000 in 5% nonfat milk in TBS-T). Signals were detected with an enhanced chemiluminescence kit (Bio-Rad) by Chemidoc XRS+ (Bio-Rad). For the analysis of tropomyosin oxidation, the density of the additional band with higher molecular weight reflecting the formation of disulfide cross bridges was normalized to densitometric values of the respective tropomyosin monomer. Antibodies against phosphorylated epitopes were removed with Pierce Stripping Buffer (Thermo Fisher Scientific) before incubation with antibodies detecting the total protein.
Statistical analysis.
Values are expressed as means ± SE. Statistical analysis was performed between groups by unpaired two-tailed t-test or by Mann-Whitney U-test by using GraphPad Prism 6 software. A P < 0.05 value was considered significant.
RESULTS
Moderately increased adiposity in prediabetic animals.
To determine the effect of high-fat diet and the single, low-dose STZ injection, we measured body weight, fat tissue volumes, and plasma lipid parameters. We found that body weights of the prediabetic animals were moderately but statistically significantly elevated from week 9 compared with the control group and that this difference reached 18% at the end of the diet period (Fig. 2, A and B). At week 20, plasma leptin level was significantly increased in prediabetes; however, CRP level was decreased, and plasma cholesterol, HDL cholesterol, triglyceride levels, and parameters of liver and kidney function were unchanged (Table 1). To characterize prediabetes-induced changes in further obesity-related molecules, we performed an adipokine array measurement, which revealed that the circulating level of TIMP-1 might be influenced by prediabetes; however, we could not confirm these results by ELISA (data not shown). CT scan showed that body fat volume of prediabetic rats was substantially increased at the end of the diet (Fig. 2, C and D). Epididymal fat tissue weight, which is an indicator of total body adiposity, was increased in the prediabetic group; however, the weight of interscapular brown adipose tissue was not changed (Fig. 2, G–I). Histological score analysis of HE- and MA-stained liver samples showed the development of hepatic steatosis in the prediabetic group (CON: 0.5 ± 0.3 vs. PRED: 2.25 ± 0.5; P < 0.05); however, no signs of hepatic fibrosis were detected (Fig. 2H). Furthermore, electron microscopy showed an increased number of lipid droplets in the myocardium of prediabetic animals compared with controls (Fig. 2, E and F). These results demonstrated a moderately increased adiposity and hepatic and cardiac fat deposits without signs of hyperlipidemia in the prediabetic group.
Fig. 2.
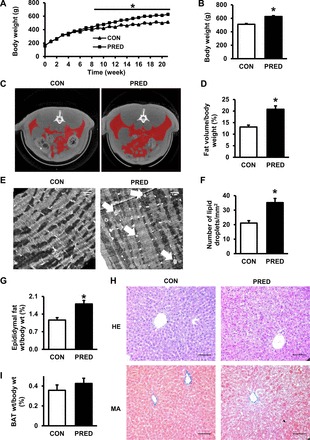
High-fat feeding with a single low-dose STZ treatment increased adiposity. Changes in body weight during the experiment (A) and body weight data after 21 wk (B) are shown. Axial representative CT slice shows the middle of the 4th lumbar spine. C: red color indicates the segmented volumes of interest (VOIs) showing the volume of fat in the control (CON) and prediabetic (PRED) rats. D: whole body fat volume:body weight ratio at week 20. Representative transmission electron micrographs of myocardial lipid droplets (E; white arrows) and the number of lipid droplets in CON and PRED cardiomyocytes (F) are shown; magnification ×7,500; scale bar = 1 μm. Epididymal fat tissue (G) and interscapular brown adipose tissue (BAT) (I) weight:body weight ratios are shown. Hematoxylin-Eosin (HE) and Masson's trichrome (MA) staining of liver sections (H) are shown; magnification ×200; scale bar = 100 μm. Data are means ± SE, n = 3–19 per group (*P < 0.05).
Table 1.
Plasma parameters at week 20
| CON | PRED | |
|---|---|---|
| Plasma leptin, ng/ml | 2.51 ± 0.33 | 5.91 ± 0.60* |
| Plasma cholesterol, mmol/l | 1.88 ± 0.06 | 1.72 ± 0.08 |
| HDL cholesterol, mmol/l | 2.75 ± 0.14 | 2.75 ± 0.10 |
| Plasma triglyceride, mmol/l | 1.20 ± 0.10 | 1.31 ± 0.09 |
| LDL cholesterol, mmol/l | 0.44 ± 0.02 | 0.47 ± 0.03 |
| Total cholesterol, mmol/l | 1.61 ± 0.05 | 1.51 ± 0.08 |
| GOT, U/l | 82.00 ± 15.00 | 58.00 ± 4.00 |
| GPT, U/l | 50.00 ± 14.00 | 49.00 ± 5.00 |
| Uric acid, mmol/l | 24.00 ± 4.00 | 16.00 ± 1.00 |
| Creatinine, μmol/l | 46.00 ± 3.00 | 40.00 ± 3.00 |
| CRP, mg/l | 109.56 ± 1.24 | 94.96 ± 3.87* |
Data are means ± SE for 12 rat per group (
P < 0.05).
HDL, high-density lipoprotein; LDL, low-density lipoprotein; GOT glutamate oxaloacetate transaminase; GPT, glutamate pyruvate transaminase; CRP, C-reactive protein; CON, control; PRED, prediabetes group.
Impaired glucose tolerance, insulin resistance, and sensory neuropathy evidence disturbed carbohydrate metabolism in prediabetes.
We aimed to characterize the glucose homeostasis in our rat model of prediabetes. At week 20 of the diet, fasting blood glucose levels were slightly elevated in prediabetes from week 10; however, they remained in the normoglycemic range (Fig. 3, A and B). OGTT and ITT demonstrated impaired glucose tolerance and insulin resistance in the prediabetic group (Fig. 3, C–F); however, there was no difference in pancreatic insulin content (Fig. 3G) or in pancreatic islet morphology (data not shown) between groups. These results demonstrate prediabetic conditions in the present model and reveal that type 1 diabetes did not develop due to the STZ treatment. Sensory neuropathy is a well-accepted accompanying symptom of diabetes (80). Accordingly, here we have found a decrease in the mechanical hind limb withdrawal threshold at week 15 (CON: 48 ± 1 g vs. PRED: 42 ± 2 g; P < 0.05) of diet in the prediabetic group, which indicates a moderate sensory neuropathy in this model of prediabetes.
Fig. 3.
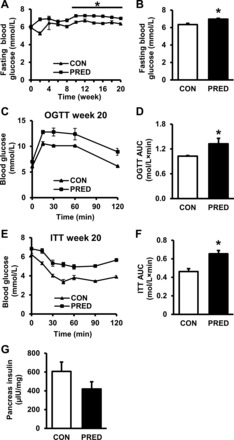
Alterations in glucose homeostasis indicate the development of prediabetes in STZ-treated and high-fat-fed rats at week 21. Fasting blood glucose levels during the experiment (A) and at week 20 (B) are shown. OGTT (C and D) and ITT (E and F) results at week 20 of the diet are shown. Insulin content of pancreas at week 21 (G) is shown. Data are means ± SE, n = 6–19 per group (*P < 0.05).
Diastolic dysfunction and hypertrophy in prediabetes with no sign of fibrosis.
To determine the cardiac effect of prediabetes, we measured morphological and functional parameters of the hearts. Heart weights were significantly increased (Fig. 4A); however, heart weight/body weight ratio was decreased in prediabetes (CON: 0.27 ± 0.01% vs. PRED: 0.24 ± 0.01%; P < 0.05), plausibly attributable to obesity. LV mass, LVAWTs, LVPWTs, and LVPWTd were increased in the prediabetic group as assessed with echocardiography; however, other cardiac dimensional parameters were unchanged (Table 2). The slope of EDPVR, which is a very early and sensitive marker of diastolic dysfunction, was significantly elevated in prediabetes although other hemodynamic parameters, including blood pressure, were unchanged, demonstrating the lack of systolic dysfunction or hypertension (Table 3, Fig. 4B). To uncover the molecular background of the observed mild diastolic dysfunction, we performed measurements on the common mechanistic contributors of heart failure (38). On HE-stained LV sections, increased cardiomyocyte diameter was detected in prediabetes (Fig. 4, C and D). To characterize components affecting diastolic function, we analyzed MHC expression. Interestingly, the gene expression of β-MHC was decreased, and α-MHC also showed a tendency to decrease (P = 0.17), the ratio of which resulted in a strong tendency to decrease in prediabetes. No increase in ANP or BNP gene expressions (Fig. 4, G and H) or in angiotensin-II level (data not shown) was detected in prediabetes. To evaluate the extent of fibrosis, MA-stained LV sections were analyzed, which revealed no difference between groups (Fig. 4E). Similarly, we found that gene expression of type I (COL1) and III (COL3) collagen isoforms were unchanged in the LV (Fig. 4F). These results indicate that mild diastolic dysfunction developed in prediabetic animals, which was associated with a mild hypertrophy (increased LV mass, LVAWT, LVPWT, and cardiomyocyte diameter) without signs of fibrosis.
Fig. 4.

Characterization of cardiac function, myocardial morphology, and fibrosis in prediabetic rats. A: quantification of heart weights after 21 wk. B: representative pressure-volume loops and slope of end diastolic pressure-volume relationship (EDPVR) in CON and PRED group. HE and MA staining of myocardial sections (C), quantification of cardiomyocyte diameter (D), and level of fibrosis (E) in control (CON) and prediabetic (PRED) rats are shown; magnification ×200; scale bar = 100 μm. Quantifications of collagen type I (COL1), COL3 (F), atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) (G), α-myosin heavy chain (α-MHC), β-MHC, and α-:β-MHC ratio (H) in CON and PRED group are shown. Data are means ± SE, n = 5–19 per group (*P < 0.05).
Table 2.
Characterization of cardiac morphology and function in prediabetes by means of echocardiography
| CON | PRED | |
|---|---|---|
| LV mass, g | 1.01 ± 0.04 | 1.22 ± 0.07* |
| LVAWTd, mm | 1.89 ± 0.12 | 2.07 ± 0.12 |
| LVAWTs, mm | 2.86 ± 0.17 | 3.42 ± 0.11* |
| LVPWTd, mm | 1.86 ± 0.07 | 2.05 ± 0.04* |
| LVPWTs, mm | 2.72 ± 0.12 | 3.25 ± 0.15* |
| LVEDD, mm | 7.71 ± 0.22 | 7.75 ± 0.17 |
| LVESD, mm | 4.93 ± 0.22 | 4.96 ± 0.31 |
| FS, % | 36.00 ± 2.20 | 37.40 ± 3.80 |
| HR, 1/min | 335.00 ± 13.00 | 348.00 ± 10.00 |
Data are means ± SE for 10 rat per group (
P < 0.05).
LV, left ventricular; LVAWTs, LV anterior wall thickness, diastolic; LVAWTs, LV anterior wall thickness, systolic; LVPWTs, LV posterior wall thickness, systolic; LVPWTd, LV posterior wall thickness, diastolic; FS%, fractional shortening %; HR, heart rate.
Table 3.
Characterization of LV hemodynamics in vivo in prediabetes by means of pressure-volume analysis
| CON | PRED | |
|---|---|---|
| MAP, mmHg | 110.100 ± 7.300 | 113.600 ± 6.100 |
| LVESP, mmHg | 116.600 ± 5.600 | 120.000 ± 6.800 |
| LVEDP, mmHg | 4.400 ± 0.400 | 4.000 ± 0.200 |
| LVEDV, μl | 292.800 ± 14.500 | 280.200 ± 9.600 |
| LVESV, μl | 130.700 ± 6.900 | 127.500 ± 4.500 |
| SV, μl | 162.100 ± 9.100 | 152.800 ± 7.700 |
| CO, ml/min | 59.500 ± 3.200 | 56.600 ± 2.300 |
| EF, % | 55.300 ± 1.300 | 54.400 ± 1.300 |
| SW, mmHg·ml | 14.500 ± 0.500 | 13.600 ± 0.600 |
| dP/dtmax, mmHg/s | 7226.000 ± 487.000 | 7387.000 ± 401.000 |
| dP/dtmin, mmHg/s | −8198.000 ± 680.000 | −8551.000 ± 545.000 |
| τ (Glantz), ms | 12.600 ± 0.300 | 12.100 ± 0.400 |
| TPR, (mmHg·min)/ml | 1.900 ± 0.190 | 2.000 ± 0.120 |
| Slope of ESPVR, mmHg/μl | 2.680 ± 0.120 | 2.710 ± 0.060 |
| Slope of EDPVR, mmHg/μl | 0.026 ± 0.001 | 0.037 ± 0.004* |
| PRSW, mmHg | 100.500 ± 5.200 | 98.900 ± 4.100 |
| Slope of dP/dtmax-EDV, (mmHg/s)/μl | 34.300 ± 2.300 | 35.200 ± 2.200 |
| Maximal power, mW | 91.800 ± 8.200 | 98.200 ± 12.500 |
Data are means ± SE for 10 rat per group (
P < 0.05).
MAP, mean arterial pressure; LVESP, LV end-systolic pressure; LVEDP, LV end-diastolic pressure; LVESV, LV end-systolic volume; LVEDV, LV end-diastolic volume; SV, stroke volume; CO, cardiac output; EF, ejection fraction; SW, stroke work; dP/dtmax, maximal slope of LV systolic pressure increment; dP/dtmin, maximal slope of LV diastolic pressure decrement; τ, time constant of LV pressure decay; TPR, total peripheral resistance; ESPVR, end-systolic pressure-volume relationship; EDPVR, end-diastolic pressure-volume relationship; PRSW, preload recruitable SW; dP/dtmax-EDV, slope of the dP/dtmax-end-diastolic volume relationship.
Elevated reactive oxygen species formation in cardiac subsarcolemmal mitochondria in prediabetic rats.
To investigate whether cardiac mitochondrial disturbances contribute to the observed diastolic dysfunction, mitochondrial morphology and enzyme activity were analyzed from LVs of prediabetic rats. Our electron microscopy results showed that there is no major difference in the number of IFM between the groups (Fig. 5, A and B). However, area (CON: 0.43 ± 0.01 vs. PRED: 0.39 ± 0.01 μm2; P < 0.05), perimeter (CON: 2.69 ± 0.02 vs. PRED: 2.63 ± 0.03 μm; P < 0.05), and sphericity (CON: 0.35 ± 0.01 vs. PRED: 0.31 ± 0.01; P < 0.05) of IFM are decreased in the prediabetes group. Previous studies indicated that IFM and SSM are affected by diabetes differentially (35, 82). Therefore, we analyzed our electron microscopy imagery containing SSM and found no difference in SSM size, perimeter, or sphericity (data not shown) although the statistical power of these analyses was not high enough (n = 2 for CON and n = 4 for PRED). Furthermore, we have not seen any major difference in mitochondrial oxygen consumption, enzyme activities (Tables 4 and 5), Ca uptake, or membrane potential (data not shown). However, we have found that hydrogen peroxide production was increased in the cardiac SSM fraction with glutamate-malate as a substrate (Fig. 5C) although there was no difference when succinate was used as substrate. Interestingly, there was no increase in reactive oxygen species (ROS) production of the IFM isolated from LV supported either with glutamate-malate or with succinate (Fig. 5, D–F). As leukocytes are one of the main sources of ROS, inflammatory mediators were measured. We could find no significant difference in TNF-α (CON: 1 ± 0.27 vs. PRED: 0.59 ± 0.07; ratio normalized to GAPDH; P > 0.05) and IL-6 (CON: 1 ± 0.27 vs. PRED: 0.69 ± 0.14; ratio normalized to GAPDH; P > 0.05) mRNA expressions between groups, which shows that, in our model, prediabetes does not elicit cardiac or systemic inflammation. Furthermore, we have not seen any difference in other markers of oxidative stress, namely the expression of p66Shc and tropomyosin oxidation between groups (Fig. 6, B–E). It is known that reactive nitrogen species have an important role in deteriorated contractile and endothelial function in diabetes (16, 59); therefore, we analyzed whether nitrative stress is influenced in prediabetes. Nitrotyrosine immunohistology indicated that protein nitrosylation is increased in prediabetes (Fig. 6A). As CaMKIIδ has been proposed to be activated in oxidative stress-associated conditions (46), we measured the levels of the active forms of the kinase that might affect the contractility and relaxation capacity of the heart (49). The phosphorylation of CaMKIIδ and of its target PLB on Thr17 was not changed by prediabetes (Fig. 6, F–H). Similarly, there was no change in the protein expression of SERCA2A in our model of prediabetes compared with control animals (Fig. 6J). On the other hand, the level of p-Ser16-PLB showed a tendency for downregulation in prediabetes (P = 0.08; Fig. 6I).
Fig. 5.

Mitochondrial morphology and function in prediabetes at week 21. Representative transmission electron micrographs (A) and number of interfibrillar mitochondria (IFM) (B) in the left ventricle are shown; magnification ×12,000, scale bar = 1 μm. Quantifications of H2O2 production in subsarcolemmal mitochondria (SSM) (C) and IFM (D) with glutamate-malate as substrate (GM) are shown. Quantification of H2O2 production in SSM (E) and IFM (F) with succinate as substrate is shown. Data are means ± SE, n = 5–9 per group (*P < 0.05).
Table 4.
Quantification of cardiac mitochondria enzyme activity in LV
| CON | PRED | |
|---|---|---|
| Citrate-synthase activity, U/mg protein | 223.12 ± 9.98 | 220.44 ± 8.32 |
| NADH:ubiquinone-oxidoreductase activity, U/mg protein | 40.52 ± 2.55 | 36.48 ± 2.99 |
| NADH:cytochrome c-oxidoreductase activity, U/mg protein | 7.85 ± 1.18 | 8.47 ± 1.31 |
| Succinate:cytochrome c-oxidoreductase activity, U/mg protein | 21.09 ± 1.49 | 23.57 ± 1.61 |
| Succinate-dehydrogenase activity, U/mg protein | 84.06 ± 5.83 | 80.09 ± 3.42 |
| Cytochrome-c-oxidase activity, U/mg protein | 38.74 ± 3.15 | 40.36 ± 2.33 |
Data are means ± SE for 5–9 rat per group.
Table 5.
Quantification of mitochondrial oxygen consumption
| CON | PRED | CON | PRED | |
|---|---|---|---|---|
| Subsarcolemmal, (pmol/ml)s | Subsarcolemmal, (pmol/ml)s | Interfibrillar, (pmol/ml)s | Interfibrillar, (pmol/ml)s | |
| Glutamate-malate | 25.08 ± 3.60 | 21.06 ± 3.53 | 58.60 ± 19.31 | 61.78 ± 21.41 |
| ADP | 203.31 ± 32.57 | 194.03 ± 42.16 | 304.23 ± 25.75 | 287.90 ± 22.35 |
| Cytochrome c | 260.80 ± 28.27 | 287.06 ± 54.91 | 335.96 ± 25.79 | 345.41 ± 28.17 |
| Succinate | 306.20 ± 25.19 | 289.74 ± 23.23 | 367.76 ± 15.74 | 393.56 ± 18.82 |
| Rotenone | 143.12 ± 18.19 | 139.70 ± 23.64 | 238.40 ± 18.44 | 220.73 ± 16.19 |
| CAT | 105.95 ± 8.89 | 106.50 ± 12.22 | 159.78 ± 6.86 | 156.60 ± 8.03 |
Data are means ± SE for 9 rat per group.
CAT, carboxyatractyloside.
Fig. 6.

Characterization of oxidative and nitrative stress in prediabetes. Representative immunostaining of nitrotyrosine in the left ventricle (A) is shown; magnification ×200; scale bar = 200 μm. Representative Western blots (B) and quantification (C) of tropomyosin oxidation are shown. Representative Western blots (D) and quantification (E) of cardiac p66Shc expression are shown. Representative Western blots (F), quantification of Ca2+/calmodulin-dependent protein kinase II (CaMKIIδ) (G), phospholamban (PLB) phosphorylation on Thr17 (H) and Ser16 (I), and sarco/endoplasmic reticulum Ca2+ ATPase II (SERCA2A) (J) expression are shown. Tm, tropomyosin; Ox. Tm, oxidized tropomyosin. Data are means ± SE, n = 6–8 per group.
Alterations in cardiac MFN2 expression and mitophagy in prediabetes.
To investigate the effect of cardiac mitochondrial dynamics, autophagy, and mitophagy in prediabetes, we analyzed protein expression changes. Cardiac expression of the mitophagy-related protein, BNIP3, was decreased in the prediabetic group in LV lysates; however, other autophagy- and mitophagy-related proteins such as Beclin-1, LC3-II, SQSTM1/p62, and Parkin were unchanged (Fig. 7A; Table 6). Upstream modulators of autophagy such as Akt, AMPKα, GSK3β, and ribosomal S6 protein (a surrogate marker of mammalian target of rapamycin complex activity) were also measured; however, expression or phosphorylation of these proteins were not different between groups (Fig. 7A; Table 6). Furthermore, the expression of a mitochondrial fusion-related protein, MFN2, was elevated; however, expression of DRP1/DLP1 and OPA1 proteins was unchanged in whole LV lysates in the prediabetic group (Fig. 7B; Table 6). Nonetheless, we measured the expression of mitochondrial dynamics- and mitophagy-related proteins from SSM and IFM isolated from LVs. No difference was found in the expression of OPA1, LC3-II, and SQSTM1/p62 in isolated cardiac SSM and IFM between groups (Fig. 7, C and D; Table 6). Our results indicate that mitochondrial dynamics and autophagy/mitophagy were not modulated substantially by prediabetes; however, the upregulation of MFN2 (increased mitochondrial fusion, tethering to endoplasmic reticulum) and the downregulation of BNIP3 (decreased mitophagy) may implicate early changes in mitochondrial homeostasis, which might lead to the accumulation of dysfunctional mitochondria.
Fig. 7.

Cardiac expression of mitochondrial dynamics, autophagy/mitophagy, heat shock proteins (HSPs), and apoptosis-related proteins in prediabetes. Representative Western blots of autophagy/mitophagy-related proteins and upstream modulators of autophagy (A) and mitochondrial fission- and fusion-related protein (B) in whole left ventricles are shown. Representative Western blots of mitochondrial dynamics- and mitophagy-related proteins in isolated SSM (C) and IFM (D) are shown. Representative Western blots of HSP-related (E) and apoptosis-related (F) proteins in whole left ventricle are shown. DRP1/DLP1, dynamin-related/like protein 1; MFN2, mitofusin-2; OPA1, optic atrophy 1; COX4, cytochrome c oxidase subunit 4, mitochondrial; LC3, 1 microtubule-associated protein 1 light chain 3; SQSTM1/p62, sequestosome 1; BNIP3, Bcl-2/adenovirus E1B 19-kDa protein-interacting protein 3; AMPKα, AMP-activated protein kinase α; GSK3β, glycogen synthase kinase-3β.
Table 6.
Quantification of HSPs, apoptosis, mitochondrial dynamics-related, and mitophagy-related protein expressions in isolated mitochondrial fractions and whole LVs
| Total LV | CON | PRED |
|---|---|---|
| BNIP3/GAPDH ratio | 0.590 ± 0.030 | 0.440 ± 0.020* |
| MFN2/GAPDH ratio | 0.270 ± 0.010 | 0.360 ± 0.020* |
| OPA1/GAPDH ratio | 1.470 ± 0.110 | 1.630 ± 0.100 |
| DRP1/GAPDH ratio | 1.160 ± 0.080 | 1.340 ± 0.140 |
| LC3-II/GAPDH ratio | 0.560 ± 0.070 | 0.560 ± 0.050 |
| p62/GAPDH ratio | 3.050 ± 0.180 | 3.450 ± 0.230 |
| Parkin/GAPDH ratio | 2.250 ± 0.140 | 2.430 ± 0.170 |
| Beclin1/GAPDH ratio | 0.810 ± 0.070 | 0.820 ± 0.070 |
| Phospho AKT(Ser473)/AKT ratio | 0.320 ± 0.040 | 0.290 ± 0.020 |
| Phospho AMPK(Thr172)/AMPK ratio | 0.120 ± 0.020 | 0.210 ± 0.060 |
| Phospho S6(Ser235/236)/S6 ratio | 2.620 ± 1.080 | 2.160 ± 0.610 |
| Phospho GSK3β(Ser9)/GSK3β ratio | 0.800 ± 0.090 | 0.720 ± 0.100 |
| Bcl-2/GAPDH ratio | 0.230 ± 0.010 | 0.200 ± 0.003* |
| Bcl-2/Bax ratio | 0.150 ± 0.020 | 0.150 ± 0.020 |
| Caspase-3/GAPDH ratio | 0.050 ± 0.001 | 0.040 ± 0.003 |
| P-HSP-27(Ser82)/T-HSP-27 ratio | 0.340 ± 0.050 | 0.280 ± 0.020 |
| HSP-60/GAPDH ratio | 0.850 ± 0.020 | 0.840 ± 0.020 |
| HSP-70/GAPDH ratio | 0.530 ± 0.010 | 0.510 ± 0.010 |
| HSP-90/GAPDH ratio | 0.460 ± 0.010 | 0.500 ± 0.020 |
| Subsarcolemmal mitochondria | ||
| OPA1/COX4 ratio | 1.340 ± 0.070 | 1.320 ± 0.060 |
| LC3-II/COX4 ratio | 0.220 ± 0.100 | 0.250 ± 0.040 |
| p62/COX4 ratio | 0.130 ± 0.020 | 0.120 ± 0.030 |
| Interfibrillar mitochondria | ||
| OPA1/COX4 ratio | 1.440 ± 0.150 | 1.520 ± 0.140 |
| LC3-II/COX4 ratio | 0.220 ± 0.090 | 0.290 ± 0.070 |
| p62/COX4 ratio | 0.350 ± 0.060 | 0.360 ± 0.040 |
Data are means ± SE for 8 rat per group (
P < 0.05).
HSP, heat shock protein; BNIP3, Bcl-2/adenovirus E1B 19-kDa protein-interacting protein 3; MFN2, mitofusin-2; OPA1, optic atrophy 1; DRP1, dynamin-related protein 1; LC3, 1 microtubule-associated protein 1 light chain 3; AMPK, AMP-activated protein kinase; GSK3β, glycogen synthase kinase-3β; COX4, cytochrome c oxidase subunit 4, mitochondrial.
Expression of cardiac Bcl-2 decreases in prediabetes.
Our study also aimed to explore the effect of prediabetes on apoptosis in the heart. Prediabetes did not affect the expression of proapoptotic caspase-3 and Bax in LVs. On the other hand, the antiapoptotic Bcl-2 was downregulated in prediabetic animals. However, the Bcl-2/Bax ratio was unchanged (Fig. 7F; Table 6).
No changes in cardiac HSPs in prediabetes.
We also characterized the effect of prediabetes on the expression and/or phosphorylation of HSPs in the LV. Our results showed no differences in the expression of HSP-60, HSP-70, and HSP-90 or in either phosphorylation or expression of HSP-27 (Fig. 7E; Table 6).
DISCUSSION
This is the first comprehensive analysis of the cardiac effects of prediabetes in a nongenetic rodent model in which we assessed cardiac functions, parameters of hypertrophy, fibrosis, oxidative and nitrative stress, inflammation, mitochondrial dynamics, autophagy, mitophagy, markers of myocardial calcium handling, apoptosis, and expression of HSPs. In this model of prediabetes, we demonstrated an impaired glucose and insulin tolerance, increased adiposity and myocardial lipid accumulation, a mild diastolic dysfunction, and sensory neuropathy despite normal fasting plasma glucose and lipid levels. We also observed elevated ROS production in the SSM, nitrative stress, elevated expression of MFN2, decreased expression of β-MHC, and phosphorylation of PLB. Furthermore, here we found early signs of dysregulated mitophagy and decreased mitochondrial size in prediabetes; however, other major markers of mitochondrial dynamics, autophagy, mitophagy, inflammation, or myocardial expression of apoptotic proteins or HSPs, were not modulated by prediabetes.
In this study, we used high-fat chow-fed Long-Evans rats treated with a single, low-dose STZ. This setting allowed us to investigate cardiac consequences of a moderate metabolic derangement, prediabetes, rather than of a severely disturbed glucose and lipid homeostasis, such as seen in genetically modified models of diabetes, e.g., in db/db or ob/ob mice (36, 67). Because it has been reported that LV hypertrophy had a higher prevalence in patients with diabetes and that 40–75% of patients with type 1 or type 2 diabetes mellitus presented with diastolic dysfunction (12, 73), we aimed to investigate whether cardiac function is affected by prediabetes. Previously, it has been shown that diastolic dysfunction was developed in several pathological conditions; however, the underlying mechanisms are still not clearly understood (38). Here, we demonstrated that the deterioration of diastolic function and sensory neuropathy occurs well before overt diabetes develops, which is accompanied by early signs of cardiac hypertrophy. These findings are in agreement with previous reports showing that neuropathy might precede the development of full-fledged diabetes (48) and that high-fat diet-induced prediabetes increased heart weights and decreased contractile function, as assessed by a diminished aortic output (23, 29). However, in contrast to our report, plasma triglycerides and insulin levels were elevated in these studies, highlighting that a substantial difference can be observed between different diet-induced models and stages of prediabetes (29). Furthermore, it has been described that obesity promoted the hypertrophy-inducing effect of diabetes regardless of hypertension (26), which could be attributed to adipokines, such as leptin and resistin (5, 39). Similarly, here we showed that even mild obesity (only 18% increase in body weight was observed in the present study) with an elevated leptin level is sufficient to induce hypertrophy even without impairment of fasting plasma glucose and lipid levels or hypertension, which is in agreement with previous reports (23, 29). However, clinical data seem to contradict these findings because no increase in the prevalence of LV hypertrophy was observed in overweight prediabetic patients with impaired fasting glucose and impaired glucose tolerance (66). Mechanistic studies on how obesity abrogates cardiac function are scarce. Increased myocardial triglyceride content is associated with diastolic dysfunction in ob/ob mice (18), which is well in line with our findings that the number of lipid particles increased in the myocardium in prediabetes. Although microRNA-451 has been demonstrated to promote cardiac hypertrophy and diminished contractile reserves in mice on high-fat diet (44), further studies are warranted to describe the relationship between cardiac dysfunction and the disturbed cardiac lipid metabolism in prediabetes. Interestingly, unlike in genetic models of prediabetes (20), diet-induced prediabetes did not result in an elevation in classical molecular markers of hypertrophy or conventional signs of fibrosis in the heart, as expected in the case of hypertrophy. Moreover, this is the first evidence on decreased β-MHC in prediabetes. Although the vast majority of publications demonstrate an increase in β-MHC in diabetes (4, 83), a small number of studies indicate a downregulation of MHC expression in animals with diverse cardiac or metabolic challenges. For instance, in cardiomyocytes from STZ-treated rats, total MHC expression was significantly decreased (25). These results indicate that, although in most cardiometabolic derangements expression of the slow MHC isoform increases, in certain conditions, such as in prediabetes, a general suppression of MHC expression might be present. The reduction in MHC expression might also contribute to the observed cardiac dysfunction in prediabetes; however, to uncover its significance and mechanism, further experiments are warranted.
Oxidative stress has a major role in the development of diabetic cardiomyopathy (10, 31); however, it has not been well described whether it is responsible for the decreased cardiac function in prediabetes. Here we found elevated hydrogen peroxide production in SSM, increased nitrotyrosine formation, and elevated cardiac expression of MFN2. These findings are in agreement with previous reports in which elevated oxidative stress, such as seen in our model of prediabetes, leads to an increase in MFN2 in rat vascular smooth muscle cells (33), and its robust overexpression induced apoptotic cell death in neonatal rat cardiomyocytes (72). Similarly, in another study, high-fat diet induced oxidative stress and MFN2 overexpression in the liver of C57BL/6 mice after 16 wk (27). However, in a previous study on diet-induced prediabetes, no sign of cardiac mitochondrial oxidative stress was shown in male Wistar rats after 16 wk (29), which may suggest that mitochondrial oxidative stress might not be present in all models and stages of prediabetes and that it might not be the primary driving force of prediabetes-induced cardiac functional alterations.
It is well established that mitochondria, especially the mitochondrial electron transport chain, is one of the main sources of ROS; however, several other intracellular components can produce ROS in mitochondria (17). For instance, it is known that p66Shc translocation to mitochondria can increase the formation of ROS (22), and NADPH oxidase 4 and monoamine oxidase also have important roles in mitochondrial ROS production (8, 43). Although here we observed a moderately increased ROS production in SSM, no difference can be seen in mitochondrial oxygen consumption between normal and prediabetic mitochondria (see Table 5), showing no impairment in mitochondrial redox chains. It is presently unknown what mechanism leads to the increased ROS production exclusively in SSM in prediabetes. In mice on a high-fat diet, cardiac mitochondrial ROS production was elevated, and, similar to our results, mitochondrial oxygen consumption did not change substantially, whereas a significant amount of cardiac lipid accumulation was observed (1). However, the source of ROS has not been identified in this study either. Thus, to reveal the direct connection between elevated ROS production and mitochondrial and cardiac dysfunction, further studies are warranted.
Molecular mechanisms that contribute to hypertrophy and cardiac dysfunction in prediabetes have not been investigated in detail. In our previous studies on diet-induced hypercholesterolemia or metabolic syndrome in ZDF rats, we have shown by DNA and miRNA microarrays that a multitude of cardiac cellular processes is modulated by these conditions (69, 79). Similarly, in this study, we showed changes in several cellular processes, suggesting that hypertrophy and deteriorated diastolic function in prediabetes may be consequences of numerous concurrent alterations in the cardiac homeostasis (see Fig. 8). Characterizing active components of the contractile apparatus and Ca2+ homeostasis, here we observed a tendency for a decrease in the Ser16 phosphorylation of PLB in prediabetes. In previous studies, decreased phosphorylation of PLB on Ser16 was demonstrated to be associated with abnormalities in contraction and relaxation in the diabetic heart (57, 85). This notion is further supported by the findings of Abdurrachim et al. (1), who demonstrated that phosphorylation of PLB was reduced in the heart of mice with diastolic dysfunction induced by a high-fat diet. Therefore, decreased phosphorylation of PLB may also contribute to the development of early diastolic dysfunction that we uncovered in prediabetes. Increased activity and expression of CaMKIIδ and reduced phosphorylation of PLB by CaMKIIδ have been found to be associated with contractile dysfunction, diabetes (47, 49), and fructose-rich diet-induced prediabetes (75). In our model, expression and phosphorylation of CaMKIIδ and phosphorylation of PLB on Thr17 were unchanged. This is in contrast with previous findings that have reported the phosphorylation of CaMKIIδ being increased in the heart of STZ-treated diabetic rats (71, 76) although, in these reports, a significant hyperglycemia was present, which was shown to facilitate the activation of CaMKIIδ (28). Apoptosis is considered to be one of the hallmarks of diabetic cardiomyopathy, and it is induced by oxidative stress in diabetes (10, 78). It has been described that experimental diabetes induces upregulation of proapoptotic and downregulation of antiapoptotic proteins (3, 84); however, no data have been available on the cardiac apoptosis in prediabetes. In the present study, we show a modest downregulation of Bcl-2; however, no change in Bcl-2/Bax ratio and caspase-3 expression was detected in prediabetic animals. Thus our data suggest an early dysregulation of pro- and antiapoptotic proteins in prediabetes; however, they do not show a gross induction of apoptosis in prediabetes. Tropomyosin is prone to loss of function by oxidative modifications that are associated with the severity of heart failure in humans (14, 15). In this study, oxidized tropomyosin content of the heart was not modulated by prediabetes. These data suggest that neither the CaMKIIδ pathway, apoptosis induction, nor tropomyosin oxidation are responsible for the diastolic dysfunction observed in prediabetes.
Fig. 8.

Schematic representation of the cardiac effects of prediabetes. ROS, reactive oxygen species.
Here we also demonstrate an early dysregulation of mitochondrial fusion and mitophagy first in the literature, as shown by elevated MFN2 and attenuated BNIP3 expression in prediabetes; however, other canonical markers of autophagy, mitophagy, and apoptosis were unaffected. Similarly, right atrial myocardial samples of type-2 diabetic patients presenting no signs of overt cardiomyopathy, the expression of the majority of mitochondrial dynamics, and autophagy-related proteins were not elevated, except for that of ATG5 and MFN1 (53). Therefore, we can assume that only major disturbances in glucose and lipid homeostasis, such as seen in untreated patients or in genetic models of diabetes, might be a powerful enough signal to extensively modulate cardiac autophagy, mitophagy, or mitochondrial dynamics, which might result in grossly deteriorated cardiac function. Moreover, experimental systemic sensory neuropathy by itself has been previously shown to cause diastolic dysfunction and global gene expression changes in the rat heart (7, 86). Therefore, prediabetes-induced sensory neuropathy observed in the present study might also contribute to the diabetic cardiomyopathy.
Furthermore, this is the first report to show that prediabetes does not modulate cardiac expression of HSP-60, HSP-70, HSP-90, and phosphorylation or expression of HSP-27. In contrast, in STZ-induced diabetes, increased levels of HSP-70 have been detected in the rat heart (77), and increased levels of circulating HSP-60 were found in diabetic patients (70), suggesting that, in advanced stages of diabetes, HSPs might be involved in the development of cardiac dysfunction. However, our data suggest no role of HSPs in prediabetes in the heart.
A limitation of this study is that the prediabetic condition was not analyzed in female rats. Because pathophysiological processes might substantially differ between sexes (see NIH notice NOT-OD-15-102), it cannot be excluded that performing the study with the inclusion of both sexes might allow different conclusions to be drawn. Furthermore, because other studies showed that MHC protein expression is influenced by certain factors such as Foxo1 (61) or miR-27a (55), assessing α- and β-MHC protein levels might have helped to understand the molecular background of the mild diastolic dysfunction and hypertrophy observed in prediabetes better.
Taken together, this study emphasizes that parallel occurrence of several abnormalities of metabolic, oxidative, and contractile functions might trigger cardiac pathological changes characteristic of prediabetes well before hyperglycemia or major metabolic derangements occur and that preventing these abnormalities might be of importance for future therapies of cardiac pathologies observed in early metabolic diseases such as prediabetes.
GRANTS
This work was supported by the European Foundation for the Study of Diabetes (EFSD) New Horizons Collaborative Research Initiative from the European Association for the Study of Diabetes (EASD) and Hungarian Scientific Research Fund (OTKA K 109737, PD100245 to T. Radovits) and Slovak Scientific Grant Agency (VEGA1/0638/12). Z. Giricz, T. Radovits, and K. Szigeti hold a “János Bolyai Research Scholarship” from the Hungarian Academy of Sciences, and Z. Varga was supported by the Rosztoczy Foundation. P. Ferdinandy is a Szentágothai Fellow of the National Program of Excellence (TAMOP 4.2.4. A/2-11-1-2012-0001). T. Baranyai is supported by the European Cooperation in Science and Technology (COST-BM1203-STSM 090515-058721). K. Boengler and R. Schreckenberg are supported by the German Research Foundation (BO-2955/2-1 and SCHU 843/9-1).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.K., Z.V.V., T.B., K.B., S.R., L.L., T. Radovits, A.O., C.M., Á.L., M.A.-K., T.K., N.B., D.M., L.D., M.B., T. Rajtík, A.A., K.S., and R. Schulz performed experiments; G.K., Z.V.V., T.B., K.B., L.L., T. Radovits, A.O., C.M., Á.L., M.A.-K., T.K., N.B., L.D., M.B., T. Rajtík, and K.S. analyzed data; G.K., T.B., T. Radovits, D.M., and Z.G. interpreted results of experiments; G.K. prepared figures; G.K., Z.V.V., T.B., M.B., A.A., and Z.G. drafted manuscript; G.K., Z.G., R. Schulz, and P.F. approved final version of manuscript; Z.V.V., K.-D.S., R. Schreckenberg, Z.G., R. Schulz, and P.F. conception and design of research; Z.V.V., T.B., S.R., L.L., K.-D.S., R. Schreckenberg, T. Radovits, A.O., C.M., x.L., T.K., N.B., L.D., M.B., T. Rajtík, A.A., K.S., P.H., Z.H., L.T., P.P., B.M., Z.G., R. Schulz, and P.F. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Melinda Károlyi-Szabó, Jenőné Benkes, Henriett Biró, Ildikó Horváth, Teréz Bagoly, Sebestyén Tuza, and Anikó Perkecz for technical assistance.
REFERENCES
- 1.Abdurrachim D, Ciapaite J, Wessels B, Nabben M, Luiken JJ, Nicolay K, Prompers JJ. Cardiac diastolic dysfunction in high-fat diet fed mice is associated with lipotoxicity without impairment of cardiac energetics in vivo. Biochim Biophys Acta 1842: 1525–1537, 2014. [DOI] [PubMed] [Google Scholar]
- 2.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care, 37: Suppl 1: S81–S90, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Amin AH, El-Missiry MA, Othman AI. Melatonin ameliorates metabolic risk factors, modulates apoptotic proteins, and protects the rat heart against diabetes-induced apoptosis. Eur J Pharmacol 747: 166–173, 2015. [DOI] [PubMed] [Google Scholar]
- 4.Aragno M, Mastrocola R, Medana C, Catalano MG, Vercellinatto I, Danni O, Boccuzzi G. Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes. Endocrinology 147: 5967–5974, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Barouch LA, Berkowitz DE, Harrison RW, O'Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 108: 754–759, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Barr EL, Zimmet PZ, Welborn TA, Jolley D, Magliano DJ, Dunstan DW, Cameron AJ, Dwyer T, Taylor HR, Tonkin AM, Wong TY, McNeil J, Shaw JE. Risk of cardiovascular and all-cause mortality in individuals with diabetes mellitus, impaired fasting glucose, and impaired glucose tolerance: The Australian Diabetes, Obesity, and Lifestyle Study (AusDiab). Circulation 116: 151–157, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Bencsik P, Kupai K, Giricz Z, Gorbe A, Huliak I, Furst S, Dux L, Csont T, Jancso G, Ferdinandy P. Cardiac capsaicin-sensitive sensory nerves regulate myocardial relaxation via S-nitrosylation of SERCA: Role of peroxynitrite. Br J Pharmacol 153: 488–496, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, Seguelas MH, Nistri S, Colucci W, Leducq N, Parini A. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 112: 3297–3305, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Bjorndal B, Burri L, Staalesen V, Skorve J, Berge RK. Different adipose depots: Their role in the development of metabolic syndrome and mitochondrial response to hypolipidemic agents. J Obes 2011: 490650, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 11: 31–39, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254, 1976. [DOI] [PubMed] [Google Scholar]
- 12.Brooks BA, Franjic B, Ban CR, Swaraj K, Yue DK, Celermajer DS, Twigg SM. Diastolic dysfunction and abnormalities of the microcirculation in type 2 diabetes. Diabetes Obes Metab 10: 739–746, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Cai L, Kang YJ. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol 3: 219–228, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Canton M, Menazza S, Sheeran FL, Polverino de Laureto P, Di Lisa F, Pepe S. Oxidation of myofibrillar proteins in human heart failure. J Am Coll Cardiol 57: 300–309, 2011. [DOI] [PubMed] [Google Scholar]
- 15.Canton M, Neverova I, Menabo R, Van Eyk J, Di Lisa F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol Heart Circ Physiol 286: H870–H877, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Cassuto J, Dou H, Czikora I, Szabo A, Patel VS, Kamath V, Belin de Chantemele E, Feher A, Romero MJ, Bagi Z. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes 63: 1381–1393, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res 114: 524–537, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology 144: 3483–3490, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Csont T, Bereczki E, Bencsik P, Fodor G, Gorbe A, Zvara A, Csonka C, Puskas LG, Santha M, Ferdinandy P. Hypercholesterolemia increases myocardial oxidative and nitrosative stress thereby leading to cardiac dysfunction in apoB-100 transgenic mice. Cardiovasc Res 76: 100–109, 2007. [DOI] [PubMed] [Google Scholar]
- 20.D'Souza A, Howarth FC, Yanni J, Dobryznski H, Boyett MR, Adeghate E, Bidasee KR, Singh J. Left ventricle structural remodelling in the prediabetic Goto-Kakizaki rat. Exp Physiol 96: 875–888, 2011. [DOI] [PubMed] [Google Scholar]
- 21.DeFronzo RA and Abdul-Ghani M. Assessment and treatment of cardiovascular risk in prediabetes: Impaired glucose tolerance and impaired fasting glucose. Am J Cardiol 108: 3b–24b, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Di Lisa F, Giorgio M, Ferdinandy P, Schulz R. New aspects of p66Shc in ischemia reperfusion injury and cardiovascular diseases. Br J Pharmacol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.du Toit EF, Smith W, Muller C, Strijdom H, Stouthammer B, Woodiwiss AJ, Norton GR, Lochner A. Myocardial susceptibility to ischemic-reperfusion injury in a prediabetic model of dietary-induced obesity. Am J Physiol Heart Circ Physiol 294: H2336–H2343, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Duncan JG. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim Biophys Acta 1813: 1351–1359, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dyntar D, Sergeev P, Klisic J, Ambuhl P, Schaub MC, Donath MY. High glucose alters cardiomyocyte contacts and inhibits myofibrillar formation. J Clin Endocrinol Metab 91: 1961–1967, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Eguchi K, Boden-Albala B, Jin Z, Rundek T, Sacco RL, Homma S, Di Tullio MR. Association between diabetes mellitus and left ventricular hypertrophy in a multiethnic population. Am J Cardiol 101: 1787–1791, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enos RT, Velazquez KT, Murphy EA. Insight into the impact of dietary saturated fat on tissue-specific cellular processes underlying obesity-related diseases. J Nutr Biochem 25: 600–612, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502: 372–376, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Essop MF, Anna Chan WY, Valle A, Garcia-Palmer FJ, Du Toit EF. Impaired contractile function and mitochondrial respiratory capacity in response to oxygen deprivation in a rat model of pre-diabetes. Acta Physiol (Oxf) 197: 289–296, 2009. [DOI] [PubMed] [Google Scholar]
- 30.Fonseca VA. Identification and treatment of prediabetes to prevent progression to type 2 diabetes. Clin Cornerstone 9: 51–59; discussion 60-51, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P. Myocardial cell death in human diabetes. Circ Res 87: 1123–1132, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Galloway CA, Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal 22: 1545–1562, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo X, Chen KH, Guo Y, Liao H, Tang J, Xiao RP. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res 101: 1113–1122, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Harms M, Seale P. Brown and beige fat: Development, function and therapeutic potential. Nat Med 19: 1252–1263, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Holloway GP, Snook LA, Harris RJ, Glatz JF, Luiken JJ, Bonen A. In obese Zucker rats, lipids accumulate in the heart despite normal mitochondrial content, morphology and long-chain fatty acid oxidation. J Physiol 589: 169–180, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsueh W, Abel ED, Breslow JL, Maeda N, Davis RC, Fisher EA, Dansky H, McClain DA, McIndoe R, Wassef MK, Rabadan-Diehl C, Goldberg IJ. Recipes for creating animal models of diabetic cardiovascular disease. Circ Res 100: 1415–1427, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Kass D. Clinical ventricular pathophysiology: a pressure-volume view. In: Ventricular Function, edited by Warltier DC. Baltimore, MD: Williams & Wilkins, 1995, pp. 131–151. [Google Scholar]
- 38.Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res 94: 1533–1542, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Kim M, Oh JK, Sakata S, Liang I, Park W, Hajjar RJ, Lebeche D. Role of resistin in cardiac contractility and hypertrophy. J Mol Cell Cardiol 45: 270–280, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41: 1313–1321, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta 1852: 252–261, 2015. [DOI] [PubMed] [Google Scholar]
- 42.Kovacs A, Olah A, Lux A, Matyas C, Nemeth BT, Kellermayer D, Ruppert M, Torok M, Szabo L, Meltzer A, Assabiny A, Birtalan E, Merkely B, Radovits T. Strain and strain rate by speckle-tracking echocardiography correlate with pressure-volume loop-derived contractility indices in a rat model of athlete's heart. Am J Physiol Heart Circ Physiol 308: H743–H748, 2015. [DOI] [PubMed] [Google Scholar]
- 43.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA 107: 15565–15570, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S, Izuhara M, Nakao T, Nishino T, Otsu K, Kita T, Kimura T, Ono K. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res 116: 279–288, 2015. [DOI] [PubMed] [Google Scholar]
- 45.Lewis CE, McTigue KM, Burke LE, Poirier P, Eckel RH, Howard BV, Allison DB, Kumanyika S, Pi-Sunyer FX. Mortality, health outcomes, and body mass index in the overweight range: A science advisory from the American Heart Association. Circulation 119: 3263–3271, 2009. [DOI] [PubMed] [Google Scholar]
- 46.Luczak ED, Anderson ME. CaMKII oxidative activation and the pathogenesis of cardiac disease. J Mol Cell Cardiol 73: 112–116, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest 123: 1262–1274, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lupachyk S, Watcho P, Obrosov AA, Stavniichuk R, Obrosova IG. Endoplasmic reticulum stress contributes to prediabetic peripheral neuropathy. Exp Neurol 247: 342–348, 2013. [DOI] [PubMed] [Google Scholar]
- 49.Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res 73: 631–640, 2007. [DOI] [PubMed] [Google Scholar]
- 50.Mansor LS, Gonzalez ER, Cole MA, Tyler DJ, Beeson JH, Clarke K, Carr CA, Heather LC. Cardiac metabolism in a new rat model of type 2 diabetes using high-fat diet with low dose streptozotocin. Cardiovasc Diabetol 12: 136, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mizushige K, Yao L, Noma T, Kiyomoto H, Yu Y, Hosomi N, Ohmori K, Matsuo H. Alteration in left ventricular diastolic filling and accumulation of myocardial collagen at insulin-resistant prediabetic stage of a type II diabetic rat model. Circulation 101: 899–907, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Mohanty JG, Jaffe JS, Schulman ES, Raible DG. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J Immunol Methods 202: 133–141, 1997. [DOI] [PubMed] [Google Scholar]
- 53.Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, Staels B. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130: 554–564, 2014. [DOI] [PubMed] [Google Scholar]
- 54.Nathan DM, Davidson MB, DeFronzo RA, Heine RJ, Henry RR, Pratley R, Zinman B. Impaired fasting glucose and impaired glucose tolerance: Implications for care. Diabetes Care 30: 753–759, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Nishi H, Ono K, Horie T, Nagao K, Kinoshita M, Kuwabara Y, Watanabe S, Takaya T, Tamaki Y, Takanabe-Mori R, Wada H, Hasegawa K, Iwanaga Y, Kawamura T, Kita T, Kimura T. MicroRNA-27a regulates beta cardiac myosin heavy chain gene expression by targeting thyroid hormone receptor beta1 in neonatal rat ventricular myocytes. Mol Cell Biol 31: 744–755, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orhan CE, Onal A, Uyanikgil Y, Ulker S. Antihyperalgesic and antiallodynic effect of sirolimus in rat model of adjuvant arthritis. Eur J Pharmacol 705: 35–41, 2013. [DOI] [PubMed] [Google Scholar]
- 57.Ouwens DM, Boer C, Fodor M, de Galan P, Heine RJ, Maassen JA, Diamant M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia 48: 1229–1237, 2005. [DOI] [PubMed] [Google Scholar]
- 58.Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protocol 3: 1422–1434, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pechanova O, Varga ZV, Cebova M, Giricz Z, Pacher P, Ferdinandy P. Cardiac NO signalling in the metabolic syndrome. Br J Pharmacol 172: 1415–1433, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pereira L, Ruiz-Hurtado G, Rueda A, Mercadier JJ, Benitah JP, Gomez AM. Calcium signaling in diabetic cardiomyocytes. Cell Calcium 56: 372–380, 2014. [DOI] [PubMed] [Google Scholar]
- 61.Qi Y, Zhu Q, Zhang K, Thomas C, Wu Y, Kumar R, Baker KM, Xu Z, Chen S, Guo S. Activation of Foxo1 by insulin resistance promotes cardiac dysfunction and beta-myosin heavy chain gene expression. Circ Heart Fail 8: 198–208, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Radovits T, Korkmaz S, Loganathan S, Barnucz E, Bomicke T, Arif R, Karck M, Szabo G. Comparative investigation of the left ventricular pressure-volume relationship in rat models of type 1 and type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol 297: H125–H133, 2009. [DOI] [PubMed] [Google Scholar]
- 63.Radovits T, Korkmaz S, Matyas C, Olah A, Nemeth BT, Pali S, Hirschberg K, Zubarevich A, Gwanmesia PN, Li S, Loganathan S, Barnucz E, Merkely B, Szabo G. An altered pattern of myocardial histopathological and molecular changes underlies the different characteristics of type-1 and type-2 diabetic cardiac dysfunction. J Diabetes Res 2015: 728741, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Radovits T, Olah A, Lux A, Nemeth BT, Hidi L, Birtalan E, Kellermayer D, Matyas C, Szabo G, Merkely B. Rat model of exercise-induced cardiac hypertrophy: hemodynamic characterization using left ventricular pressure-volume analysis. Am J Physiol Heart Circ Physiol 305: H124–H134, 2013. [DOI] [PubMed] [Google Scholar]
- 65.Reffelmann T, Kloner RA. Transthoracic echocardiography in rats. Evaluation of commonly used indices of left ventricular dimensions, contractile performance, and hypertrophy in a genetic model of hypertrophic heart failure (SHHF-Mcc-facp-Rats) in comparison with Wistar rats during aging. Basic Res Cardiol 98: 275–284, 2003. [DOI] [PubMed] [Google Scholar]
- 66.Rerkpattanapipat P, D'Agostino RB Jr, Link KM, Shahar E, Lima JA, Bluemke DA, Sinha S, Herrington DM, Hundley WG. Location of arterial stiffening differs in those with impaired fasting glucose versus diabetes: Implications for left ventricular hypertrophy from the Multi-Ethnic Study of Atherosclerosis. Diabetes 58: 946–953, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Russell JC, Proctor SD. Small animal models of cardiovascular disease: Tools for the study of the roles of metabolic syndrome, dyslipidemia, and atherosclerosis. Cardiovasc Pathol 15: 318–330, 2006. [DOI] [PubMed] [Google Scholar]
- 68.Ryden L, Grant PJ, Anker SD, Berne C, Cosentino F, Danchin N, Deaton C, Escaned J, Hammes HP, Huikuri H, Marre M, Marx N, Mellbin L, Ostergren J, Patrono C, Seferovic P, Uva MS, Taskinen MR, Tendera M, Tuomilehto J, Valensi P, Zamorano JL, Zamorano JL, Achenbach S, Baumgartner H, Bax JJ, Bueno H, Dean V, Deaton C, Erol C, Fagard R, Ferrari R, Hasdai D, Hoes AW, Kirchhof P, Knuuti J, Kolh P, Lancellotti P, Linhart A, Nihoyannopoulos P, Piepoli MF, Ponikowski P, Sirnes PA, Tamargo JL, Tendera M, Torbicki A, Wijns W, Windecker S, De Backer G, Sirnes PA, Ezquerra EA, Avogaro A, Badimon L, Baranova E, Baumgartner H, Betteridge J, Ceriello A, Fagard R, Funck-Brentano C, Gulba DC, Hasdai D, Hoes AW, Kjekshus JK, Knuuti J, Kolh P, Lev E, Mueller C, Neyses L, Nilsson PM, Perk J, Ponikowski P, Reiner Z, Sattar N, Schachinger V, Scheen A, Schirmer H, Stromberg A, Sudzhaeva S, Tamargo JL, Viigimaa M, Vlachopoulos C, Xuereb RG. ESC guidelines on diabetes, prediabetes, and cardiovascular diseases developed in collaboration with the EASD: the Task Force on Diabetes, Prediabetes, and Cardiovascular Diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD). Eur Heart J 34: 3035–3087, 2013. [DOI] [PubMed] [Google Scholar]
- 69.Sarkozy M, Zvara A, Gyemant N, Fekete V, Kocsis GF, Pipis J, Szucs G, Csonka C, Puskas LG, Ferdinandy P, Csont T. Metabolic syndrome influences cardiac gene expression pattern at the transcript level in male ZDF rats. Cardiovasc Diabetol 12: 16, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shamaei-Tousi A, Stephens JW, Bin R, Cooper JA, Steptoe A, Coates AR, Henderson B, Humphries SE. Association between plasma levels of heat shock protein 60 and cardiovascular disease in patients with diabetes mellitus. Eur Heart J 27: 1565–1570, 2006. [DOI] [PubMed] [Google Scholar]
- 71.Shao CH, Wehrens XH, Wyatt TA, Parbhu S, Rozanski GJ, Patel KP, Bidasee KR. Exercise training during diabetes attenuates cardiac ryanodine receptor dysregulation. J Appl Physiol 106: 1280–1292, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen KH, Xiao RP. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem 282: 23354–23361, 2007. [DOI] [PubMed] [Google Scholar]
- 73.Shivalkar B, Dhondt D, Goovaerts I, Van Gaal L, Bartunek J, Van Crombrugge P, Vrints C. Flow mediated dilatation and cardiac function in type 1 diabetes mellitus. Am J Cardiol 97: 77–82, 2006. [DOI] [PubMed] [Google Scholar]
- 74.Soetkamp D, Nguyen TT, Menazza S, Hirschhauser C, Hendgen-Cotta UB, Rassaf T, Schluter KD, Boengler K, Murphy E, Schulz R. S-nitrosation of mitochondrial connexin 43 regulates mitochondrial function. Basic Res Cardiol 109: 433, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sommese L, Valverde CA, Blanco P, Castro MC, Rueda OV, Kaetzel M, Dedman J, Anderson ME, Mattiazzi A, Palomeque J. Ryanodine receptor phosphorylation by CaMKII promotes spontaneous Ca(2+) release events in a rodent model of early stage diabetes: The arrhythmogenic substrate. Int J Cardiol 202: 394–406, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tuncay E, Zeydanli EN, Turan B. Cardioprotective effect of propranolol on diabetes-induced altered intracellular Ca2+ signaling in rat. J Bioenerg Biomembr 43: 747–756, 2011. [DOI] [PubMed] [Google Scholar]
- 77.Ugurlucan M, Erer D, Karatepe O, Ziyade S, Haholu A, Gungor Ugurlucan F, Filizcan U, Tireli E, Dayioglu E, Alpagut U. Glutamine enhances the heat shock protein 70 expression as a cardioprotective mechanism in left heart tissues in the presence of diabetes mellitus. Expert Opin Ther Targets 14: 1143–1156, 2010. [DOI] [PubMed] [Google Scholar]
- 78.Varga ZV, Giricz Z, Liaudet L, Hasko G, Ferdinandy P, Pacher P. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta 1852: 232–242, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Varga ZV, Kupai K, Szucs G, Gaspar R, Paloczi J, Farago N, Zvara A, Puskas LG, Razga Z, Tiszlavicz L, Bencsik P, Gorbe A, Csonka C, Ferdinandy P, Csont T. MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J Mol Cell Cardiol 62: 111–121, 2013. [DOI] [PubMed] [Google Scholar]
- 80.Verkhratsky A, Fernyhough P. Mitochondrial malfunction and Ca2+ dyshomeostasis drive neuronal pathology in diabetes. Cell Calcium 44: 112–122, 2008. [DOI] [PubMed] [Google Scholar]
- 81.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 27: 1047–1053, 2004. [DOI] [PubMed] [Google Scholar]
- 82.Williamson CL, Dabkowski ER, Baseler WA, Croston TL, Alway SE, Hollander JM. Enhanced apoptotic propensity in diabetic cardiac mitochondria: Influence of subcellular spatial location. Am J Physiol Heart Circ Physiol 298: H633–H642, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yagi K, Kim S, Wanibuchi H, Yamashita T, Yamamura Y, Iwao H. Characteristics of diabetes, blood pressure, and cardiac and renal complications in Otsuka Long-Evans Tokushima Fatty rats. Hypertension 29: 728–735, 1997. [DOI] [PubMed] [Google Scholar]
- 84.Yu W, Zha W, Guo S, Cheng H, Wu J, Liu C. Flos Puerariae extract prevents myocardial apoptosis via attenuation oxidative stress in streptozotocin-induced diabetic mice. PLoS One 9: e98044, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhong Y, Ahmed S, Grupp IL, Matlib MA. Altered SR protein expression associated with contractile dysfunction in diabetic rat hearts. Am J Physiol Heart Circ Physiol 281: H1137–H1147, 2001. [DOI] [PubMed] [Google Scholar]
- 86.Zvara A, Bencsik P, Fodor G, Csont T, Hackler L Jr, Dux M, Furst S, Jancso G, Puskas LG, Ferdinandy P. Capsaicin-sensitive sensory neurons regulate myocardial function and gene expression pattern of rat hearts: A DNA microarray study. FASEB J 20: 160–162, 2006. [DOI] [PubMed] [Google Scholar]