

Abstract
Toll-like receptors (TLRs) are pattern recognition receptors that participate in host defense by recognizing pathogen-associated molecular patterns alongside inflammatory processes by recognizing damage associated molecular patterns. Given constant exposure to pathogens from gut, strict control of TLR-associated signaling pathways is essential in the liver, which otherwise may lead to inappropriate production of pro-inflammatory cytokines and interferons and may generate a predisposition to several autoimmune and chronic inflammatory diseases. The liver is considered to be a site of tolerance induction rather than immunity induction, with specificity in hepatic cell functions and distribution of TLR. Recent data emphasize significant contribution of TLR signaling in chronic liver diseases via complex immune responses mediating hepatocyte (i.e., hepatocellular injury and regeneration) or hepatic stellate cell (i.e., fibrosis and cirrhosis) inflammatory or immune pathologies. Herein, we review the available data on TLR signaling, hepatic expression of TLRs and associated ligands, as well as the contribution of TLRs to the pathophysiology of hepatic diseases.
Keywords: Toll-like receptors, Innate immunity, Liver disease, Pathophysiology, Signaling
Core tip: Toll-like receptors (TLRs) are known to be pattern recognition receptors that recognize pathogen- and damage-associated molecular pattern molecules and thus participate in the activation of innate immune system. TLR signaling plays a significant role in liver diseases, whereas inflammatory or immune pathologies targeting distinct liver cells are based on complex immune responses. Herein, we review the current data on TLR signaling, hepatic expression of TLRs and associated ligands, as well as the contribution of TLRs to the pathophysiology of hepatic diseases.
INTRODUCTION
Liver, main filter organ acting as a first line of defense, is continuously exposed to massive gut-derived antigenic load via the portal vein, whereas inflammatory signs occur under normal conditions owing to highly specific immune properties leading to immune tolerance[1-7].
Pathogen-associated molecular patterns (PAMP) are specific signature molecules essential to entire categories of microorganisms[8-11]. Innate immune system recognizes PAMPs via pattern recognition receptors (PRRs)[7-9,12,13] and consequent downstream signaling cascades for proper host recognition and prevention of immune system hyperactivation[7-9,14].
Toll-like receptors (TLRs) are a family of PRRs that induce innate immune system by recognizing PAMPs and damage-associated molecular pattern molecules (DAMPs)[15-18]. Although the recognition of PAMPs enables a prompt and effective protection against invading pathogens[5,11,12], TLRs also contribute to the activation of adaptive immune responses, epithelial regeneration and carcinogenesis and regulation of sterile inflammation[5,19,20].
Consistent with their extensive hetapocellular expression[7,18,21,22], TLRs have recently been recognized as principal elements of the hepatic immune system that also play a crucial role in liver physiology and pathophysiology[11,15,23]. Despite being constantly exposed to gut-derived PAMPs, healthy liver is free of inflammation risk due to presence of “liver tolerance” in which modulation of TLR signals also plays a role[5,15,23-25]. A tight regulation of TLR activation occurs at many levels involving the receptor itself, the signaling cascade and a distinct compartmentalization of TLRs[24,26,27]. Acute and chronic liver diseases are highly associated with triggering TLR signaling by gut-derived microbiota in the breakdown of the tolerance and sterile insult-associated products of damaged cells[28].
Ligand mediated stimulation of TLRs activates downstream adaptor molecules, including myeloid differentiation primary response protein 88 (MyD88), myeloid toll/interleukin (IL)-1 receptor (TIR)-domain-containing adaptor-inducing interferon-β (TRIF) and TRIF-related adaptor molecule (TRAM). This triggers signaling cascades that converge on nuclear factor-κB (NF-κB), interferon (IFN) response factors (IRFs) and mitogen-activated protein (MAP) kinases[23,29-32]. As a result, transcription of certain proinflammatory agents including IL-6, IL-12, IL-23, and tumor necrosis factor α (TNF-α) is induced[23,29-32].
TLR-mediated inflammatory-signaling pathways are shown to be associated with entire spectrum of liver diseases, from hepatitis, liver fibrosis and cirrhosis to alcoholic and nonalcoholic liver disease, ischemia/reperfusion injury, liver regeneration and hepatocellular carcinoma[4,5,7,8,15,18,23,33].
Herein, we review the available literature on TLR signaling, hepatic expression of TLRs and associated ligands, as well as the contribution of TLRs to the pathophysiology of hepatic diseases.
TLR FAMILY, DISTRIBUTION, LIGANDS
TLRs are a group of evolutionarily conserved type I transmembrane proteins responsible for innate immune and inflammatory responses[34-38]. They comprise an extracellular domain with receptor specific leucine-rich repeat motifs and a highly conserved cytosolic domain alike to the IL-1 receptor called TIR[13,29,36,37].
Of 13 TLRs exist in mammals, only TLRs 1-10 exist in humans[9,26,39-41]. The presence of multiple widely expressed TLRs enables recognition of different pathogens and thus initiation of appropriate immunologic response by the innate immunity system[30,42,43]. PAMPs include microbial molecular structures such as Gram-negative related lipopolysaccharide (LPS); Gram-positive bacteria related lipoteichoic acid and peptidoglycan (PGN); lipoglycans, lipoarabinomannan, lipopeptides and lipomannans from mycobacteria; zymosan from yeast; and DNA from viruses and bacteria[34,44].
DAMP include extracellular matrix and plasma membrane components, nuclear and cytosolic proteins and elements of damaged organelles[9,34,45,46].
Each TLR is able to recognize a particular molecular pattern[29]. TLR1, TLR2, TLR4, TLR5 and TLR6 bind to molecules associated with bacterial membrane such as LPS, lipoprotein and PGN, whereas TLR3, TLR7, TLR8 and TLR9 detect viral and bacterial or endogenous nucleic acids, including ssRNA, dsRNA, and unmethylated cytosine phosphate guanine (CpG)-containing DNA[29]. TLR4 along with TLR2 can recognize antigens from bacteria, fungi, parasites, viruses and DAMPs[47,48]. TLR10 is the only family member among humans with no definite ligand, function or localization[9,13].
Given their ability to detect wide range of non-microbial host-derived stimuli and their extensive expression in various cell types, TLRs are considered to participate in development, progression and resolution of several noninfectious inflammatory and immune diseases[37,49].
TLR SIGNALING PATHWAYS
Healthy liver contains low mRNA levels of TLRs and shows no activation of TLR-signaling pathways[5,50,51]. However, in the case of a breakdown in TLR tolerance against endogenous ligands under pathologic conditions, the TLR-related immune response induces TLR-ligand complex activated expression of proinflammatory/anti-inflammatory cytokines and interferons[7,9,27,52].
The differential host cell response after TLR ligand stimulation is associated with the fact that TLRs selectively use four main adaptor molecules, including MyD88, TIR domain-containing adaptor protein (TIRAP, or MyD88 adaptor-like), TIR domain-containing adaptor protein inducing interferon-b (TRIF) and TRAM[7,9,27,30,52].
Signal transduction pathways following ligand-induced receptor dimerization involve one or more TIR-containing adaptor molecules, such as IL-1 receptor-associated kinase (IRAK)-1, IRAK-4, TNF receptor-associated factor (TRAF)-6 and TANK binding kinase (TBK)-1, MAP kinases and IκB kinase (IKK). This leads to activation of the nuclear transcriptional factor kappa-B (NF-κB), interferon (IFN) regulatory factor 3 (IRF-3) and activator protein (AP)-1[37,53].
Upon binding with their ligand, all superfamily receptors except TLR3 use MyD88 to initiate signaling which may also act along with other adaptors, such as TIRAP, in the response induced by TLR4, TLR1/2, and TLR2/6. Activation of TLRs 5, 7, 8 and 9 also leads to NF-κb and AP-1 production, with no need for TIRAP to stimulate MyD88. TLRs 7 and 9 act through IRAK-1, 4 and TRAF-6, phosphorylate IRF-7 and lead to type 1 interferon mRNA expression. TLR3-mediated signaling uses only the TRIF adaptor molecule, which is also recruited by TLR4 in concert with another adaptor called TRAM[9,12,23,32,39,54] (Figure 1).
Figure 1.
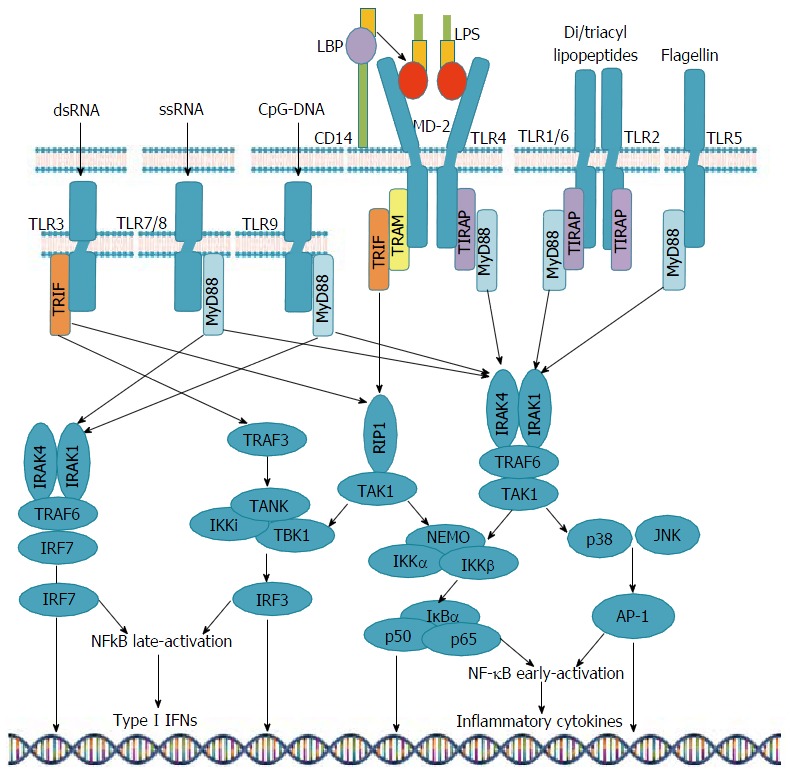
Toll-like receptors signaling pathways. TLR: Toll-like receptors; LPS: Lipopolysaccharide; NF-κB: Nuclear factor; IFNs: Interferons; LBP: LPS-binding protein; TIRF: Toll/interleukin-1 receptor-domain-containing adaptor-inducing interferon-β; MyD88: Myeloid differentiation primary response protein 88; TRAM: TRIF-related adaptor molecule; TIRAP: TIR domain-containing adaptor protein; IRAK: IL-1 receptor-associated kinase; TRAF: Tumor necrosis factor receptor-associated factor; TBK1: TANK binding kinase-1; IKK: IκB kinase; AP: Activator protein; JNK: c-Jun N-terminal kinase.
Hence, while intracellular signaling is similar, the final outcome of TLR activation differs depending on the nature of PAMPs, concomitantly activated TLRs and PRRs, the level of cytokines, and the cell stimulated[13,27,55-57]. Moreover, chronically activated signaling pathways is likely to induce transcription of oncogenic factors, which adds a further level of complexity to the intracellular signaling for these receptors[13,27,58].
TLR EXPRESSION AND SIGNALING IN HEPATIC CELL POPULATIONS
Under constant exposure to gut-derived microbiota, strict regulation of TLR signaling pathways is crucial in the liver, which otherwise may lead to inappropriate production of proinflammatory cytokines and interferons creating a predisposition to several autoimmune and chronic inflammatory diseases[9].
Liver cells are classified as parenchymal or non-parenchymal cells. Hepatocytes comprise 60%-80% of the parenchymal cells, whereas the remaining population of non-parenchymal cells include Kupffer cells (KCs), sinusoidal endothelial cells (SECs), hepatic stellate cells (HSCs), dendritic cells (DCs), biliary epithelial cells (BECs) and intrahepatic lymphocytes[1,9,33].
Besides distinct function of liver cells with a highly specific distribution of TLR[1,33], liver comprises many populations of cells with immune competence that may respond to TLR signals, indicating the complexity of immune responses underlying inflammatory or immune pathologies associated with the liver cells[10].
mRNA levels of TLR1, TLR2, TLR4, TLR6, TLR7, TLR8, TLR9, TLR10 and signaling molecules such as MD-2 and MyD88 are lower in liver as compared with the levels observed in other organs[50,51,59]. This discrepancy indicates the high tolerance to TLR ligands from the intestinal microbiota in liver[11], whereas no specific liver cell population is considered central in TLR-mediated pathologies, with the different effects of TLR ligation varying from cell to cell[10] (Table 1).
Table 1.
| TLR subfamily | Members | Expression of cell population in the liver (protein level) | Location | Ligand (origin) | Signaling | Final product-effect |
| TLR2 subfamily | TLR1/2 | NK cells, DCs (h) | Plasma membrane | Bacterial lipoproteins Triacylated lipopeptides | TIRAP-MyD88-NF-κB/AP-1/IRF5 pathway | Pro- and anti-inflammatory cytokines excluding type 1 IFNs; the apoptotic cascade via recruiting FADD leading to caspase-8 activation |
| TLR2/6 | Hepatocytes, Kupffer cells, NK cells, B cells, activated T cells, DCs (m), biliary epithelial cells | Diacylated lipopeptides LPS of Gram-positive bacteria Fungal zymosan Mycoplasma lipopeptides | TIRAP-MyD88-NF-κB/AP-1 pathway | |||
| TLR10 | Unknown | ND | ||||
| TLR3 subfamily | TLR3 | Hepatocytes, LSECs, Kupffer cells, NK cells, NKT cells, activated T cells, cDCs (m), biliary epithelial cells | Endosome | Double-stranded RNA (viruses) | PI3K/TRIF-IRF3 pathway TRAM-TRIF-NF-κB pathway PI3K/TRIF-RIP1-NF-κB pathway | Production of type 1 IFNs; the apoptotic cascade via recruiting FADD leading to caspase 8 activation; DC maturation |
| TLR4 subfamily | TLR41 | Hepatocytes, LSECs, Kupffer cells, NK cells, B cells, activated T cells, DCs (m), biliary epithelial cells, HSCs | Plasma membrane | LPS of Gram-negative bacteria; fusion protein (respiratory syncytial virus), envelope protein (mouse mammary-tumor virus); HMGB1, hyaluronan, HSP60, free fatty acids (endogenous ligands); HSP72 (cells during stress and injury) surfactant protein A; fibrinogen; fibronectin extra domain A | TIRAP-MyD88-NF-κB/AP-1 pathway TRAM-TRIF-NF-κB/IRF3 pathway | Pro- and anti-inflammatory cytokines excluding type 1 IFNs; the apoptotic cascade via recruiting FADD leading to caspase 8 activation; DC maturation; activating caspase-1 through adaptor molecule apoptosis associated speck-like protein2 |
| TLR5 subfamily | TLR5 | Biliary epithelial cells | Plasma membrane | Flagellin protein (bacteria) | MyD88-NF-κB/IRF5 pathway | Pro- and anti-inflammatory cytokines excluding type 1 IFNs |
| TLR9 subfamily | TLR7/8 | NK cells, B cells, DCs (h), DCs (m) | Endosome | Single-stranded RNA (viruses), double-stranded, shortinterfering RNA (siRNA) | MyD88 and endosomal acidification (maturation)-IRF7 pathway; MyD88- NF-κB pathway | High levels of type 1 IFN production in pDCs; proinflammatory cytokine production |
| TLR9 | LSECs, Kupffer cells, NK cells, B in mDCs and macrophages | Imidazoquinoline CpG-containing viral or bacterial DNA Endogenous host-DNA |
TLR4 requires LPS-binding protein (LBP), CD14 and MD2 to recognize LPS;
Containing a caspase recruitment domain (ASC)[33]. RIP1: Receptor-interacting protein 1; FADD: Fas-associated death domain; TLR: Toll-like receptors; LPS: Lipopolysaccharide; DCs: Dendritic cells; HSCs: Hepatic stellate cells; LSECs: Liver sinusoidal endothelial cells; IFNs: Interferons; DC: Dendritic cell; MyD88: Myeloid differentiation primary response protein 88.
Hepatocytes
Constituting 60% of liver cells, hepatocytes are the principal site for PRR production[5,33]. They express mRNA for all TLRs and are responsive to multiple PAMPs, while respond fairly weakly to TLR2 and TLR4 ligands[5,9,33]. While TLR4 expression in hepatocytes is not upregulated by proinflammatory mediators, hepatocytes show increased responsiveness to TLR2 ligands under inflammatory conditions leading to up-regulation of TLR2 expression by LPS, TNF-alpha, bacterial lipoprotein, and IL-1β in an NF-κB-dependent manner[5,11,33,60,61].
Kupffer cells
Accounting for approximately 20% of non-parenchymal cells, KCs play a significant role in host defense by orchestrating the inflammatory response via functional properties, including phagocytosis, antigen processing and presentation, and secretion of proinflammatory mediators such as cytokines, prostanoids, nitric oxide, and reactive oxygen intermediates[5,9,11,33,62].
KCs express TLRs 2, 3, 4 and 9 and have a higher threshold for activation when compared with other immune cells given their milieu[5,9,33,63].
KCs are less responsive to “LPS tolerance” in the physiological environment, whereas upon activation, they produce several pro-inflammatory (IL-6, IL-12, IL-18 and TNFα) and anti-inflammatory (IL-10) mediators[33,64-66]. Additionally, KCs produce IFN-β, upregulate the expression of MHC-II/costimulatory molecules and promote T cell proliferation and IFN-γ production; when stimulated with TLR3/TLR4 ligands; TLR1/TLR8 ligands and TLR1/2/4/6 ligands, respectively[22,33].
Hepatic stellate cells
Constituting < 1% of non-parenchymal cells, HSCs undergo an activation process after liver injury and become the main liver cell type that produce extracellular matrix, contributing onset of liver fibrosis[67-70].
HSCs express TLRs 4 and 9, whereas expression of TLR2 is induced by TLR4 stimulation in HSCs[68-70]. Activated HSCs express TLR4 and CD14 and respond to LPS upon the activation of IKK/NF-κB and c-Jun N-terminal kinase (JNK) as well as the secretion of proinflammatory cytokines such as transforming growth factor (TGF)-β, IL-6, IL-8 and several chemokines such as MCP-1, MIP-2, intercellular cell adhesion molecule 1, vascular cell adhesion molecule 1, and E-selectin[9,33,70]. TLR4 enhances TGF-β signaling, and stellate cell activation was shown to promote hepatic fibrosis[71]. In chimeric C3H/HeJ mice with TLR4 mutation in HSC or KCs, amelioration of hepatic fibrosis by LPS indicated a cardinal role for KCs and HSC in hepatic inflammation and fibrosis[9,72]. LPS was shown to downregulate the TGF-β pseudoreceptor BAMBI in quiescent HSCs to induce TGF-β signaling and stellate cell activation[71]. Additionally, TLR9 signaling activated via DNA from apoptotic hepatocytes was shown to modulate liver fibrosis via its effects on HSC differentiation through increased collagen production and inhibited HSC migration[73]. Hence, LPS and other TLR ligands are suggested to facilitate fibrogenic responses in the liver via their direct effects on HSCs[9,11,33].
Biliary epithelial cells
Accounting for approximately 5% of non-parenchymal cell population in the liver, BECs are commonly exposed to several gut-derived microbes[74,75]. BECs mainly express TLRs 2, 3, 4 and 5, which are upregulated by IFN-γ stimulation[74,75]. TLR2 and TLR4 activation results in increased IRAK-M expression and provide negative feedback in human intrahepatic BECs[76].
Under normal conditions, increased IRAK-M expression is critical in preventing undesired induction of the TLR signaling cascade, while in case of inflammatory conditions, upregulation of BEC-associated TLRs leads to IFN-c and TNF-α exposure, participating in biliary pathogenic responses[9,75].
Sinusoidal endothelial cells
Making up 50% of the non-parenchymal cells, SECs function in hepatic perfusion and nutrient supply[66,77-79]. They express TLR3, 4 and 9 and show increased NF-κB activation and CD54 expression alongside a limited ability to trigger leukocyte adhesion after LPS stimulation[66,77-79]. Although these effects indicate a scavenging role and thus the likelihood of SECs acting as antigen presenting cells, the exact role of the TLR signaling in inflammatory process in SEC remains inconclusive[9,11,33,66,77-79].
Isolated SECs from WT mice were shown to respond to TLR1, 2, 6 and 9 ligands via producing TNF-α; to TLR3 ligands by producing TNF-α, IL-6 and IFN-β; and to TLR4 ligands via production of TNF-α and IL-6[22,33]. Upon TLR8 ligand binding, SECs leads to TNF-α production alongside upregulation of major histocompatibility complex (MHC)-II and co-stimulatory molecules. Stimulation of SECs by TLR1, 2 or 6 ligands is suggested to be associated with activation of allogeneic T cells, as evaluated by the mixed lymphocyte reaction[22,33]. The SEC immune response is also modulated by LPS tolerance, which appears to be based on prostanoid expression rather than regulation at the level of TLR4 surface expression[78]. Although SECs have been suggested to be involved in the hepatic uptake of LPS in some studies, several studies have not confirmed such a role[33,80,81].
Hepatic dendritic cells
Comprising < 1% of non-parenchymal cells, hepatic DCs are recruited into the liver sinusoids during inflammation and then they may migrate to periportal and pericentral areas[5,33,82,83]. Plasmacytoid DCs (pDCs), myeloid DCs, lymphoid DCs, mixed lymphoid + myeloid DCs and natural killer DCs are amongst the DC subsets, whereas lymphoid and myeloid DCs are considered conventional DCs[33,82,83].
Each DC subset show distinct TLR expression pattern in humans with TLR1, 7 and 9 expression via pDCs, while expression of all TLRs excluding TLR9 by other DC subsets[20,33,84]. Cytokines TNF-α, IL-6 and IL-12 TLR7 are produced by hepatic pDCs upon TLR7 and TLR9 activation, whereas TNF-α and IL-6 in response to TLR2, TLR3 and TLR4 activation[50,85].
TLRs IN THE PATHOPHYSIOLOGY OF LIVER DISEASES
Increasing evidence suggests that TLRs have significant contribution to the pathogenesis and progression of several liver diseases, i.e., non-alcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), viral hepatitis, autoimmune liver disease and hepatic inflammation-fibrosis-carcinoma (IFC) sequence including hepatic fibrosis and/or cirrhosis and hepatocarcinoma[9,11,13,15,23,33].
LPS/TLR4 and TLR2 signaling have been suggested to be principal actors in the human hepatic IFC sequence associated with viral chronic hepatitis[86], while the participation of TLR3 in the pathophysiology of several liver diseases has also been suggested in the recent studies[11,15,23,87] (Figure 2).
Figure 2.
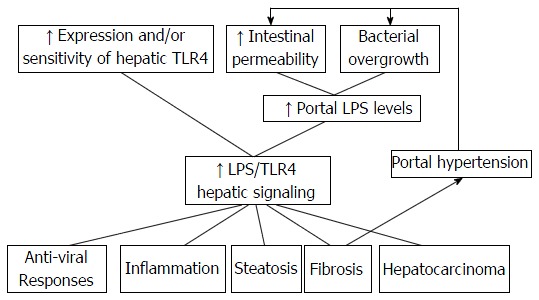
Enhanced lipopolysaccharide/toll-like receptors 4 signaling in chronic liver diseases. Induction of anti-viral responses, inflammation, steatosis, fibrosis, and hepatocarcinoma via LPS/TLR4 signaling alongside hepatic fibrosis mediated portal hypertension which further increases bacterial overgrowth and intestinal permeability, creating a positive feedback process. TLR4: Toll-like receptors 4; LPS: Lipopolysaccharide.
NAFLD and steatohepatitis
NAFLD and steatohepatitis is characterized by a pathologic spectrum that ranges from fatty liver (hepatic steatosis) to cirrhosis with intervening non-alcoholic steatohepatitis (NASH) and usually occurs in association with obesity and insulin resistance[13,72,88-90].
Increased serum PAMP levels were observed in both experimental models and in NAFLD patients[9,18,91-96]. A shift in microbial populations to adopt an “obese” phenotype in NAFLD is referred to as “metabolic endotoxaemia”, in which a high-fat diet is associated with elevated levels of LPS translocation[27,90,97].
While TLR2, TLR4 and TLR9 participate in the development of NASH and NAFLD, LPS-TLR4 is considered to be the main pathway for the progression of NAFLD[98-100]. The role of bacterial overgrowth has also been associated with development of NASH, emphasizing the interaction between bacterial overgrowth, gut permeability and liver injury[90,101,102].
While the role of adipose tissue macrophages in the development of NAFLD is not yet clear, KCs are known to play a pivotal role in the development of NAFLD alongside accompanying hepatic inflammation and related complications[18,98].
When inflammation occurs in NAFLD, NF-κB and transcriptional factor AP1 are activated, stimulating the production of TNF-α and IL-10, in particular, by KCs[23,103]. Studies in animal models indicated the likelihood of TLRs 2, 4 and 9 to participate in NAFLD onset or progression[9,18,91,104]. LPS/TLR4 and TLR9 signaling in KCs have been associated with both onset and progression of NAFLD by inducing reactive oxygen species (ROS)-dependent activation of X-box binding protein-1 and IL-1b, respectively, whereas induction of hepatic steatosis occurs independent of TLR2 signaling in KCs[18,104-106] (Figure 2).
While free fatty acids and denatured host DNA are considered to be potential candidates to activate TLR2, TLR4 and TLR9 signals, no clear-cut evidence exists to confirm their capacity to activate TLRs in NAFLD[18]. TLR4 signaling has been considered to play a major role in the pathogenesis of NAFLD that operates via KCs stimulation and increased ROS and TNF-α production[13].
ALD
ALD is described along a disease spectrum ranging from steatosis and steatohepatitis to fibrosis and cirrhosis and potential development of hepatocellular carcinoma (HCC)[90,107].
Despite a strong association between alcohol and hepatotoxicity, the exact pathogenesis has not yet been elucidated[90]. Involvement of the gut microbiota via a “leaky” gut has been indicated in the development of ALD[18], whereas the role of alcohol has also been suggested in increasing gut permeability by disrupting tight junctions[108,109]. Increased plasma LPS levels and hepatic endotoxin levels, which leads to increased TLR4 signaling on KCs, HSC, LSECs and hepatocytes and thus the release of pro-inflammatory cytokines have been associated with inflammation and liver damage[9,107,108,110].
Recent studies indicate significant contribution of TLR4 signaling and thus the crucial role of both KCs and HSCs in development of gut-derived endotoxin related effects in ALD[18]. Chronic alcohol consumption is also associated with the increased expression of TLR1, TLR2, TLR4 and TLR6-TLR9, which further potentiates the secretion of the pro-inflammatory TNF-α in response to LPS[111].
KCs produce pro-inflammatory cytokines (TNF-α, IL-1, IL-6 and IL-8, chemokines) and profibrogenic factors (TGF-β) under post-LPS mediated TLR4-dependent stimulation, and consequent liver inflammation and stellate cell activation induce liver fibrosis[9,15,112,113]. The TLR4-dependent downstream signaling cascade in ALD was shown to proceed via the MyD88-independent pathway, possibly via adapter molecule TRIF[114]. Nonetheless, increased expression of not only TLR4 but also other TLRs such as TLR1, 2, 6, 7, 8 and 9 was shown in an experimental chronic alcohol model[115].
Although activation of KCs via TLR4 signaling is a key event in the pathogenesis of alcohol-induced liver injury[18], recent data emphasize the activation of TLR4 signaling in HSCs as well, indicating the their contribution to alcohol-induced hepatocyte injury, steatosis, inflammation, and fibrogenesis[18,116]. In HSCs, activated TLR4 signaling downregulates TGF-β pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI), resulting in enhancement of TGF-β signaling, whereas BAMBI downregulation is dependent on MyD88 but not TRIF[18,110]. The TLR4-TRIF-IRF3-dependent pathway associated with bone marrow-derived cells including KCs is considered to be more important than the TLR4-MyD88-dependent pathway in the development of alcoholic steatohepatitis[18,110,114].
Acting through upregulation of TLR4 and MD-2 and induction of a Th1-type immune response, bacterial DNA recognition by TLR9 was also shown to be associated with LPS induced liver injury[117], indicating the likelihood of TLR9 signaling to contribute to pathogenesis of ALD[18].
Hepatic fibrosis and cirrhosis
The development of hepatic fibrosis and consequent cirrhosis upon continued liver insults may occur in any type of chronic hepatic injury, including viral hepatitis, alcohol, autoimmune and metabolic disease[9,67].
Prolonged or repeated liver injury leads to a maladaptive interplay of hepatocytes, HSCs and KCs in association with TLR expression, eventually resulting in abnormal extracellular matrix protein deposition in the liver[35,67,118].
LPS-TLR4 activation is considered essential for hepatic fibrogenesis, whereas TLR4 is expressed on KCs and HSCs, the key mediators of hepatic fibrogenesis[27,75,80,81].
KCs express the highest levels of TLR4 and act as the principal target of LPS leading to release of several pro-inflammatory and pro-fibrogenic mediators[5,27,71,114,119]. However, HSCs are crucial in the pathogenesis of fibrosis and cirrhosis given their myofibroblastic phenotype and ability to produce collagen, the principal component of fibrotic tissue[9,120].
Activation of HSC occurs either via pro-inflammatory cytokines and growth factors secreted by LPS-TLR4-stimulated KCs, or directly via LPS-TLR4-dependent HSC stimulation[9,71]. LPS/TLR4 signaling in HSCs is essential for development of liver fibrosis and acts via stimulating production of chemokines that recruit KCs alongside enabling unrestricted activation of HSCs by KCs-derived profibrogenic cytokine TGF-β[11,13,103,121] (Figure 2).
TLR4 activation in HSCs is considered to be the main step for collagen production and the main mediator of fibrosis and cirrhosis[9,11,67,70,71].
KCs induce fibrogenesis by means of proinflammatory and profibrogenic cytokine secretion, whereas HSCs are the leading source of extracellular matrix production in the fibrotic liver[11,67].
TLR9 signaling-associated metabolic pathways are also considered important in the genesis of hepatic fibrosis in vivo, leading to activation of pathways such as IL-1 production and thus HSCs by upregulating profibrogenic genes, such as procollagen type I and tissue inhibitor metalloproteinase-1[16,69,103,104].
Moreover, a deficiency of TLR3-mediated NK cell-dependent apoptosis of HSCs has been linked to the progression of alcohol-induced liver fibrosis[122,123]. Upregulation of TLR2 was shown to promote liver inflammation and fibrogenesis in NASH[106] and HSCs activation and inflammation response during carbon tetrachloride-induced liver fibrosis mediated via MAPK and NF-jB signaling pathways[124], whereas TLR5 was also shown to be directly involved in the progression of fibrosis via activation of the NF-κB and MAPK signaling pathways[52].
Hepatitis B
Hepatitis B virus (HBV) is a DNA virus responsible for acute hepatitis, which is self-limiting in 80%-90% of adults and chronic in 10%-20% of cases[5,125]. Hepatitis B is associated with an increased risk of developing cirrhosis, hepatic decompensation and HCC, but prognosis shows interpersonal variation depending on the viral susceptibility and induction of antiviral immune response[126,127].
Indicating the role of TLRs in HBV infection, the activation of TLR3, TLR7 and TLR9 as well as TLR4 and TLR5, has been associated with blockage of viral replication via IFN-dependent inhibition of HBV[76,128,129]. Moreover, HBV leads to TLR downregulation alongside restriction of receptor activity, increasing the likelihood of persistent infection[27].
In vitro HBV studies on TLR expression in HepG2 cells revealed elevated expression of TLRs 2, 3, 4, 5, 6, 7 and 9 mRNA upon ligand binding along with an induced IFN response and abolished HBV DNA replication and RNA transcription, whereas no or very limited expression of TLRs 1, 8 and 10[9,130]. Furthermore, transfection of HBV-positive cell lines with TLR adaptor molecules was shown to be associated with elevated TLR activity and a consequent reduction in HBV DNA and mRNA levels[131], whereas HBV replication was completely abolished after injection of TLR3, TLR4, TLR5, TLR7 and TLR9 ligands into HBV transgenic mice[129].
TLR1, TLR2, TLR4 and TLR6 were shown to be downregulated in HBV-infected peripheral blood monocytes along with a decreased cytokine response to TLR2 and TLR4 ligands[132]. Downregulation of TLR2 on hepatocytes and hepatic KCs was demonstrated in HBeAg-positive CHB-infected patients, whereas upregulation of TLR2 and cytokine expression was observed in HBeAg-negative CHB patients[133]. Hence, HBeAg-induced downregulation of TLR2 via precore protein has been accused for the accelerated progression of disease in HBeAg-positive patients[9,133].
Although HBV is able to downregulate TLRs and thus avoid anti-viral pathways, prolonged infection and loss of HBeAg is considered likely to upregulate TLR signaling pathways such as TLR2 that are not primarily involved in anti-HBV responses while trigger hepatic inflammation and disease progression[11].
In vitro analysis of HBV-Met cells revealed that TLR-treated KCs and SECs to have a modulatory effect on HBV replication[134]. TLR3- and TLR4-stimulated KCs and TLR3-activated SECs were shown to affect HBV replication via MyD88-independent pathway[66]. HBV-suppressing effect was mediated by IFN-β in case of TLR3 ligand activation, whereas by cytokines of an undefined nature in case of TLR4-activated KCs[66].
HBV is a stealth virus and thus does not induce an IFN response during the early phase of infection, whereas its recognition by liver resident cells is considered likely to activate innate immune responses without IFN induction[107,135]. Notably, HBV was shown to be recognized by hepatic NPCs, mainly by KCs, leading to NF-κB-dependent induction of the release of the inflammatory cytokines IL-6, IL-8, TNF-α and IL-1β as well as reduced expression of transcription factors essential for HBV gene expression and replication including hepatocyte nuclear factor (HNF) 1α and HNF4α[136].
Hepatitis C
Hepatitis C virus (HCV) is a hepatotropic virus responsible for development of chronic hepatitis and related complications such as liver cirrhosis, liver failure or HCC[137,138].
Similarly to HBV, current evidence indicates that HCV selectively impairs activation of TLR signaling controlling HCV replication, while it concomitantly stimulates TLR pathways that generate a chronic inflammatory state leading to persistent liver injury[11,27,139,140].
HCV-induced inhibition of TLR signaling contributes to its chronicity related to virus dissemination, inflammation and eventual progression to fibrosis and cirrhosis[9,11].
Regulation of HCV replication by non-parenchymal liver cells occurs through the production of IFN-β upon their stimulation by TLR3 and TLR4[141]. The inhibitory effect of HCV proteins on TLR7 and TLR9, is also likely to prevent virus clearance[27]. Furthermore, activation of TLR2 along with TLR1 and TLR6 and possibly TLR4 by HCV core protein and NS3 promotes hepatic inflammation and injury[142-145].
In the presence of HCV, significantly decreased TLR7 expression along with TLR7-independent activation of IRF-7 pathway was demonstrated both in vitro and in vivo[146].
The NS3/4A serine protease of HCV, HCV NS3 protein and HCV NS5A act via three signaling pathways including the TLR3-TRIF-TBK1-IRF-3, TLRMyD88, and RIG-I/MDA5-IPS-1 pathways to enable HCV to evade innate immune signaling[33]. Moreover, LPS, the HCV core protein and IFN-γ have been suggested to amplify inflammatory monocyte/macrophage activation via formation of MyD88-IRAK complexes, increased NF-κB activation and increased production of TNF-α, leading to the loss of TLR tolerance[147].
Based on these findings, both host- and virus derived factors have been considered likely to act on macrophages to induce persistent inflammation during chronic HCV infection[53,107].
Hepatocarcinoma
Diseases associated with uncontrolled innate immunity related to TLR ligand exposure in the liver (fibrosis, hepatitis B and C infection, ALD and NASH) are also among the etiologies for HCC. Therefore, it appears likely that TLRs play a role in the development of inflammation-associated liver cancer and are involved in the progression of HCC[18,107]. Hence, chronic hepatic inflammation and fibrosis, as regulated by TLR activation, promotes HCC formation in approximately 10% of cases of cirrhosis[9,54].
TLRs, TLR4 in particular, are considered to play a significant role in associating hepatic chronic inflammation and hepatocarcinoma[13]. A significant regression in liver tumors in TLR4 and MyD88 deficient mice indicates a prominent contribution of TLR signaling to hepatocarcinogenesis[23,148].
HCC has been indicated to be promoted via gut microbiota and TLR4 in association with increased production of proinflammatory cytokines (TNF-α, IL-6), hepatomitogen epiregulin expression and prevention of apoptosis, whereas a reduction in the development of HCC was shown via gut sterilization, germ-free status or TLR4 inactivation[18,149,150].
Activation of KCs via TLRs is considered to be involved in the process of tumorigenesis[18] by inducing proinflammatory cytokines and hepatomitogens responsible for enhanced development of HCC[150,151], whereas TLR4 expression on non-marrow-derived resident liver cells is considered to be required for the promotion of HCC[149].
TLR4 contributes significantly to hepatic inflammation and fibrosis, whereas upregulation of inflammatory factors such as COX-2 and NF-κB by TLR4 as well as the TLR adaptor protein Myd-88 is also important in hepatocarcinogenesis[148,152-155]. TLR3 expression is suggested to contribute to hepatocarcinoma via proapoptotic activity, while activation of TLR9 via CpG DNA of HBV has been associated with malignant transformation in liver cells[27,156,157].
Although, TLR2 binding with ligands such as HMGB1 and HSPA1A is associated with tumor enhancement, the effect of TLR2 activation is considered likely to differ according to the phase of HCC carcinogenesis, with anti-oncogenic potential slowing down the onset and development of HCC in earlier phases, whereas pro-oncogenic potential during later stages that promotes the progression of inflammation and fibrosis[158].
Activation of the NF-κB and JNK pathways and higher expression levels of IKKα and IKKβ are considered critical in the production of the cytokines related to TLR-induced liver damage and HCC progression[107].
Recently, spontaneous HCC development was demonstrated in hepatocyte-specific TAK1 deleted (TAK1DHEP) mice along with a resistance for HCC development that occurs via deletion of MyD88, TLR4 or TLR9 signaling[159].
Alcohol and HCV are suggested to interact in causing progression of liver disease and malignancy, whereas TLR4, TLR4 downstream gene Nanog and activated LPS-TLR4 are also considered to contribute to this synergy via triggering proliferative and anti-apoptotic signals to non-marrow-derived resident liver cells and thus HCC progression[9,149,150,160].
Ischemic/reperfusion injury and liver allograft rejection
Ischemia-reperfusion (I/R) injury in partial hepatectomy and liver transplantation is associated with the release of various endogenous ligands for hepatic tissue TLRs and thus the activation of complex signaling pathways that induce neutrophilic and T-lymphocytic tissue inflammation and injury[53,161,162].
Among the most studied TLRs in hepatic I/R, TLR4 was shown to participate in certain acute sterile injury models, including liver I/R, by mobilizing the immune system upon detection of endogenous ligands, whereas limited data are available on TLR2 and TLR9[163,164].
MyD88-independent activation of TLR4 by DAMPs is considered central to the inflammatory process observed in I/R lesions[165-167], whereas HSP, heparan sulfate, fibronectin, fibrinogen, hyaluronan and HMGB1 are known to act as endogenous ligands for TLR4 activation in hepatic I/R injury[5,163].
Release of HMGB1 activates the cell surface TLR4 on KCs and leads to a subsequent release of cytotoxic mediators (TNF-α, IL-6 and chemokine IP-10), alongside an inappropriate activation of the pro-apoptotic protein kinase JNK and stress-responsive NF-κB, all of which are mediators of cell injury[5,163,168,169]. Cellular expression of TLR4 is further upregulated via newly synthesized mediators such as TNF-α, leading to formation of a vicious cycle of proinflammatory cytokine production[61,163,170].
Downstream TLR4 signaling pathways in I/R injury seems to be independent of MyD88 signaling, whereas TRIF-dependent activation of the interferon response and IRF1 expression is considered critical for mediating I/R injury in hepatocytes in terms of releasing the danger signal HMGB1[164,171,172]. Hence, TLR4, IRF1 and HMGB1 are considered three important and interacting mediators of I/R injury[164].
Albeit not consistent, available data suggest that besides lack of TLR4, downregulation of TLR2 expression in the donor organ also suppress I/R injury[27,165,173]. Accordingly, given the amelioration of liver injury in I/R via non-selective inhibition of TLR2 and TLR4 activation by certain molecules such as bicyclol or N-acetylcysteine, role of TLRs in I/R lesion has been emphasized[27,174,175].
TLR9, which shows affinity toward both pathogen-derived and endogenous host DNA, is considered to play a crucial role in non-pathogen-induced hepatic I/R injury by causing neutrophil activation, liver necrosis, and inflammatory cytokine release[163,176,177].
Although TLR signaling dependent early activation of the innate immune system is consistently reported in the setting of I/R injury, additional studies are required to fully explore the roles of other TLRs and TLR signaling pathways in I/R injury[163,164].
Liver regeneration after partial hepatectomy
Recognizing the mechanism of liver regeneration is important not only for managing acute liver failure and post-transplant hepatic dysfunction but also for disturbed liver regeneration in NASH or NAFLD and advanced liver fibrosis[178]. The deposition of excessive amounts of extracellular matrix, the presence of persistent inflammation, the transformation of SECs and HSCs, portal blood flow reduction and increased JNK activity are considered among the factors associated with the regenerative ability of fibrotic livers[178,179].
TLR/MyD88-mediated pathways are associated with onset of liver regeneration after partial hepatectomy (PH) via activation of NF-κB, release of TNF-α and IL-6 and the expression of the immediate early genes for cell replication in hepatocytes, whereas distinct TLR ligands responsible for the priming process have not yet been clarified[33,178]. No contribution of TLR2, TLR4 or TLR9 to MyD88-mediated pathways and no influence of TLR2 or TLR4 on proinflammatory cytokine production or gene replication have been reported for liver regeneration after PH[33,180,181].
In fact, given the inhibition of regenerative process via excessive TLR signaling produced by LPS injection after PH, the magnitude of TLR signaling is considered critical for intact liver regeneration[178,182].
TLR3 signaling, which utilizes a distinct adaptor protein, TRIF, is considered to attenuate the initiation of liver regeneration via TLR3-dependent NF-κB activation in hepatocytes and TLR3-induced IFN-γ through STAT1 and consequent induction of the IRF-1 and p21 pathways[178,183,184].
In addition, although a non-TLR MyD88-dependent pathway with IL-1 and IL-18 has been suggested to play a role in allograft rejection initially, findings on the existence of normal liver regeneration after PH in caspase 1-deficient mice indicate unremarkable participation of IL-1β and IL-18 in liver regeneration[178].
Hepatic autoimmune disorders
Although antibody formation against self-antigens is key to the development of autoimmune hepatic diseases, including autoimmune hepatitis, primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC)[185], recently the influence of gut microbiota on the propagation of these diseases has been indicated[90].
Given that the liver is considered a classical immunoprivileged site, TLR signals may act as an important promoter for overcoming this immunoprivilege and in–ducing hepatic autoimmune disease[11,13,186].
Previous studies have suggested regulator role of gut-derived products on T cell function within the liver[90], based on the connection between TLR4 signaling and the trapping of CD8+ T cells in the murine liver[187], as well as contribution of TLR9 to the homing and stimulation of hepatic NKT cells via a KC and IL-12 dependent process[188]. The role of LPS/TLR4 signaling has been indicated in the pathogenesis of PBC and PSC[13]. Monocytes from PBC patients have been suggested to show increased sensitivity to activation of selective TLRs (TLR2, TLR4, TLR3, TLR5 and TLR9), while the subsequent release of proinflammatory cytokines has been associated with development of self-tolerance and autoimmune progression[189] (Figure 2).
LPS was shown to accumulate in significant amounts in the biliary epithelia of PBC patients, whereas positivity for IgM antibodies against lipid A, an immunogenic and toxic component of LPS, is confirmed in 64% of PBC sera[190,191]. TLR4 expression is significantly elevated in BECs, periportal hepatocytes and blood monocytes of PBC patients[192,193], whereas LPS/TLR4 signaling has been associated with an increased release of proinflammatory cytokines such as IL-1b, IL-6, IL-8 and TNF-α[189]. TLR4 ligand-stimulated NK cells have been suggested to be associated with BEC damage in the presence of TLR3 ligand-activated monocytes among PBC patients[194]. Despite similar levels of TLRs in BECs isolated from livers from patients and controls, stimulation via TLR3 agonist poly I:C and co-culture with liver-infiltrating mononuclear cells resulted in elevated chemokine levels in livers from patients[195]. Moreover, when compared to patients with autoimmune hepatitis and Hepatitis C, patients with PBC showed higher levels of TLR3 and IFN-α/β in portal tracts and liver parenchyma[196]. Furthermore, TLR9 ligand (CpG) stimulation of peripheral blood monocytes from PBC patients was demonstrated to activate IgM-producing B cells and to increase TLR9 expression on these cells[197,198]. These findings emphasize the role of innate immunity not only in the pathogenesis and progression of PBC but also in the regulation of adaptive immune responses[9].
The role of TLRs in PSC has not been extensively studied[11]. Abnormal LPS accumulation was demonstrated in BECs in PSC[190]. Stimulating isolated BECs with anti-BEC antibodies from patients with PSC leads to increased expression of TLR4 along with higher levels of inflammatory cytokines in the presence of LPS[199].
Accordingly, increased LPS accumulation and TLR4 expression in BECs has been suggested to induce breakdown of self-tolerance and onset of bile duct damage in PBC and PSC thorough their stimulatory effects on selective pro-inflammatory cytokines with a critical role[13]. Given the signs of inflammatory bowel disease to exist in most patients with PSC and the likelihood of gut factors to induce response onset per se with no preceding immune cell dysfunction, future investigations are needed addressing the role of gut microbiota in conjunction with PSC and PBC to provide a better understanding of the mechanisms and treatment of these complex diseases[90].
CONCLUSION
TLRs have been recognized as key regulators of innate and adaptive immune responses in the liver, although growing evidence suggests the critical role of TLR dysregulation in the pathogenesis and progression of many liver diseases[9,107]. TLRs, mainly TLR4 and TLR2, play a fundamental role in the inflammation and fibrosis of the liver and promote the progression of chronic liver diseases[27,35,86]. Indeed, LPS/TLR4 signaling is enhanced and essential in liver diseases such as ALD, NAFLD, PSC, CBP and fibrosis, and inhibition of TLR4 has been associated with amelioration of liver injury, emphasizing the contribution of LPS/TLR4 signaling to the pathogenesis of liver diseases[13].
The local innate immune system represented by liver cells participates in tolerance induction or inflammation alongside its interaction with the adaptive immune system, whereas suppression of the TLR system in the liver by pathogens enhance chronicity of infection[107]. Therefore, targeting TLR signaling at different levels of cascade appears to offer therapeutic potential in the management of chronic liver disease[11].
LPS/TLR4 signaling pathway has been recognized as an important pharmacological target in chronic liver diseases. Suppression of TLR4 signaling via modulation of LPS production, TLR and co-receptor expression and downstream signaling molecules has been shown to ameliorate liver injury, indicating the contribution of LPS/TLR4 signaling to the pathogenesis of chronic liver diseases. Given the likelihood of systemic suppression of TLR4 to disable responding pattern of TLR4 to invading pathogens, modulation of intestinal microbiota via probiotics and symbiotics become a preferred therapeutic strategy for liver diseases, associated with favorable tolerability and safety[13,23]. Besides, certain synthetic ligands of TLRs have been considered to act as target molecules for drug development given their effects on regulation of innate and adaptive immune responses, including TLR activators (for infections and certain cancers), TLR inhibitors (for inflammatory diseases and sepsis) as well as TLR neutralizing antibodies[34,37]. Further investigation of the role of TLR pathways in liver diseases addressing the downstream mediators and regulation of TLR signaling, the specific cell populations involved, the role of TLR polymorphisms and the mechanisms underlying liver tumorigenesis is needed to transfer knowledge on TLR pathophysiology into clinical practice in treating human liver diseases[5,23].
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Turkey
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: Author declares no conflict of interests for this article.
Peer-review started: June 17, 2016
First decision: July 11, 2016
Article in press: September 22, 2016
P- Reviewer: Arias J, Balaban YH, Skrypnyk IN S- Editor: Qiu S L- Editor: A E- Editor: Li D
References
- 1.Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 2.Tiegs G, Lohse AW. Immune tolerance: what is unique about the liver. J Autoimmun. 2010;34:1–6. doi: 10.1016/j.jaut.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Crispe IN. Immune tolerance in liver disease. Hepatology. 2014;60:2109–2117. doi: 10.1002/hep.27254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henao-Mejia J, Elinav E, Thaiss CA, Licona-Limon P, Flavell RA. Role of the intestinal microbiome in liver disease. J Autoimmun. 2013;46:66–73. doi: 10.1016/j.jaut.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology. 2006;130:1886–1900. doi: 10.1053/j.gastro.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 6.Doherty DG. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J Autoimmun. 2016;66:60–75. doi: 10.1016/j.jaut.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 7.Norberto C, Chávez-Tapia, Leticia González-Rodríguez, MinSeung Jeong, Yanine López-Ramírez, Varenka Barbero-Becerra, Eva Juárez-Hernández, Juan L. Romero-Flores, Marco Arrese, Nahúm Méndez-Sánchez, Misael Uribe. Current evidence on the use of probiotics in liver diseases. J Functional Foods. 2015;17:137–151. [Google Scholar]
- 8.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–1407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kesar V, Odin JA. Toll-like receptors and liver disease. Liver Int. 2014;34:184–196. doi: 10.1111/liv.12315. [DOI] [PubMed] [Google Scholar]
- 10.Bigorgne AE, Crispe IN. TLRs in Hepatic Cellular Crosstalk. Gastroenterol Res Pract. 2010:2010. doi: 10.1155/2010/618260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–720. doi: 10.1136/gut.2008.156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 13.Soares JB, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int. 2010;4:659–672. doi: 10.1007/s12072-010-9219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvalho FA, Aitken JD, Vijay-Kumar M, Gewirtz AT. Toll-like receptor-gut microbiota interactions: perturb at your own risk! Annu Rev Physiol. 2012;74:177–198. doi: 10.1146/annurev-physiol-020911-153330. [DOI] [PubMed] [Google Scholar]
- 15.Szabo G, Dolganiuc A, Mandrekar P. Pattern recognition receptors: a contemporary view on liver diseases. Hepatology. 2006;44:287–298. doi: 10.1002/hep.21308. [DOI] [PubMed] [Google Scholar]
- 16.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto M, Takeda K. Current views of toll-like receptor signaling pathways. Gastroenterol Res Pract. 2010;2010:240365. doi: 10.1155/2010/240365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol. 2013;28 Suppl 1:38–42. doi: 10.1111/jgh.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rakoff-Nahoum S, Medzhitov R. Role of toll-like receptors in tissue repair and tumorigenesis. Biochemistry (Mosc) 2008;73:555–561. doi: 10.1134/s0006297908050088. [DOI] [PubMed] [Google Scholar]
- 20.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 21.Hösel M, Broxtermann M, Janicki H, Esser K, Arzberger S, Hartmann P, Gillen S, Kleeff J, Stabenow D, Odenthal M, et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology. 2012;55:287–297. doi: 10.1002/hep.24625. [DOI] [PubMed] [Google Scholar]
- 22.Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, Bucchi A, Krux F, Dittmer U, Yang D, et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology. 2010;129:363–374. doi: 10.1111/j.1365-2567.2009.03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 24.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 25.Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology. 2008;47:729–736. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
- 26.Hopkins PA, Sriskandan S. Mammalian Toll-like receptors: to immunity and beyond. Clin Exp Immunol. 2005;140:395–407. doi: 10.1111/j.1365-2249.2005.02801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pimentel-Nunes P, Soares JB, Roncon-Albuquerque R, Dinis-Ribeiro M, Leite-Moreira AF. Toll-like receptors as therapeutic targets in gastrointestinal diseases. Expert Opin Ther Targets. 2010;14:347–368. doi: 10.1517/14728221003642027. [DOI] [PubMed] [Google Scholar]
- 28.Szabo G, Billiar TR, Machida K, Crispe IN, Seki E. Toll-like receptor signaling in liver diseases. Gastroenterol Res Pract. 2010;2010:971270. doi: 10.1155/2010/971270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohammad Hosseini A, Majidi J, Baradaran B, Yousefi M. Toll-Like Receptors in the Pathogenesis of Autoimmune Diseases. Adv Pharm Bull. 2015;5:605–614. doi: 10.15171/apb.2015.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 31.Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 32.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y, Sun R. Toll-like receptors in acute liver injury and regeneration. Int Immunopharmacol. 2011;11:1433–1441. doi: 10.1016/j.intimp.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Song E, Bai B, Vanhoutte PM. Toll-like receptors mediating vascular malfunction: Lessons from receptor subtypes. Pharmacol Ther. 2016;158:91–100. doi: 10.1016/j.pharmthera.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Huebener P, Schwabe RF. Regulation of wound healing and organ fibrosis by toll-like receptors. Biochim Biophys Acta. 2013;1832:1005–1017. doi: 10.1016/j.bbadis.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Booth J, Wilson H, Jimbo S, Mutwiri G. Modulation of B cell responses by Toll-like receptors. Cell Tissue Res. 2011;343:131–140. doi: 10.1007/s00441-010-1031-3. [DOI] [PubMed] [Google Scholar]
- 37.Lin Q, Li M, Fang D, Fang J, Su SB. The essential roles of Toll-like receptor signaling pathways in sterile inflammatory diseases. Int Immunopharmacol. 2011;11:1422–1432. doi: 10.1016/j.intimp.2011.04.026. [DOI] [PubMed] [Google Scholar]
- 38.De Nardo D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine. 2015;74:181–189. doi: 10.1016/j.cyto.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 39.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 40.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 41.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 42.Barton GM, Medzhitov R. Control of adaptive immune responses by Toll-like receptors. Curr Opin Immunol. 2002;14:380–383. doi: 10.1016/s0952-7915(02)00343-6. [DOI] [PubMed] [Google Scholar]
- 43.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 44.Babu S, Blauvelt CP, Kumaraswami V, Nutman TB. Cutting edge: diminished T cell TLR expression and function modulates the immune response in human filarial infection. J Immunol. 2006;176:3885–3889. doi: 10.4049/jimmunol.176.7.3885. [DOI] [PubMed] [Google Scholar]
- 45.Murad S. Toll-like receptor 4 in inflammation and angiogenesis: a double-edged sword. Front Immunol. 2014;5:313. doi: 10.3389/fimmu.2014.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–519. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- 47.Kaisho T, Akira S. Pleiotropic function of Toll-like receptors. Microbes Infect. 2004;6:1388–1394. doi: 10.1016/j.micinf.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 48.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 49.Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. 2010;14:2592–2603. doi: 10.1111/j.1582-4934.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Creus A, Abe M, Lau AH, Hackstein H, Raimondi G, Thomson AW. Low TLR4 expression by liver dendritic cells correlates with reduced capacity to activate allogeneic T cells in response to endotoxin. J Immunol. 2005;174:2037–2045. doi: 10.4049/jimmunol.174.4.2037. [DOI] [PubMed] [Google Scholar]
- 51.Lichtman SN, Wang J, Lemasters JJ. LPS receptor CD14 participates in release of TNF-alpha in RAW 264.7 and peritoneal cells but not in kupffer cells. Am J Physiol. 1998;275:G39–G46. doi: 10.1152/ajpgi.1998.275.1.G39. [DOI] [PubMed] [Google Scholar]
- 52.Shu M, Huang DD, Hung ZA, Hu XR, Zhang S. Inhibition of MAPK and NF-κB signaling pathways alleviate carbon tetrachloride (CCl4)-induced liver fibrosis in Toll-like receptor 5 (TLR5) deficiency mice. Biochem Biophys Res Commun. 2016;471:233–239. doi: 10.1016/j.bbrc.2016.01.119. [DOI] [PubMed] [Google Scholar]
- 53.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 54.Maeda S. NF-κB, JNK, and TLR Signaling Pathways in Hepatocarcinogenesis. Gastroenterol Res Pract. 2010;2010:367694. doi: 10.1155/2010/367694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chuang T, Ulevitch RJ. Identification of hTLR10: a novel human Toll-like receptor preferentially expressed in immune cells. Biochim Biophys Acta. 2001;1518:157–161. doi: 10.1016/s0167-4781(00)00289-x. [DOI] [PubMed] [Google Scholar]
- 56.Palazzo M, Gariboldi S, Zanobbio L, Dusio GF, Selleri S, Bedoni M, Balsari A, Rumio C. Cross-talk among Toll-like receptors and their ligands. Int Immunol. 2008;20:709–718. doi: 10.1093/intimm/dxn027. [DOI] [PubMed] [Google Scholar]
- 57.Re F, Strominger JL. Heterogeneity of TLR-induced responses in dendritic cells: from innate to adaptive immunity. Immunobiology. 2004;209:191–198. doi: 10.1016/j.imbio.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 58.Chen R, Alvero AB, Silasi DA, Mor G. Inflammation, cancer and chemoresistance: taking advantage of the toll-like receptor signaling pathway. Am J Reprod Immunol. 2007;57:93–107. doi: 10.1111/j.1600-0897.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 59.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 60.Matsumura T, Degawa T, Takii T, Hayashi H, Okamoto T, Inoue J, Onozaki K. TRAF6-NF-kappaB pathway is essential for interleukin-1-induced TLR2 expression and its functional response to TLR2 ligand in murine hepatocytes. Immunology. 2003;109:127–136. doi: 10.1046/j.1365-2567.2003.01627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsumura T, Ito A, Takii T, Hayashi H, Onozaki K. Endotoxin and cytokine regulation of toll-like receptor (TLR) 2 and TLR4 gene expression in murine liver and hepatocytes. J Interferon Cytokine Res. 2000;20:915–921. doi: 10.1089/10799900050163299. [DOI] [PubMed] [Google Scholar]
- 62.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175–1186. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 63.Thobe BM, Frink M, Hildebrand F, Schwacha MG, Hubbard WJ, Choudhry MA, Chaudry IH. The role of MAPK in Kupffer cell toll-like receptor (TLR) 2-, TLR4-, and TLR9-mediated signaling following trauma-hemorrhage. J Cell Physiol. 2007;210:667–675. doi: 10.1002/jcp.20860. [DOI] [PubMed] [Google Scholar]
- 64.Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Büschenfelde KH, Gerken G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol. 1995;22:226–229. doi: 10.1016/0168-8278(95)80433-1. [DOI] [PubMed] [Google Scholar]
- 65.Su GL, Klein RD, Aminlari A, Zhang HY, Steinstraesser L, Alarcon WH, Remick DG, Wang SC. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology. 2000;31:932–936. doi: 10.1053/he.2000.5634. [DOI] [PubMed] [Google Scholar]
- 66.Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A, Krux F, Dittmer U, Roggendorf M, Gerken G, et al. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology. 2007;46:1769–1778. doi: 10.1002/hep.21897. [DOI] [PubMed] [Google Scholar]
- 67.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brun P, Castagliuolo I, Pinzani M, Palù G, Martines D. Exposure to bacterial cell wall products triggers an inflammatory phenotype in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G571–G578. doi: 10.1152/ajpgi.00537.2004. [DOI] [PubMed] [Google Scholar]
- 69.Gäbele E, Mühlbauer M, Dorn C, Weiss TS, Froh M, Schnabl B, Wiest R, Schölmerich J, Obermeier F, Hellerbrand C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem Biophys Res Commun. 2008;376:271–276. doi: 10.1016/j.bbrc.2008.08.096. [DOI] [PubMed] [Google Scholar]
- 70.Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. [DOI] [PubMed] [Google Scholar]
- 71.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 72.Guo J, Friedman SL. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair. 2010;3:21. doi: 10.1186/1755-1536-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, Mehal WZ. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007;46:1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 74.Harada K, Ohira S, Isse K, Ozaki S, Zen Y, Sato Y, Nakanuma Y. Lipopolysaccharide activates nuclear factor-kappaB through toll-like receptors and related molecules in cultured biliary epithelial cells. Lab Invest. 2003;83:1657–1667. doi: 10.1097/01.lab.0000097190.56734.fe. [DOI] [PubMed] [Google Scholar]
- 75.Harada K, Isse K, Nakanuma Y. Interferon gamma accelerates NF-kappaB activation of biliary epithelial cells induced by Toll-like receptor and ligand interaction. J Clin Pathol. 2006;59:184–190. doi: 10.1136/jcp.2004.023507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harada K, Isse K, Sato Y, Ozaki S, Nakanuma Y. Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 2006;26:935–942. doi: 10.1111/j.1478-3231.2006.01325.x. [DOI] [PubMed] [Google Scholar]
- 77.Martin-Armas M, Simon-Santamaria J, Pettersen I, Moens U, Smedsrød B, Sveinbjørnsson B. Toll-like receptor 9 (TLR9) is present in murine liver sinusoidal endothelial cells (LSECs) and mediates the effect of CpG-oligonucleotides. J Hepatol. 2006;44:939–946. doi: 10.1016/j.jhep.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 78.Uhrig A, Banafsche R, Kremer M, Hegenbarth S, Hamann A, Neurath M, Gerken G, Limmer A, Knolle PA. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J Leukoc Biol. 2005;77:626–633. doi: 10.1189/jlb.0604332. [DOI] [PubMed] [Google Scholar]
- 79.Lohse AW, Knolle PA, Bilo K, Uhrig A, Waldmann C, Ibe M, Schmitt E, Gerken G, Meyer Zum Büschenfelde KH. Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and Kupffer cells. Gastroenterology. 1996;110:1175–1181. doi: 10.1053/gast.1996.v110.pm8613007. [DOI] [PubMed] [Google Scholar]
- 80.Van Bossuyt H, De Zanger RB, Wisse E. Cellular and subcellular distribution of injected lipopolysaccharide in rat liver and its inactivation by bile salts. J Hepatol. 1988;7:325–337. doi: 10.1016/s0168-8278(88)80005-9. [DOI] [PubMed] [Google Scholar]
- 81.Mimura Y, Sakisaka S, Harada M, Sata M, Tanikawa K. Role of hepatocytes in direct clearance of lipopolysaccharide in rats. Gastroenterology. 1995;109:1969–1976. doi: 10.1016/0016-5085(95)90765-3. [DOI] [PubMed] [Google Scholar]
- 82.Hsu W, Shu SA, Gershwin E, Lian ZX. The current immune function of hepatic dendritic cells. Cell Mol Immunol. 2007;4:321–328. [PubMed] [Google Scholar]
- 83.Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–163. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 84.Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, Akira S, Reis e Sousa C. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol. 2003;33:827–833. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- 85.Shu SA, Lian ZX, Chuang YH, Yang GX, Moritoki Y, Comstock SS, Zhong RQ, Ansari AA, Liu YJ, Gershwin ME. The role of CD11c(+) hepatic dendritic cells in the induction of innate immune responses. Clin Exp Immunol. 2007;149:335–343. doi: 10.1111/j.1365-2249.2007.03419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soares JB, Pimentel-Nunes P, Afonso L, Rolanda C, Lopes P, Roncon-Albuquerque R, Gonçalves N, Boal-Carvalho I, Pardal F, Lopes S, et al. Increased hepatic expression of TLR2 and TLR4 in the hepatic inflammation-fibrosis-carcinoma sequence. Innate Immun. 2012;18:700–708. doi: 10.1177/1753425912436762. [DOI] [PubMed] [Google Scholar]
- 87.Yin S, Gao B. Toll-like receptor 3 in liver diseases. Gastroenterol Res Pract. 2010:2010. doi: 10.1155/2010/750904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bedogni G, Miglioli L, Masutti F, Tiribelli C, Marchesini G, Bellentani S. Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology. 2005;42:44–52. doi: 10.1002/hep.20734. [DOI] [PubMed] [Google Scholar]
- 89.Younossi ZM, Diehl AM, Ong JP. Nonalcoholic fatty liver disease: an agenda for clinical research. Hepatology. 2002;35:746–752. doi: 10.1053/jhep.2002.32483. [DOI] [PubMed] [Google Scholar]
- 90.Vaikunthanathan T, Safinia N, Lombardi G, Lechler RI. Microbiota, immunity and the liver. Immunol Lett. 2016;171:36–49. doi: 10.1016/j.imlet.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 91.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571–579. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, Tripathi G, Ashour E, Abdalla MS, Sharada HM, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (Lond) 2010;7:15. doi: 10.1186/1476-9255-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thuy S, Ladurner R, Volynets V, Wagner S, Strahl S, Königsrainer A, Maier KP, Bischoff SC, Bergheim I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr. 2008;138:1452–1455. doi: 10.1093/jn/138.8.1452. [DOI] [PubMed] [Google Scholar]
- 94.Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G518–G525. doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]
- 95.Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, Desimone C, Song XY, Diehl AM. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 96.Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, Keshavarzian A. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008;28:1026–1033. doi: 10.1111/j.1478-3231.2008.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 98.Miura K, Ohnishi H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:7381–7391. doi: 10.3748/wjg.v20.i23.7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li D, Wang X, Lan X, Li Y, Liu L, Yi J, Li J, Sun Q, Wang Y, Li H, et al. Down-regulation of miR-144 elicits proinflammatory cytokine production by targeting toll-like receptor 2 in nonalcoholic steatohepatitis of high-fat-diet-induced metabolic syndrome E3 rats. Mol Cell Endocrinol. 2015;402:1–12. doi: 10.1016/j.mce.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 100.Miura K, Seki E, Ohnishi H, Brenner DA. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic Fatty liver disease. Gastroenterol Res Pract. 2010;2010:362847. doi: 10.1155/2010/362847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–211. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 103.Federico A, Dallio M, Godos J, Loguercio C, Salomone F. Targeting gut-liver axis for the treatment of nonalcoholic steatohepatitis: translational and clinical evidence. Transl Res. 2016;167:116–124. doi: 10.1016/j.trsl.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 104.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–324.e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ye D, Li FY, Lam KS, Li H, Jia W, Wang Y, Man K, Lo CM, Li X, Xu A. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut. 2012;61:1058–1067. doi: 10.1136/gutjnl-2011-300269. [DOI] [PubMed] [Google Scholar]
- 106.Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 2013;57:577–589. doi: 10.1002/hep.26081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Broering R, Lu M, Schlaak JF. Role of Toll-like receptors in liver health and disease. Clin Sci (Lond) 2011;121:415–426. doi: 10.1042/CS20110065. [DOI] [PubMed] [Google Scholar]
- 108.Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–644. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet. 1984;1:179–182. doi: 10.1016/s0140-6736(84)92109-3. [DOI] [PubMed] [Google Scholar]
- 110.Petrasek J, Mandrekar P, Szabo G. Toll-like receptors in the pathogenesis of alcoholic liver disease. Gastroenterol Res Pract. 2010:2010. doi: 10.1155/2010/710381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Testro AG, Visvanathan K. Toll-like receptors and their role in gastrointestinal disease. J Gastroenterol Hepatol. 2009;24:943–954. doi: 10.1111/j.1440-1746.2009.05854.x. [DOI] [PubMed] [Google Scholar]
- 112.Machida K. TLRs, Alcohol, HCV, and Tumorigenesis. Gastroenterol Res Pract. 2010;2010:518674. doi: 10.1155/2010/518674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gao B, Seki E, Brenner DA, Friedman S, Cohen JI, Nagy L, Szabo G, Zakhari S. Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300:G516–G525. doi: 10.1152/ajpgi.00537.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, Szabo G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224–1231. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, Franchimont D, Louis H, Devière J, Le Moine O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43:989–1000. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- 116.Inokuchi S, Tsukamoto H, Park E, Liu ZX, Brenner DA, Seki E. Toll-like receptor 4 mediates alcohol-induced steatohepatitis through bone marrow-derived and endogenous liver cells in mice. Alcohol Clin Exp Res. 2011;35:1509–1518. doi: 10.1111/j.1530-0277.2011.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Romics L, Dolganiuc A, Kodys K, Drechsler Y, Oak S, Velayudham A, Mandrekar P, Szabo G. Selective priming to Toll-like receptor 4 (TLR4), not TLR2, ligands by P. acnes involves up-regulation of MD-2 in mice. Hepatology. 2004;40:555–564. doi: 10.1002/hep.20350. [DOI] [PubMed] [Google Scholar]
- 118.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 119.Seki E, Tsutsui H, Nakano H, Tsuji N, Hoshino K, Adachi O, Adachi K, Futatsugi S, Kuida K, Takeuchi O, et al. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J Immunol. 2001;166:2651–2657. doi: 10.4049/jimmunol.166.4.2651. [DOI] [PubMed] [Google Scholar]
- 120.Forbes SJ, Parola M. Liver fibrogenic cells. Best Pract Res Clin Gastroenterol. 2011;25:207–217. doi: 10.1016/j.bpg.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 121.Isayama F, Hines IN, Kremer M, Milton RJ, Byrd CL, Perry AW, McKim SE, Parsons C, Rippe RA, Wheeler MD. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1318–G1328. doi: 10.1152/ajpgi.00405.2005. [DOI] [PubMed] [Google Scholar]
- 122.Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology. 2008;134:248–258. doi: 10.1053/j.gastro.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Aoyama T, Paik YH, Seki E. Toll-like receptor signaling and liver fibrosis. Gastroenterol Res Pract. 2010:Epub 2010 Jul 25. doi: 10.1155/2010/192543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ji L, Xue R, Tang W, Wu W, Hu T, Liu X, Peng X, Gu J, Chen S, Zhang S. Toll like receptor 2 knock-out attenuates carbon tetrachloride (CCl4)-induced liver fibrosis by downregulating MAPK and NF-κB signaling pathways. FEBS Lett. 2014;588:2095–2100. doi: 10.1016/j.febslet.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 125.Lee WM. Hepatitis B virus infection. N Engl J Med. 1997;337:1733–1745. doi: 10.1056/NEJM199712113372406. [DOI] [PubMed] [Google Scholar]
- 126.McQuillan GM, Coleman PJ, Kruszon-Moran D, Moyer LA, Lambert SB, Margolis HS. Prevalence of hepatitis B virus infection in the United States: the National Health and Nutrition Examination Surveys, 1976 through 1994. Am J Public Health. 1999;89:14–18. doi: 10.2105/ajph.89.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Keskinen P, Nyqvist M, Sareneva T, Pirhonen J, Melén K, Julkunen I. Impaired antiviral response in human hepatoma cells. Virology. 1999;263:364–375. doi: 10.1006/viro.1999.9983. [DOI] [PubMed] [Google Scholar]
- 128.McClary H, Koch R, Chisari FV, Guidotti LG. Relative sensitivity of hepatitis B virus and other hepatotropic viruses to the antiviral effects of cytokines. J Virol. 2000;74:2255–2264. doi: 10.1128/jvi.74.5.2255-2264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol. 2005;79:7269–7272. doi: 10.1128/JVI.79.11.7269-7272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xia C, Lu M, Zhang Z, Meng Z, Zhang Z, Shi C. TLRs antiviral effect on hepatitis B virus in HepG2 cells. J Appl Microbiol. 2008;105:1720–1727. doi: 10.1111/j.1365-2672.2008.03896.x. [DOI] [PubMed] [Google Scholar]
- 131.Dalpke AH, Lehner MD, Hartung T, Heeg K. Differential effects of CpG-DNA in Toll-like receptor-2/-4/-9 tolerance and cross-tolerance. Immunology. 2005;116:203–212. doi: 10.1111/j.1365-2567.2005.02211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen Z, Cheng Y, Xu Y, Liao J, Zhang X, Hu Y, Zhang Q, Wang J, Zhang Z, Shen F, et al. Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin Immunol. 2008;128:400–408. doi: 10.1016/j.clim.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 133.Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R, Rodgers S, Kurtovic J, Chang J, Lewin S, et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology. 2007;45:102–110. doi: 10.1002/hep.21482. [DOI] [PubMed] [Google Scholar]
- 134.Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R, Bucchi A, Sowa JP, Dittmer U, Yang D, et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology. 2009;49:1132–1140. doi: 10.1002/hep.22751. [DOI] [PubMed] [Google Scholar]
- 135.Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci USA. 2004;101:6669–6674. doi: 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hösel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, Tedjokusumo R, Esser K, Arzberger S, Kirschning CJ, et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology. 2009;50:1773–1782. doi: 10.1002/hep.23226. [DOI] [PubMed] [Google Scholar]
- 137.Besinger R. [Postpartum hemorrhage: perspectives in the United States] J Gynecol Obstet Biol Reprod (Paris) 1997;26:34–38. [PubMed] [Google Scholar]
- 138.Di Bisceglie AM. Hepatitis C. Lancet. 1998;351:351–355. doi: 10.1016/S0140-6736(97)07361-3. [DOI] [PubMed] [Google Scholar]
- 139.Ishii S, Koziel MJ. Immune responses during acute and chronic infection with hepatitis C virus. Clin Immunol. 2008;128:133–147. doi: 10.1016/j.clim.2008.03.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 141.Broering R, Wu J, Meng Z, Hilgard P, Lu M, Trippler M, Szczeponek A, Gerken G, Schlaak JF. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol. 2008;48:914–922. doi: 10.1016/j.jhep.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 142.Chang S, Dolganiuc A, Szabo G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J Leukoc Biol. 2007;82:479–487. doi: 10.1189/jlb.0207128. [DOI] [PubMed] [Google Scholar]
- 143.Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 2004;127:1513–1524. doi: 10.1053/j.gastro.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 144.Düesberg U, von dem Bussche A, Kirschning C, Miyake K, Sauerbruch T, Spengler U. Cell activation by synthetic lipopeptides of the hepatitis C virus (HCV)--core protein is mediated by toll like receptors (TLRs) 2 and 4. Immunol Lett. 2002;84:89–95. doi: 10.1016/s0165-2478(02)00178-5. [DOI] [PubMed] [Google Scholar]
- 145.Machida K, Cheng KT, Sung VM, Levine AM, Foung S, Lai MM. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol. 2006;80:866–874. doi: 10.1128/JVI.80.2.866-874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chang S, Kodys K, Szabo G. Impaired expression and function of toll-like receptor 7 in hepatitis C virus infection in human hepatoma cells. Hepatology. 2010;51:35–42. doi: 10.1002/hep.23256. [DOI] [PubMed] [Google Scholar]
- 147.Dolganiuc A, Norkina O, Kodys K, Catalano D, Bakis G, Marshall C, Mandrekar P, Szabo G. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology. 2007;133:1627–1636. doi: 10.1053/j.gastro.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 149.Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, Tang L, Lin Y, He YQ, Zou SS, et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology. 2010;52:1322–1333. doi: 10.1002/hep.23845. [DOI] [PubMed] [Google Scholar]
- 151.Maeda S, Omata M. Inflammation and cancer: role of nuclear factor-kappaB activation. Cancer Sci. 2008;99:836–842. doi: 10.1111/j.1349-7006.2008.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hu KQ. Rationale and feasibility of chemoprovention of hepatocellular carcinoma by cyclooxygenase-2 inhibitors. J Lab Clin Med. 2002;139:234–243. doi: 10.1067/mlc.2002.122281. [DOI] [PubMed] [Google Scholar]
- 153.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 154.Prieto J. Inflammation, HCC and sex: IL-6 in the centre of the triangle. J Hepatol. 2008;48:380–381. doi: 10.1016/j.jhep.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 155.Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu X, Xu Q, Chen W, Cao H, Zheng R, Li G. Hepatitis B virus DNA-induced carcinogenesis of human normal liver cells by virtue of nonmethylated CpG DNA. Oncol Rep. 2009;21:941–947. doi: 10.3892/or_00000307. [DOI] [PubMed] [Google Scholar]
- 157.Yoneda K, Sugimoto K, Shiraki K, Tanaka J, Beppu T, Fuke H, Yamamoto N, Masuya M, Horie R, Uchida K, et al. Dual topology of functional Toll-like receptor 3 expression in human hepatocellular carcinoma: differential signaling mechanisms of TLR3-induced NF-kappaB activation and apoptosis. Int J Oncol. 2008;33:929–936. [PubMed] [Google Scholar]
- 158.Lopes JA, Borges-Canha M, Pimentel-Nunes P. Innate immunity and hepatocarcinoma: Can toll-like receptors open the door to oncogenesis? World J Hepatol. 2016;8:162–182. doi: 10.4254/wjh.v8.i3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, Miyai K, Akira S, Brenner DA, Seki E. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci USA. 2010;107:844–849. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM, Asahina K, Govindarajan S, Ray R, Ou JH, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA. 2009;106:1548–1553. doi: 10.1073/pnas.0807390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, Kupiec-Weglinski JW. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 162.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chang WJ, Toledo-Pereyra LH. Toll-like receptor signaling in liver ischemia and reperfusion. J Invest Surg. 2012;25:271–277. doi: 10.3109/08941939.2012.687802. [DOI] [PubMed] [Google Scholar]
- 164.Evankovich J, Billiar T, Tsung A. Toll-like receptors in hepatic ischemia/reperfusion and transplantation. Gastroenterol Res Pract. 2010:Epub 2010 Aug 5. doi: 10.1155/2010/537263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shen XD, Ke B, Zhai Y, Gao F, Tsuchihashi S, Lassman CR, Busuttil RW, Kupiec-Weglinski JW. Absence of toll-like receptor 4 (TLR4) signaling in the donor organ reduces ischemia and reperfusion injury in a murine liver transplantation model. Liver Transpl. 2007;13:1435–1443. doi: 10.1002/lt.21251. [DOI] [PubMed] [Google Scholar]
- 166.Wang H, Li ZY, Wu HS, Wang Y, Jiang CF, Zheng QC, Zhang JX. Endogenous danger signals trigger hepatic ischemia/reperfusion injury through toll-like receptor 4/nuclear factor-kappa B pathway. Chin Med J (Engl) 2007;120:509–514. [PubMed] [Google Scholar]
- 167.Zhai Y, Qiao B, Shen XD, Gao F, Busuttil RW, Cheng G, Platt JL, Volk HD, Kupiec-Weglinski JW. Evidence for the pivotal role of endogenous toll-like receptor 4 ligands in liver ischemia and reperfusion injury. Transplantation. 2008;85:1016–1022. doi: 10.1097/TP.0b013e3181684248. [DOI] [PubMed] [Google Scholar]
- 168.King LA, Toledo AH, Rivera-Chavez FA, Toledo-Pereyra LH. Role of p38 and JNK in liver ischemia and reperfusion. J Hepatobiliary Pancreat Surg. 2009;16:763–770. doi: 10.1007/s00534-009-0155-x. [DOI] [PubMed] [Google Scholar]
- 169.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, Lotze MT, Geller DA, Billiar TR. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661–7668. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 170.Liu S, Salyapongse AN, Geller DA, Vodovotz Y, Billiar TR. Hepatocyte toll-like receptor 2 expression in vivo and in vitro: role of cytokines in induction of rat TLR2 gene expression by lipopolysaccharide. Shock. 2000;14:361–365. doi: 10.1097/00024382-200014030-00021. [DOI] [PubMed] [Google Scholar]
- 171.Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, Takayanagi H, Ohba Y, Taniguchi T, Honda K. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc Natl Acad Sci USA. 2006;103:15136–15141. doi: 10.1073/pnas.0607181103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ueki S, Dhupar R, Cardinal J, Tsung A, Yoshida J, Ozaki KS, Klune JR, Murase N, Geller DA. Critical role of interferon regulatory factor-1 in murine liver transplant ischemia reperfusion injury. Hepatology. 2010;51:1692–1701. doi: 10.1002/hep.23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang JX, Wu HS, Wang H, Zhang JH, Wang Y, Zheng QC. Protection against hepatic ischemia/reperfusion injury via downregulation of toll-like receptor 2 expression by inhibition of Kupffer cell function. World J Gastroenterol. 2005;11:4423–4426. doi: 10.3748/wjg.v11.i28.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yao XM, Chen H, Li Y. Protective effect of bicyclol on liver injury induced by hepatic warm ischemia/reperfusion in rats. Hepatol Res. 2009;39:833–842. doi: 10.1111/j.1872-034X.2009.00504.x. [DOI] [PubMed] [Google Scholar]
- 175.Jin X, Wang L, Wu HS, Zhang L, Wang CY, Tian Y, Zhang JH. N-acetylcysteine inhibits activation of toll-like receptor 2 and 4 gene expression in the liver and lung after partial hepatic ischemia-reperfusion injury in mice. Hepatobiliary Pancreat Dis Int. 2007;6:284–289. [PubMed] [Google Scholar]
- 176.Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology. 2010;51:621–632. doi: 10.1002/hep.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, Bauer S, Hochrein H, Wagner H. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J Immunol. 2005;174:6129–6136. doi: 10.4049/jimmunol.174.10.6129. [DOI] [PubMed] [Google Scholar]
- 178.Iimuro Y, Fujimoto J. TLRs, NF-κB, JNK, and Liver Regeneration. Gastroenterol Res Pract. 2010:Epub 2010 Sep 26. doi: 10.1155/2010/598109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–359. doi: 10.1053/j.gastro.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Seki E, Tsutsui H, Iimuro Y, Naka T, Son G, Akira S, Kishimoto T, Nakanishi K, Fujimoto J. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology. 2005;41:443–450. doi: 10.1002/hep.20603. [DOI] [PubMed] [Google Scholar]
- 181.Campbell JS, Riehle KJ, Brooling JT, Bauer RL, Mitchell C, Fausto N. Proinflammatory cytokine production in liver regeneration is Myd88-dependent, but independent of Cd14, Tlr2, and Tlr4. J Immunol. 2006;176:2522–2528. doi: 10.4049/jimmunol.176.4.2522. [DOI] [PubMed] [Google Scholar]
- 182.Akita K, Okuno M, Enya M, Imai S, Moriwaki H, Kawada N, Suzuki Y, Kojima S. Impaired liver regeneration in mice by lipopolysaccharide via TNF-alpha/kallikrein-mediated activation of latent TGF-beta. Gastroenterology. 2002;123:352–364. doi: 10.1053/gast.2002.34234. [DOI] [PubMed] [Google Scholar]
- 183.Zorde-Khvalevsky E, Abramovitch R, Barash H, Spivak-Pohis I, Rivkin L, Rachmilewitz J, Galun E, Giladi H. Toll-like receptor 3 signaling attenuates liver regeneration. Hepatology. 2009;50:198–206. doi: 10.1002/hep.22973. [DOI] [PubMed] [Google Scholar]
- 184.Sun R, Park O, Horiguchi N, Kulkarni S, Jeong WI, Sun HY, Radaeva S, Gao B. STAT1 contributes to dsRNA inhibition of liver regeneration after partial hepatectomy in mice. Hepatology. 2006;44:955–966. doi: 10.1002/hep.21344. [DOI] [PubMed] [Google Scholar]
- 185.Washington MK. Autoimmune liver disease: overlap and outliers. Mod Pathol. 2007;20 Suppl 1:S15–S30. doi: 10.1038/modpathol.3800684. [DOI] [PubMed] [Google Scholar]
- 186.Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, Odermatt B, Conrad C, Ittner LM, Bauer S, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11:138–145. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 187.Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 188.Lebeis SL, Powell KR, Merlin D, Sherman MA, Kalman D. Interleukin-1 receptor signaling protects mice from lethal intestinal damage caused by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun. 2009;77:604–614. doi: 10.1128/IAI.00907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mao TK, Lian ZX, Selmi C, Ichiki Y, Ashwood P, Ansari AA, Coppel RL, Shimoda S, Ishibashi H, Gershwin ME. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology. 2005;42:802–808. doi: 10.1002/hep.20859. [DOI] [PubMed] [Google Scholar]
- 190.Sasatomi K, Noguchi K, Sakisaka S, Sata M, Tanikawa K. Abnormal accumulation of endotoxin in biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. J Hepatol. 1998;29:409–416. doi: 10.1016/s0168-8278(98)80058-5. [DOI] [PubMed] [Google Scholar]
- 191.Ballot E, Bandin O, Chazouilleres O, Johanet C, Poupon R. Immune response to lipopolysaccharide in primary biliary cirrhosis and autoimmune diseases. J Autoimmun. 2004;22:153–158. doi: 10.1016/j.jaut.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 192.Wang AP, Migita K, Ito M, Takii Y, Daikoku M, Yokoyama T, Komori A, Nakamura M, Yatsuhashi H, Ishibashi H. Hepatic expression of toll-like receptor 4 in primary biliary cirrhosis. J Autoimmun. 2005;25:85–91. doi: 10.1016/j.jaut.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 193.Honda Y, Yamagiwa S, Matsuda Y, Takamura M, Ichida T, Aoyagi Y. Altered expression of TLR homolog RP105 on monocytes hypersensitive to LPS in patients with primary biliary cirrhosis. J Hepatol. 2007;47:404–411. doi: 10.1016/j.jhep.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 194.Shimoda S, Harada K, Niiro H, Shirabe K, Taketomi A, Maehara Y, Tsuneyama K, Nakanuma Y, Leung P, Ansari AA, et al. Interaction between Toll-like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology. 2011;53:1270–1281. doi: 10.1002/hep.24194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shimoda S, Harada K, Niiro H, Yoshizumi T, Soejima Y, Taketomi A, Maehara Y, Tsuneyama K, Nakamura M, Komori A, et al. Biliary epithelial cells and primary biliary cirrhosis: the role of liver-infiltrating mononuclear cells. Hepatology. 2008;47:958–965. doi: 10.1002/hep.22102. [DOI] [PubMed] [Google Scholar]
- 196.Takii Y, Nakamura M, Ito M, Yokoyama T, Komori A, Shimizu-Yoshida Y, Nakao R, Kusumoto K, Nagaoka S, Yano K, et al. Enhanced expression of type I interferon and toll-like receptor-3 in primary biliary cirrhosis. Lab Invest. 2005;85:908–920. doi: 10.1038/labinvest.3700285. [DOI] [PubMed] [Google Scholar]
- 197.Kikuchi K, Lian ZX, Yang GX, Ansari AA, Ikehara S, Kaplan M, Miyakawa H, Coppel RL, Gershwin ME. Bacterial CpG induces hyper-IgM production in CD27(+) memory B cells in primary biliary cirrhosis. Gastroenterology. 2005;128:304–312. doi: 10.1053/j.gastro.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 198.Kikuchi K, Lian ZX, Kimura Y, Selmi C, Yang GX, Gordon SC, Invernizzi P, Podda M, Coppel RL, Ansari AA, et al. Genetic polymorphisms of toll-like receptor 9 influence the immune response to CpG and contribute to hyper-IgM in primary biliary cirrhosis. J Autoimmun. 2005;24:347–352. doi: 10.1016/j.jaut.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 199.Karrar A, Broomé U, Södergren T, Jaksch M, Bergquist A, Björnstedt M, Sumitran-Holgersson S. Biliary epithelial cell antibodies link adaptive and innate immune responses in primary sclerosing cholangitis. Gastroenterology. 2007;132:1504–1514. doi: 10.1053/j.gastro.2007.01.039. [DOI] [PubMed] [Google Scholar]