

Summary
Alzheimer's disease (AD) is an age‐associated disease. Mutations in the amyloid precursor protein (APP) may be causative or protective of AD. The presence of two functionally redundant APP‐like genes (APLP1/2) has made it difficult to unravel the biological function of APP during aging. The nematode Caenorhabditis elegans contains a single APP family member, apl‐1. Here, we assessed the function of APL‐1 on C. elegans’ lifespan and found tissue‐specific effects on lifespan by overexpression of APL‐1. Overexpression of APL‐1 in neurons causes lifespan reduction, whereas overexpression of APL‐1 in the hypodermis causes lifespan extension by repressing the function of the heterochronic transcription factor LIN‐14 to preserve youthfulness. APL‐1 lifespan extension also requires signaling through the FOXO transcription factor DAF‐16, heat‐shock factor HSF‐1, and vitamin D‐like nuclear hormone receptor DAF‐12. We propose that reinforcing APL‐1 expression in the hypodermis preserves the regulation of heterochronic lin‐14 gene network to improve maintenance of somatic tissues via DAF‐16/FOXO and HSF‐1 to promote healthy aging. Our work reveals a mechanistic link of how a conserved APP‐related protein modulates aging.
Keywords: Alzheimer's disease, APL‐1, APP, FOXO transcription factor DAF‐16, heat‐shock factor HSF‐1, heterochronic gene LIN‐14, lifespan, vitamin D‐like nuclear hormone receptor DAF‐12
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder affecting over five million Americans and is the sixth leading cause of death (Alzheimer's Association 2015). The etiology of Alzheimer's disease is unclear. The highest risk factor for developing Alzheimer's disease is age, whereby 96% of patients with AD develop the disease at 65 years or older (Alzheimer's Association 2015). The disease is characterized by the deposition of amyloid plaques in brain tissue; the major component of the plaques is the beta‐amyloid peptide, a cleavage product of the amyloid precursor protein (APP) (reviewed Selkoe & Hardy, 2016). Mutations in APP and in genes that produce the presenilin enzymes that are part of the γ‐secretase complex that cleaves APP are correlated with cases of familial Alzheimer's disease (Tanzi, 2012; reviewed Selkoe & Hardy, 2016).
In mammals, the functional analysis of APP is complicated by the presence of two APP‐related proteins, APLP1 and APLP2 (reviewed Shariati & De Strooper, 2013). Whereas knockouts of individual APP family members are viable, double knockouts of APP and APLP2 or APLP1 and APLP2 result in postnatal lethality (von Koch et al., 1997; Herms et al., 2000, 2004), indicating an essential role for the APP family and functional overlap among family members that is not dependent on the beta‐amyloid peptide. Knockout of the entire APP gene family results not only in postnatal lethality, but also defects in neuronal migration and type II lissencephaly (Weyer et al., 2011). Interestingly, duplication of the APP gene has also been associated with families that develop early‐onset Alzheimer's disease (Cabrejo et al., 2006; Rovelet‐Lecrux et al., 2006; Sleegers et al., 2006). More strikingly, patients with Down syndrome, who have a trisomy of chromosome 21, have a high incidence of early‐onset Alzheimer's disease (Korbel et al., 2009; reviewed in Shariati & De Strooper, 2013). These findings suggest that higher levels of APP, whether it be higher levels of the beta‐amyloid peptide, the extracellular or cytoplasmic fragments, and/or full‐length APP, are correlated with early‐onset Alzheimer's disease. However, the cellular functions of APP and related proteins remain unclear.
The functions of genes correlated with Alzheimer's disease, including the presenilin genes, were first identified in genetic screens using the nematode Caenorhabditis elegans [reviewed (Ewald & Li, 2010)]. We have been examining the role of APL‐1, the C. elegans orthologue of mammalian APP. Similar to human APP, C. elegans APL‐1 is a single transmembrane‐spanning protein with a large extracellular and a small intracellular domain, both of which share sequence homology to human APP (Daigle & Li, 1993). Analogous to the postnatal lethality of APP family‐knockout mice, loss of apl‐1 in C. elegans results in a completely penetrant larval lethality that is rescued by the reintroduction of either full‐length APL‐1 or only the extracellular domain of APL‐1 (Hornsten et al., 2007). This finding was later extended to mammals, whereby knockin of sAPPα rescued the postnatal lethality of the APP‐APLP2‐double‐knockout mice (Weyer et al., 2011). Furthermore, overexpression of APL‐1 in C. elegans also causes an early developmental lethality, but the lethality shows an incomplete penetrance that is correlated with higher levels of APL‐1 (Hornsten et al., 2007). Similarly, overexpression of human or mouse APP in mice causes early developmental lethality in the absence of amyloid plaques (Hsiao et al., 1995). This correlation suggests that findings in C. elegans can provide insights into the rudimentary conserved functions of APP (Ewald & Li, 2012).
Here, we examined the effects of overexpressing APL‐1 during adulthood on lifespan in C. elegans. Overexpressing APL‐1 in neurons shortens lifespan, whereas overexpressing APL‐1 in hypodermis extends lifespan. This lifespan extension is independent from longevity mediated by reduced insulin/IGF‐1 signaling (rIIS) or by reduced germ stem cell numbers (rGSC). However, genes that are essential for rIIS and rGSC longevity, such as the FOXO transcription factor DAF‐16, heat‐shock factor HSF‐1, and vitamin D‐like nuclear hormone receptor DAF‐12, are required for APL‐1‐dependent longevity, suggesting that APL‐1 interacts with these genes in parallel to rIIS and rGSC. We found that hypodermal APL‐1 represses the transcriptional function of the heterochronic transcription factor LIN‐14, which has been shown to regulate longevity through HSF‐1 and DAF‐16. Repressing LIN‐14 during aging is required for hypodermal‐expressed APL‐1 to extend lifespan and higher levels of LIN‐14 are sufficient to abrogate the APL‐1‐induced longevity. These findings suggest that APL‐1 promotes longevity by regulating heterochronic processes.
Results
Ectopic expression of APL‐1 during adulthood increases lifespan
High levels of APL‐1 cause larval lethality (Hornsten et al., 2007). To bypass the developmental effects of APL‐1 and to determine the role of APL‐1 during aging, we used transgenic animals (ynIs14) that have the hsp‐16.2 promoter fused with an apl‐1 cDNA [Phsp‐16.2::APL‐1; (Ewald et al., 2012a)] and induced APL‐1 expression in adults in all tissues via heat shock. Surprisingly, a single heat shock for 3.5 hours in day one adults mildly increased lifespan (5%; Fig. 1a). Moreover, two hours of heat shock administered every second day (Fig. 1b) or applying moderate heat continuously (Fig. 1c; 25°C) robustly increased lifespan (12–30%) of ynIs14 [Phsp‐16.2::APL‐1] animals compared to wild‐type. As a control, the same animals were assayed in parallel at 15 °C, a temperature at which the hsp‐16.2 promoter is not active, and no change in lifespan was observed (Fig. 1d; Table S1). These results suggest that inducing ubiquitous overexpression of APL‐1 exclusively during adulthood is sufficient to extend lifespan.
Figure 1.
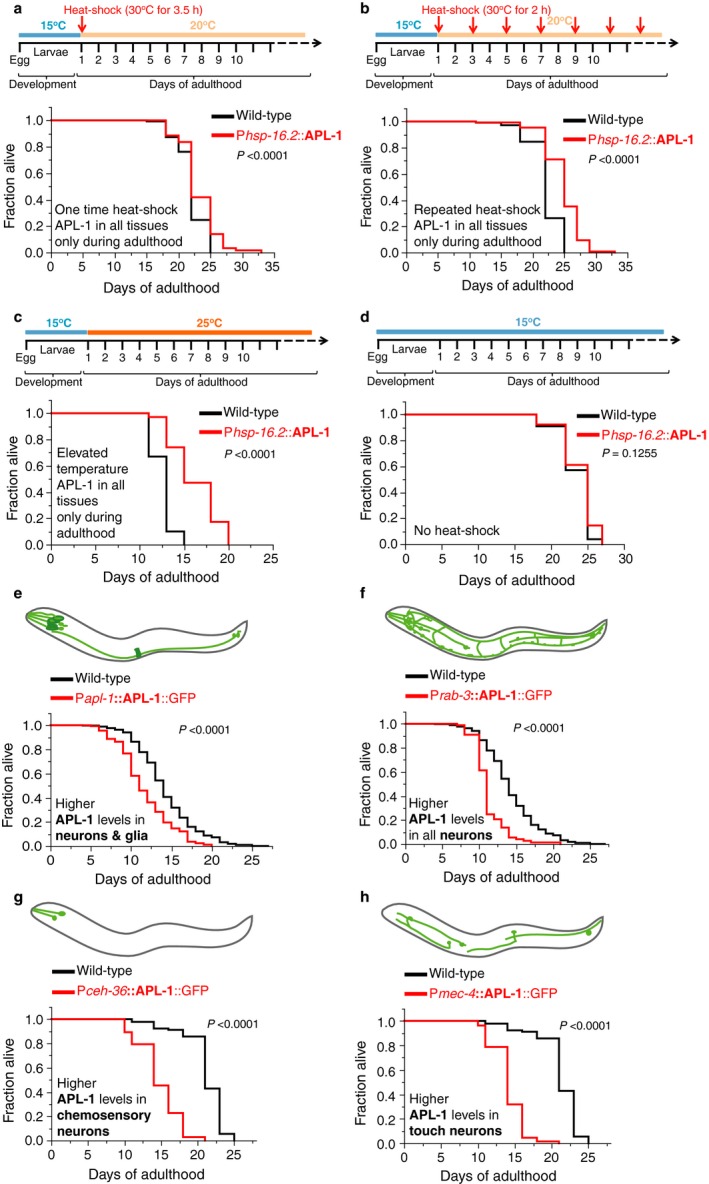
Overexpression of APL‐1 in all tissues during adulthood is sufficient to increase lifespan. (a–d) Wild‐type (N2) and transgenic ynIs14 [Phsp‐16.2::APL‐1] animals that express APL‐1 driven by a heat‐shock promoter (Phsp‐16.2) were maintained at 15°C and shifted to different temperatures as indicated in the schematics. (a) A single heat shock during early adulthood was sufficient to increase the lifespan of ynIs14 [Phsp‐16.2::APL‐1] animals. (b) Repeated heat shocks every other day during adulthood robustly increased the lifespan of ynIs14 [Phsp‐16.2::APL‐1] animals. (c) Elevated temperatures were sufficient to drive APL‐1 expression from hsp‐16.2 promoter (not shown; G. O'Connor, personal comm.) and increased lifespan of ynIs14 [Phsp‐16.2::APL‐1] animals. (d) At lower temperatures, the hsp‐16.2 promoter does not drive APL‐1 expression. The lifespan of ynIs14 [Phsp‐16.2::APL‐1] animals was not extended. (e–h) Overexpression of APL‐1 with cell‐specific promoters and its own promoter in wild‐type animals. (e) Pan‐neuronal APL‐1 overexpression of ynIs104 [Prab‐3::APL‐1::GFP] animals resulted in a shortened lifespan. (f) Transgenic ynEx214 [Pceh‐36::APL‐1::GFP] animals that overexpress APL‐1 in chemosensory neurons showed a shortened lifespan. (g) Transgenic ynIs113 [Pmec‐4::APL‐1::GFP] animals that overexpress APL‐1 in touch mechanosensory neurons showed a shortened lifespan. (h) Driving APL‐1 overexpression from its endogenous promoter results in higher APL‐1 levels in neurons, glia cells, and vulva muscles [Papl‐1::APL‐1::GFP]. Transgenic ynIs79 [Papl‐1::APL‐1::GFP] animals showed a shortened lifespan. For (a–d) Lifespan assays were run in parallel at the same time. P‐value was determined by log rank. Statistics and additional lifespan data are in Table S1. For (e–f). Lifespans are shown in cumulative form. For (e–h) Schematic representation of the expression pattern of APL‐1 with different promoters is from Ewald et al. (2012a>). Light green indicates neurons and dark green indicates other tissues. P‐value was determined by log rank. Statistics and additional lifespan data are in Tables S1 and S2.
Overexpressing APL‐1 in neurons shortens lifespan
The finding that ubiquitous adulthood expression of APL‐1 increased lifespan was surprising. To gain some mechanistic insights into how this lifespan extension is mediated, we investigated in which tissue apl‐1 expression is necessary for lifespan extension. C. elegans expresses apl‐1 strongly in neurons throughout its life cycle (Hornsten et al., 2007) and neurons regulate aging and longevity in a conserved spectrum of animals, including C. elegans (Kenyon, 2010). To investigate whether APL‐1 expressed in neurons increases lifespan, we examined transgenic animals carrying multicopy chromosomal arrays where apl‐1 is driven by different promoters: its endogenous promoter, the pan‐neuronal rab‐3 promoter that drives expression only in neurons, or the ceh‐36 or mec‐4 promoters that drive expression in a subset of neurons. Overexpression of APL‐1 with its own promoter, which includes strong expression in neurons and glial cells ([Papl‐1::apl‐1::GFP]; Fig. 1e; Table S2), shortened lifespan. Similarly, overexpressing APL‐1 in all neurons with the rab‐3 (Prab‐3::APL‐1::GFP; Fig. 1f; Table S2), or a subset of sensory neurons (Pceh‐36::APL‐1::GFP; Fig. 1g and Pmec‐4::APL‐1::GFP; Fig. 1h; Table S1), also shortened lifespan. Collectively, these results indicate that neuronal overexpression of APL‐1 shortens lifespan and is in stark contrast to the extended lifespan induced by heat‐shock overexpression of APL‐1 in all tissues, including neurons, in adults (Fig. 1). Therefore, we hypothesized that expression of APL‐1 in tissues other than neurons must activate a protective pathway to circumvent the detrimental effects of APL‐1 expression in neurons.
Overexpression of APL‐1 in multiple tissue types, including neurons, increases lifespan
To identify the cells in which apl‐1 expression counteracts the detrimental effects of pan‐neuronal apl‐1 overexpression, we used the snb‐1 promoter, which drives expression strongly in neurons and somatic gonad, but weakly in the hypodermis during adulthood (Fig. 2a; Ewald et al., 2012a>). APL‐1 expression was tracked by fusion to green fluorescent protein (GFP) at the C‐terminus (Psnb‐1::APL‐1 cDNA::GFP), which did not interfere with rescuing apl‐1(null) larval lethality (Hornsten et al., 2007). The most prominent expression in ynIs109 [Psnb‐1::apl‐1 cDNA::GFP] transgenic animals was in neurons and somatic gonad at 20°C (Fig. 2a). At 25°C, there were also high levels of APL‐1 expression in the hypodermis (Fig. 2b). ynIs109 [Psnb‐1::apl‐1 cDNA::GFP] lived 28% longer than control animals at 20°C (Fig. 2c); this lifespan extension was not affected by the presence of the GFP tag, as animals carrying a transgene without a GFP tag (ynIs12 [Psnb‐1::apl‐1 cDNA] and ynIs13 [Psnb‐1::apl‐1 cDNA]) showed a comparable lifespan extension of 24% (average of eight individual trials) and 21% (average of five individual trials) longer than wild‐type, respectively (Table S2). Knockdown of apl‐1 by RNA interference (RNAi) suppressed the increased longevity in ynIs13 [Psnb‐1::apl‐1 cDNA] animals (Fig. 2d; Table S3), suggesting that the increased lifespan is specific to apl‐1 expression. Furthermore, ynIs13 [Psnb‐1::apl‐1 cDNA] animals showed a slowed aging process based on less accumulation of age‐pigments and enhanced muscle functions (Fig. 2e–f and S1). These results suggest that overexpressing APL‐1 in tissues other than neurons is beneficial for extending health and lifespan.
Figure 2.
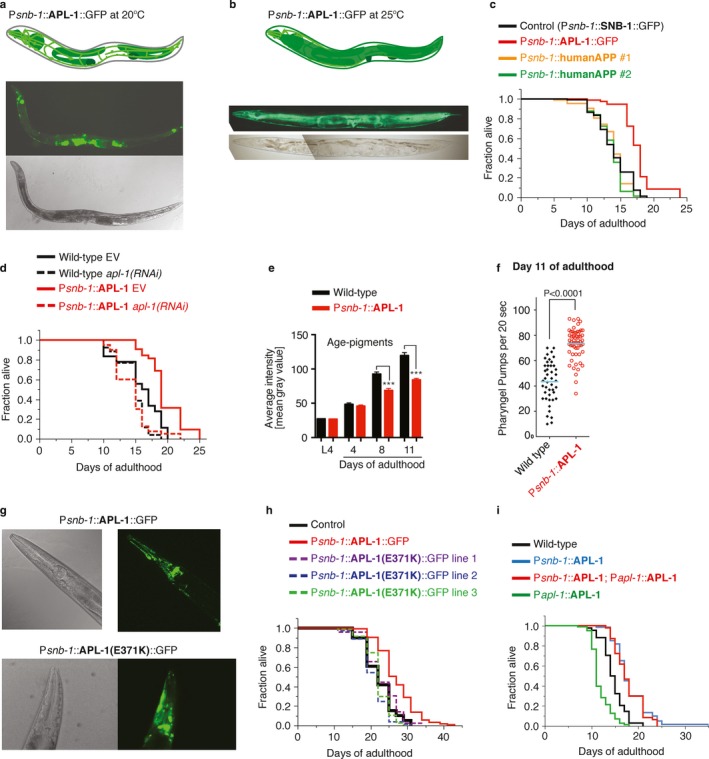
Overexpression of APL‐1 in neurons and other tissues extends lifespan. Using the snb‐1 promoter, apl‐1 [Psnb‐1::APL‐1::GFP] is strongly expressed in neurons, pharynx, and somatic gonad and more weakly expressed in hypodermal cells during adulthood at the normal cultivation temperature of 20°C. (a, b) Top panel: Schematic representation of apl‐1 expression. Middle panel: APL‐1::GFP expression driven by the snb‐1 promoter. Bottom panel: Corresponding bright field. (a) Day 3 adults lon‐2(e678) apl‐1(yn10) ynEx109 [Psnb‐1::APL‐1::GFP] are shown with anterior to the left and ventral side down; animals were grown at 20°C. (b) At higher temperatures (25°C), the hypodermal APL‐1 expression becomes apparent in ynIs109 [Psnb‐1::APL‐1::GFP] animals. Day 1 ynIs109 [Psnb‐1::APL‐1::GFP] adults were shifted from 20°C to 25°C and day 3 adults are shown with anterior to the left and ventral side down. (c) Transgenic ynIs109 [Psnb‐1::APL‐1::GFP] showed extended lifespan compared to jsIs1 [Psnb‐1::SNB‐1::GFP] control animals. Driving human APP with the snb‐1 promoter had no effect on lifespan. P‐value was determined by log rank. Statistics are in Table S3. (d) Knockdown of apl‐1 by RNAi mildly shortened lifespan of wild‐type (N2) and suppressed longevity of ynIs13 [Psnb‐1::APL‐1] animals. P‐value was determined by log rank. Statistics are in Table S3. (e) Long‐lived ynIs12 [Psnb‐1::APL‐1] animals showed lower lipofuscin (age‐pigment) levels at days 8 and 11 compared to wild‐type (N2). Data were represented as mean ± SEM P‐values were determined by unpaired two‐tailed t‐test (***P < 0.0001). For scoring and additional strains, see Fig. S1a,b. (f) Long‐lived ynIs13 [Psnb‐1::APL‐1] animals showed pharyngeal pumping rates at day 11 compared to wild‐type (N2). Data represented as mean ± SEM P‐values were determined by unpaired two‐tailed t‐test. For additional strains and time points, see Fig. S1c–f. (g) Mutant or wild‐type APL‐1 expression driven with the snb‐1 promoter shows similar GFP levels during adulthood at the normal cultivation temperature (20°C). Top panel shows head of a day 3 adult lon‐2(e678) apl‐1(yn10) ynEx109 [Psnb‐1::APL‐1::GFP] animal with bright field on the left and corresponding GFP on the right. Bottom panel shows head of a day 3 adult ynEx236 [Psnb‐1::APL‐1(E371K)::GFP] animal with bright field on the left and corresponding GFP on the right. (h) Transgenic animals that express a mutated APL‐1(E371K) with the snb‐1 promoter failed to increase lifespan. Lines 1, 2, and 3 are ynEx236, ynEx237, and ynEx238, respectively. Statistics and additional lifespan data are in Table S1. (i) The double transgenic ynIs12; ynIs86 animals, which carry two transgenes [Psnb‐1::APL‐1] and [Papl‐1::APL‐1], that drive APL‐1 overexpression with its endogenous and snb‐1 promoter, respectively, showed increased lifespan. Statistics and additional lifespan data are in Tables S2 and S3.
Overexpression of functional APL‐1 is required to increase lifespan
There are several possible mechanisms by which Psnb‐1::apl‐1 cDNA overexpression could increase lifespan. For example, lowering food intake results in caloric restriction that increases lifespan (Kenyon, 2010). However, pharyngeal pumping rates, a measure of food intake, were unaltered in young transgenic APL‐1 animals (Fig. S1c). Another hallmark of caloric‐restricted animals is lower fat content (Heestand et al., 2013). The fat content of long‐lived Psnb‐1::APL‐1 animals was not lower compared to wild‐type. Furthermore, the fat content of short‐lived apl‐1(yn5) and Papl‐1::APL‐1 animals was also lower compared to wild‐type animals (Fig. S2), suggesting that the effects of APL‐1 on fat content do not correlate with its effect on lifespan and are, therefore, uncoupled from longevity. Lifespan extension can also be induced in C. elegans by mobilizing the unfolded protein response (UPR) (Taylor & Dillin, 2013). APL‐1 is a glycosylated, transmembrane protein that is presumably processed in the endoplasmic reticulum (ER). Overexpression of APL‐1 could overload the ER causing ER stress, thereby activating the UPR. However, APL‐1 overexpression does not cause ER stress or induce the unfolded protein response (Fig. S3a–c). The increased lifespan, however, was dependent on a functional APL‐1 protein. The missense mutation apl‐1(yn32) causes an APL‐1 E371K substitution that presumably interferes with alpha secretase cleavage at the cell membrane. Expression of the apl‐1(yn32) transgene did not rescue the apl‐1(null) larval lethality (Hornsten et al., 2007; Hoopes et al., 2010), suggesting that APL‐1(yn32) is not sufficient for rescue. Animals carrying the Psnb‐1::APL‐1(E371K)::GFP transgene (ynEx236, 237, or 238) showed no lifespan extension (Fig. 2h), suggesting that only functional APL‐1 can increase lifespan and that nonfunctional APL‐1 has no adverse effects. To further exclude that any protein structurally similar to APL‐1 (e.g., human APP) driven by the snb‐1 promoter could extend lifespan, we measured the lifespan of animals carrying the Psnb‐1::humanAPP transgene, which had no effect on lifespan (Fig. 2c; Table S3). Human APP is unable to rescue the apl‐1(null) larval lethality (Hornsten et al., 2007; Wiese et al., 2010), suggesting that human APP is not functional in C. elegans. Taken together, these results suggest that overexpression of functional APL‐1 increases lifespan.
APL‐1 protein levels do not correlate with changes in lifespan
Another possibility for APL‐1 lifespan effects is that levels of the APL‐1 protein determine lifespan. However, levels of APL‐1 protein did not correlate with changes in lifespan, as ynIs79 [Papl‐1::apl‐1::GFP] animals expressed the highest levels of APL‐1::GFP, but showed a shortened lifespan (Figs. 1e and S3d–f). Instead, we postulate that the increased lifespan is context dependent; that is, where APL‐1 is overexpressed determines the APL‐1 effect on longevity. Hence, as long as APL‐1 is overexpressed in the relevant tissues, lifespan will be increased. We tested this model by mating short‐lived ynIs86 [Papl‐1::APL‐1] transgenic animals that show a 177‐fold increase in APL‐1 protein (Hornsten et al., 2007) with long‐lived ynIs12 [Psnb‐1::APL‐1] transgenic animals that show a 71‐fold increase in APL‐1 protein (Hornsten et al., 2007). The resultant double transgenic strain showed a 199‐fold increase in APL‐1 levels compared to wild‐type and was long‐lived (Figs. 2i and S3d–e; Table S3), demonstrating that the tissue in which apl‐1 is expressed and not the overall levels of APL‐1 protein determines lifespan.
Overexpression of APL‐1 in hypodermal cells is sufficient to increase lifespan
The long‐lived ynIs109 [Psnb‐1::APL‐1::GFP] transgenic animals showed prominent APL‐1::GFP expression in neurons and the somatic gonad and slightly weaker expression in the hypodermis (Fig. 2). The somatic gonad has previously been shown to regulate lifespan via hormonal signaling (Kenyon, 2010; Antebi, 2013). However, driving APL‐1 expression exclusively in the somatic gonad or in combination with neuronal APL‐1 did not alter lifespan (Fig. 3a; Table S1). Lastly, we tested whether hypodermal APL‐1 expression could increase lifespan. We generated transgenic animals that overexpress APL‐1 exclusively in the hypodermis (ynEx234 [Pcol‐10::APL‐1::GFP]). These transgenic lines showed an 11–22% increase in lifespan and fully recapitulated the increase in longevity seen in ynIs109 [Psnb‐1::APL‐1::GFP] animals (Fig 3b; Tables S1 and S4). Hence, apl‐1 overexpression in the hypodermis is critical for the APL‐1‐dependent increase in lifespan.
Figure 3.
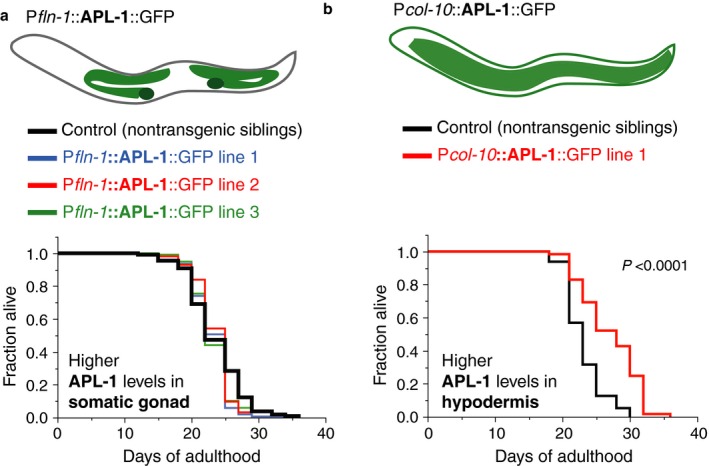
Ectopic hypodermal APL‐1 expression extends lifespan. (a) Driving APL‐1 expression in the somatic gonad with the fln‐1 promoter was not sufficient to increase lifespan. Lines 1, 2, and 3 are ynEx242, ynEx243, and ynEx244, respectively. (b) Driving APL‐1 expression in the hypodermis with the col‐10 promoter was sufficient to increase lifespan. Line 1 is ynEx234. For (a, b) P‐value was determined by log rank. Statistics and additional lifespan data are in Table S1.
Overexpression of the extracellular domain of APL‐1 is sufficient to affect lifespan in a tissue‐dependent manner
Similar to other APP family members, C. elegans APL‐1 is a single transmembrane‐spanning protein with a large extracellular and a small intracellular domain (Fig. 4a; Daigle & Li, 1993). The extracellular domain of APL‐1 (APL‐1EXT), which is analogous to sAPL‐1, is necessary and sufficient to rescue the apl‐1(null) lethality (Hornsten et al., 2007; Wiese et al., 2010). Furthermore, the apl‐1(yn5) allele completely lacks the genomic region encoding the transmembrane and intracellular domains, but is viable (Hornsten et al., 2007). To determine whether the extracellular domain of APL‐1 modulates lifespan, we examined apl‐1(yn5) mutants and transgenic animals that overexpress the extracellular domain of APL‐1 (APL‐1EXT) in a wild‐type background. Both apl‐1(yn5) mutants and transgenic animals that overexpressed APL‐1EXT from the apl‐1 promoter (ynIs71 [Papl‐1::APL‐1EXT]) showed shortened lifespans, similar to the shortened lifespan when full‐length APL‐1 was overexpressed (Fig. 4b,c). By contrast, when APL‐1EXT expression was driven with the snb‐1 promoter (ynIs105 [Psnb‐1::APL‐1EXT]), the animals showed an extended lifespan compared to wild‐type (Fig. 4d). Furthermore, expressing APL‐1EXT in the hypodermis increased lifespan (ynEx241 [Pcol‐10::APL‐1EXT]; Fig. 4e; Table S1). By contrast, expressing APL‐1EXT in the hypodermis is not sufficient to rescue the lethality seen in apl‐1(yn10) null alleles (0/3 lines rescued). Thus, the extracellular domain of APL‐1 modulates aging in a tissue‐specific manner.
Figure 4.
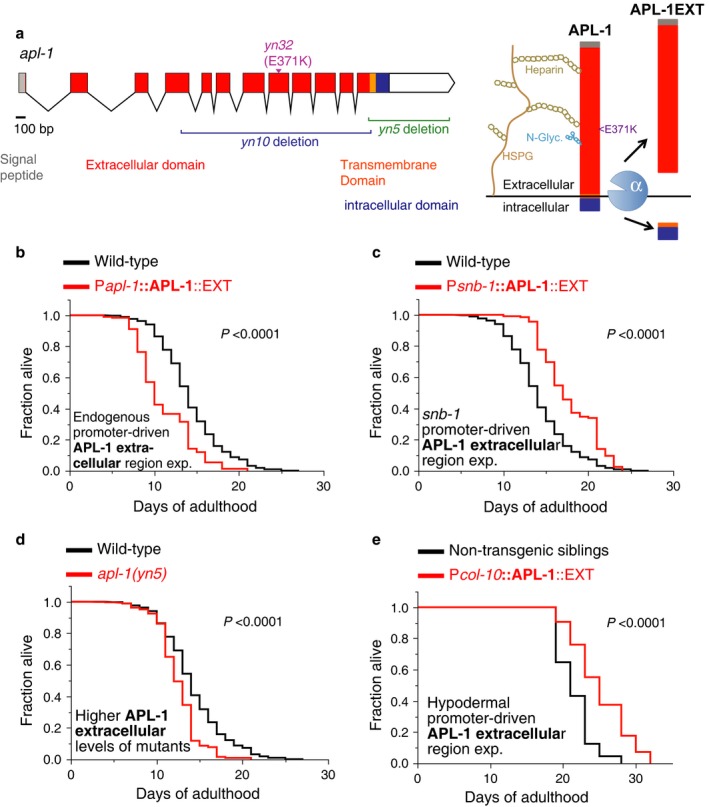
The extracellular domain of APL‐1 modulates lifespan depending on the tissues in which it is expressed. (a) Schematic of genomic region (left panel) and model of APL‐1 cleavage (right panel). α = alpha secretase, HSPG = heparan sulfate proteoglycans, N‐Glyc. = N‐glycosylation. The extracellular domain of APL‐1 has two potential HS binding domains. (b) Transgenic ynIs71 [Papl‐1::APL‐1EXT] animals that overexpress the extracellular domain of APL‐1 (APL‐1EXT) driven by its endogenous promoter showed a shortened lifespan. (c) Transgenic ynIs105 [Psnb‐1::APL‐1EXT] animals that overexpress the extracellular domain of APL‐1 (APL‐1EXT) driven by the snb‐1 promoter showed an extended lifespan. (d) apl‐1(yn5) mutants, which contain higher levels of the extracellular domain of APL‐1 compared to wild‐type (Fig. S3d,e), showed a shortened lifespan. (e) Transgenic ynEx241 [Pcol‐10::APL‐1EXT] animals that overexpress the extracellular domain of APL‐1 (APL‐1EXT) in the hypodermis showed an extended lifespan. For (b‐d) Lifespans are shown in cumulative form. Statistics and additional lifespan data are in Table S2.
Extended lifespan of Psnb‐1::APL‐1 transgenic animals requires daf‐16 and daf‐12 activity
We have previously demonstrated that overexpression of APL‐1EXT slows developmental progression via the FOXO transcription factor DAF‐16 and vitamin D‐like nuclear hormone receptor DAF‐12 VDR (Ewald et al., 2012b>). Both DAF‐16 FOXO and DAF‐12 VDR activities are essential to mobilize mechanisms that extend lifespan (Kenyon, 2010; Antebi, 2013). Thus, we investigated whether overexpression of APL‐1 modulates lifespan via daf‐16 and daf‐12. The longevity of ynIs12 [Psnb‐1::apl‐1 cDNA], ynIs109 [Psnb‐1::apl‐1 cDNA::GFP], and ynIs105 [Psnb‐1::APL‐1EXT] was completely abolished in daf‐16(null) and daf‐12 mutants (Fig. 5a,b; Tables S1 and S3), suggesting that activity of DAF‐16 FOXO and DAF‐12 VDR is required to mediate longevity by APL‐1 overexpression. Furthermore, we assessed whether other known transcriptional regulators essential for extending lifespan are required for the Psnb‐1::APL‐1EXT‐mediated longevity (Fig. S3g). We found that hsf‐1 (heat‐shock factor 1) was completely and skn‐1 (Nrf1,2,3 homologue) was partially required for the lifespan extension of ynIs105 [Psnb‐1::APL‐1EXT] animals (Table S3).
Figure 5.
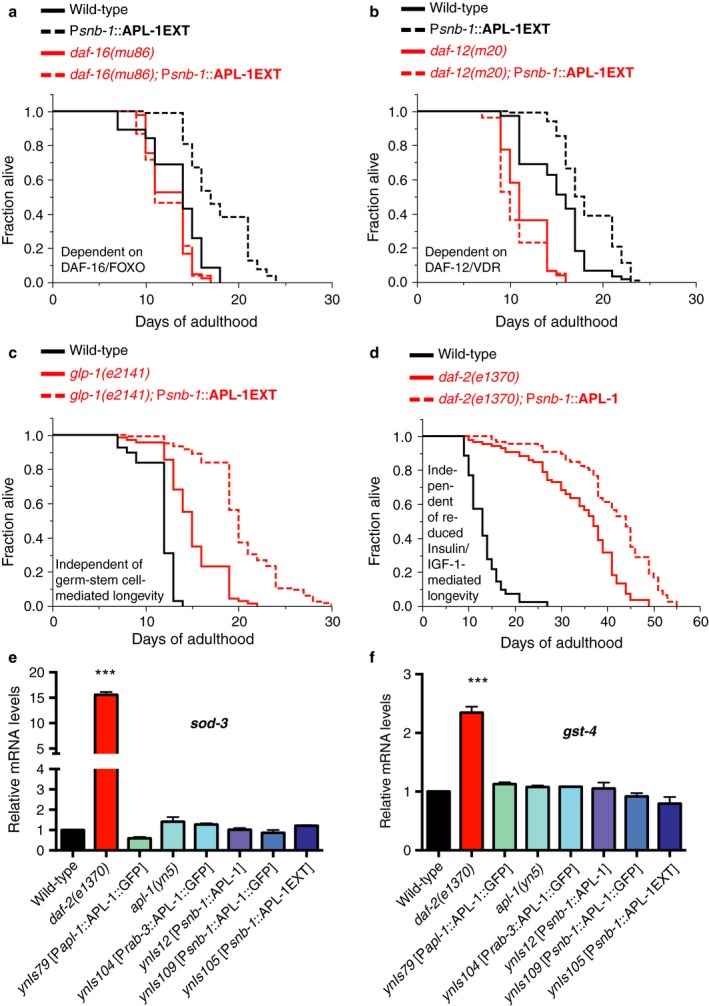
The APL‐1‐induced longevity requires DAF‐16 and DAF‐12. (a) The longevity of transgenic ynIs105 [Psnb‐1::APL‐1EXT] animals was suppressed by a null mutation in daf‐16, which encodes a FOXO transcription factor. (b) The longevity of transgenic ynIs105 [Psnb‐1::APL‐1EXT] animals was suppressed by a mutation in daf‐12, which encodes a vitamin D‐like nuclear hormone receptor (VDR). (c) The longevity of transgenic ynIs105 [Psnb‐1::APL‐1EXT] animals was additive to the longevity of animals with reduced germ stem cell numbers (glp‐1(e2141)). (d) The longevity of transgenic ynIs12 [Psnb‐1::APL‐1] animals was additive to the longevity of animals with reduced Insulin/IGF‐1 signaling (daf‐2(e1370)). (e–f) mRNA levels of canonical DAF‐16 target genes (sod‐3 and gst‐4) under reduced insulin/IGF‐1 conditions are not altered in animals that overexpress APL‐1. Three independent trials of N > 1000 of mixed‐stage animals per trial were analyzed. Data are represented as mean ± SEM P‐value ***<0.0001 relative to wild‐type (N2) was determined by one‐sample t‐test, two‐tailed, hypothetical mean of 1. For (a–d) P‐value was determined by log rank. Statistics and additional lifespan data are in Tables S2 and S3.
Many reproductive and metabolic disruptions increase lifespan. For instance, loss of germline stem cells, such as with a glp‐1(e2141) mutation, or reducing insulin/IGF‐1 signaling, such as with a daf‐2(e1370) mutation, extends lifespan via daf‐16 and/or daf‐12 (Kenyon, 2010). We investigated how APL‐1EXT released from hypodermal cells acts to increase lifespan. When germline stem cells were removed with a glp‐1 mutation, the longevity of ynIs105 [Psnb‐1::APL‐1EXT] animals was further extended (Fig. 5c; Table S3). In addition, RNAi of kri‐1 and tcer‐1, genes required for the glp‐1 mutant‐mediated longevity, did not suppress the longevity of ynIs105 [Psnb‐1::APL‐1EXT] animals (Table S3), suggesting that APL‐1 overexpression increases lifespan in a pathway independent of the longevity pathway mediated by the loss of germline stem cells. Similarly, the longevity of ynIs12 [Psnb‐1::APL‐1] was further extended when insulin/IGF‐1 signaling was reduced (rIIs; Fig. 5d; Tables S1 and S3), again suggesting that the APL‐1‐mediated longevity is independent of the reduced rIIs‐mediated longevity pathway. Furthermore, mRNA levels of sod‐3 and gst‐4, genes that are transcriptionally upregulated under reduced insulin/IGF‐1 conditions (Murphy et al., 2003; Ewald et al., 2015), were not altered when APL‐1 was overexpressed (Fig. 5e–f). Taken together, these results suggest that hypodermal APL‐1 interacts with DAF‐16 and DAF‐12 in pathways parallel to the insulin/IGF‐1 and germline stem cells pathways to increase lifespan.
Hypodermal APL‐1 longevity is mediated by the heterochronic transcription factor LIN‐14
Recently, daf‐16 FOXO and daf‐12 VDR activities have been shown to form a regulatory circuit with lin‐14 to regulate longevity (Shen et al., 2012). LIN‐14 is a heterochronic transcription factor that controls the transition from the first to second larval stage during development (Hristova et al., 2005). At the first larval stage (L1), LIN‐14 protein levels are high, thereby regulating genes that retain cells in the L1 stage (Ambros & Horvitz, 1984, 1987). During the first to second larval stage transition and later in development, the microRNA lin‐4 prevents lin‐14 translation by an RNA–RNA interaction (Lee et al., 1993). However, this inhibition is gradually lost during aging, when lin‐4 miRNA levels decrease, thereby allowing LIN‐14 protein levels to increase (Boehm & Slack, 2005; Ibáñez‐Ventoso et al., 2006; Ibáñez‐Ventoso & Driscoll, 2009). Interestingly, knocking down lin‐14 levels by RNAi in adults or by a temperature‐sensitive lin‐14 mutation during the larval‐to‐adult transition is sufficient to increase lifespan via daf‐16 FOXO and heat‐shock factor HSF‐1 (Boehm & Slack, 2005). Thus, we investigated whether the Psnb‐1::APL‐1 transgene expression mediates lifespan extension by acting in the same pathway as LIN‐14. The Psnb‐1::APL‐1 transgene‐mediated longevity was not further increased by either RNAi knockdown of lin‐14 or by a lin‐14(n179) mutation (Fig. 6a,b; Table S4). Furthermore, RNAi knockdown of lin‐14 was not additive to the longevity of hypodermal APL‐1 (ynEx234 [Pcol‐10::APL‐1::GFP; Fig. 6a; Table S4], suggesting that hypodermal APL‐1 and lin‐14 act in the same pathway to mediate longevity. Conversely, overexpressing LIN‐14 protein in ynIs109 [Psnb‐1::APL‐1::GFP] abolished the Psnb‐1::APL‐1‐mediated longevity (Fig. 6b; Table S4), suggesting that APL‐1 activity represses the activity of LIN‐14. Interestingly, increasing LIN‐14 protein levels in ynIs109 [Psnb‐1::APL‐1::GFP] animals results in a shortened lifespan (Fig. 6b; Table S4). We hypothesize that repression of lin‐14 mobilizes a protective response against the detrimental effects of neuronal APL‐1. We tested this hypothesis by increasing levels of LIN‐14 in hypodermal APL‐1 overexpression lines and found the suppression of hypodermal APL‐1‐induced longevity, but no lifespan reduction compared to wild‐type (Fig. 6c; Table S4). To determine whether hypodermal APL‐1 represses LIN‐14 activity during aging, we collected mRNA of postreproductive 8‐day‐old adult animals and measured the expression levels of three canonical LIN‐14 target genes, cyp‐13, ins‐33, and C15C7.5. During the first larval stage when LIN‐14 expression is high, C15C7.5 is upregulated, while cyp‐13 and ins‐33 are downregulated (Hristova et al., 2005). If APL‐1 acts to repress LIN‐14 activity, we predicted that genes that are normally repressed by LIN‐14 are upregulated and genes that are normally upregulated by LIN‐14 are downregulated. Consistent with this hypothesis, cyp‐13 and ins‐33 were upregulated and C15C7.5 gene was downregulated in ynIs109 [Psnb‐1::APL‐1::GFP] animals compared to wild‐type at day 8 of adulthood (Fig. 6d), suggesting that hypodermal APL‐1 is repressing LIN‐14 activity during aging. Taken together, we propose that hypodermal APL‐1 represses LIN‐14 levels during aging, which prolongs lifespan via hsf‐1, daf‐12, and daf‐16 (Fig. 6e).
Figure 6.
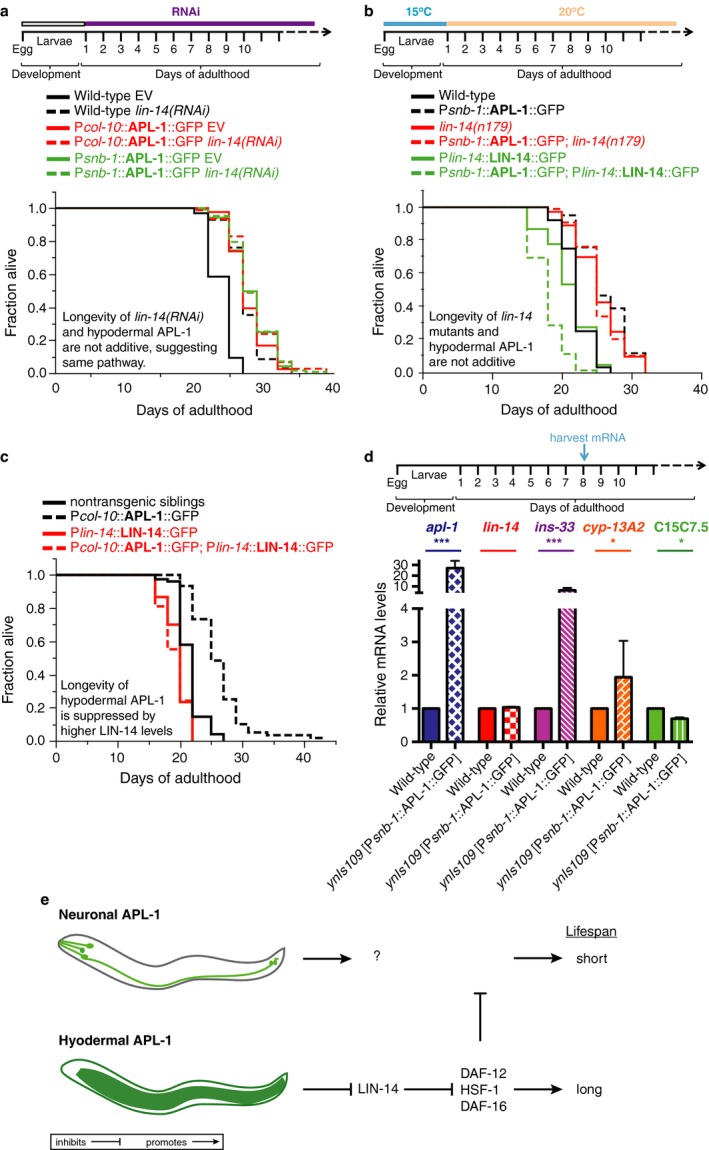
The APL‐1‐dependent longevity is mediated via the heterochronic transcription factor LIN‐14. (a) The longevity of transgenic ynEx234 [Pcol‐10::APL‐1::GFP] and ynIs109 [Psnb‐1::APL‐1::GFP] animals was not additive to the longevity of animals treated with lin‐14 RNAi. (b) Higher LIN‐14 protein levels of zaIs2 [LIN‐14::GFP] transgenic animals that contain the lin‐4 microRNA binding sites in the 3′UTR were sufficient to suppress the longevity of ynIs109 [Psnb‐1::APL‐1::GFP] animals. The longevity of transgenic ynIs109 [Psnb‐1::APL‐1::GFP] animals was not additive to the longevity of animals carrying a mutation in the lin‐14 gene (lin‐14(n179)). (c) Higher LIN‐14 protein levels suppressed the longevity of ynEx234 [Pcol‐10::APL‐1::GFP] animals. (d) mRNA levels of canonical LIN‐14 target genes were altered in day 8 of transgenic ynIs109 [Psnb‐1::APL‐1::GFP] adults compared to wild‐type. Three independent trials of N = 200 day 8 adult animals per trial were analyzed. Data are represented as mean ± SEM P‐value *<0.05 and ***<0.0001 relative to wild‐type (N2) was determined by one‐sample t‐test, two‐tailed, hypothetical mean of 1. (e) Model of how APL‐1 modulates lifespan. Neuronal APL‐1 overexpression shortens lifespan through an unknown mechanism (upper panel). Hypodermal APL‐1 overexpression represses LIN‐14 accumulation during aging, thereby releasing the inhibition of DAF‐12, HSF‐1, and DAF‐16 activity to promote longevity (lower panel). Repression of LIN‐14 and activation of DAF‐12, HSF‐1, and DAF‐16 are sufficient to protect against the adverse effects of higher APL‐1 levels in neurons. For (a–c) P‐value was determined by log rank. Statistics and additional lifespan data are in Table S4.
Discussion
Mammalian APP is expressed in all tissue types (Tanzi et al., 1987), but an unresolved question remains as to whether APP has a similar function in all cell types or whether its functions are context dependent. As this question is difficult to address in mammalian systems, we turned to C. elegans where the use of cell‐specific promoters is routine. Because higher levels of APP have been correlated with Alzheimer's disease and Down syndrome, we asked whether levels of APL‐1, the C. elegans APP‐related protein, have an effect on lifespan. Longevity in any organism is the result of a combination of environmental effects and genetic factors. C. elegans is constantly monitoring its surroundings and integrating this information to regulate its metabolic and reproductive processes, which ultimately determine the animal's lifespan. Several tissues, such as neurons, intestine, and gonad, have been implicated in regulating lifespan in C. elegans (Kenyon, 2010). For instance, ablation of certain amphidial sensory neurons can either lead to lifespan enhancement or reduction; similarly, ablation of germline precursor cells leads to an increased lifespan. These effects on lifespan are dependent in DAF‐16 FOXO and DAF‐12 VDR activity (Kenyon, 2010).
The effects of APL‐1 overexpression on lifespan were context dependent (Fig. 6e). Overexpressing apl‐1 under its own promoter caused accelerated aging and lifespan reduction. By contrast, ubiquitous expression of apl‐1 in adults caused lifespan extension. These results suggest that where APL‐1 is expressed impacts its effects on lifespan. Because apl‐1 expression in neurons, but not in hypodermal cells, is sufficient to rescue the lethality of apl‐1 null mutants, we hypothesized that neuronal apl‐1 expression was responsible for lifespan extension. Unexpectedly, the converse results were found; namely, apl‐1 overexpression only in neurons led to lifespan reduction. However, apl‐1 overexpression in hypodermal tissue is critical for lifespan extension and this lifespan extension is additive to the longevity induced by reduction in insulin signaling or by the removal of germline stem cells. Furthermore, we showed that expression of only the extracellular domain of APL‐1 in these tissues was sufficient to cause these lifespan effects and lifespan extension is dependent on the activity of daf‐16 FOXO and DAF‐12 VDR. By contrast, hypodermal expression of apl‐1 or APL‐1EXT is not sufficient to rescue the lethality of apl‐1 null mutants. Hence, the target of neuronal release of sAPL‐1 vs. hypodermal release of sAPL‐1 appears different. We hypothesize that migration of sAPL‐1 is limited or confined by its local environment. For instance, sAPL‐1 has two heparin binding domains (Fig. 4a), suggesting that the secreted APL‐1 could interact locally with the extracellular matrix to limit its diffusion. Deleting the heparin binding domains of APL‐1EXT resulted in a failure to rescue the apl‐1 null lethality (unpublished observation). Furthermore, overexpression of APL‐1EXT lacking the heparin binding domains had no effect on lifespan (Table S3), suggesting that interactions between sAPL‐1 and the extracellular matrix are essential for APL‐1 function. Collectively, these results indicate that APL‐1 has multiple functions: expression of apl‐1 in neurons is required for viability, whereas expression of apl‐1 in hypodermal cells regulates longevity.
The lifespan effects due to apl‐1 overexpression were not dependent on downregulating metabolic pathways via insulin/IGF‐1 signaling or decreasing numbers of germline stem cells, but were influenced by the heterochronic pathway. apl‐1 has been previously linked to the let‐7 microRNA and heterochronic regulatory network (Niwa et al., 2008), which is composed of a regulatory cascade of protein regulators and microRNAs (miRNAs) that coordinate the onset of stereotypical stage‐specific developmental events (Ambros & Horvitz, 1987; Rougvie & Moss, 2013). In particular, while levels of the LIN‐14 transcription factor are negatively regulated by the lin‐4 miRNA to allow transition from the first to the second larval stage (Lee et al., 1993), levels of lin‐4 miRNA decrease with age, allowing LIN‐14 levels to increase, thereby limiting the lifespan of C. elegans (Ibáñez‐Ventoso & Driscoll, 2009). We propose that hypodermal apl‐1 expression represses levels of LIN‐14, resulting in an increased lifespan relative to wild‐type animals (Fig. 6e).
In C. elegans, maintenance of somatic tissues is lost during the first day of adulthood (Labbadia & Morimoto, 2014). This is consistent with the disposable soma theory that predicts that resources are allocated from the somatic tissue to the germ line (Kirkwood & Holliday, 1979), which happens when animals become reproductive during the L4‐to‐adult transition. Interestingly, using a transcriptional reporter of the endogenous promoter of apl‐1 expressing GFP, apl‐1 has been shown by Niwa and colleagues to be transiently expressed under the control of let‐7 miRNA network during the L4‐to‐adult transition in the hypodermal seam cells (Niwa et al., 2008). However, this hypodermal APL‐1 expression is lost in adulthood and we show here that ectopically expressing APL‐1 during adulthood in the hypodermis leads to increased health‐ and lifespan. We speculate that reinforcing APL‐1 expression in the hypodermis is a reprogramming of developmental networks via heterochronic LIN‐14 to improve the maintenance of somatic tissues via DAF‐16/FOXO and HSF‐1 that leads to improved organismal lifespan. The implication of an APP‐related protein functioning in a miRNA and heterochronic gene network to promote healthy aging might point toward a possible entry to use miRNAs as a therapeutic intervention to modulate age‐related consequences of APP function.
Material and methods
Strains
Caenorhabditis elegans strains were grown and maintained on MYOB or NGM plates containing OP50 Escherichia coli bacteria at 20°C, unless noted. All mutations are described in WormBase (www.wormbase.org) and include LGI: daf‐16(mu86); LGII: tcer‐1(tm1452); LGIII: daf‐2(e1370), glp‐1(e2141); LGX: daf‐12(m20), lon‐2(e678), apl‐1(yn5 and yn10) (Hornsten et al., 2007), dpy‐8(e130), and lin‐14(n179). Construction of the APL‐1 transgenes and the resulting transgenic lines are described (Hornsten et al., 2007) and (Ewald et al., 2012a>), except as indicated. Nonintegrated transgenic lines used were ynEx30 [Papl‐1::APL‐1, sur‐5::GFP], ynEx65 [Psnb‐1::APL‐1(yn5)(cDNA), sur‐5::GFP], ynEx93 [Prab‐3::APL‐1(cDNA)::GFP, pRF4 rol‐6(su1006gf)], ynEx109 [Psnb‐1::APL‐1(cDNA)::GFP], ynEx212 [Pmec‐4::APL‐1 (cDNA)::GFP, Pmyo‐3::mCherry], ynEx213 [Pmec‐4::APL‐1 (cDNA)::GFP, Pmyo‐3::mCherry], ynEx214 [Pceh‐36::APL‐1 (cDNA)::GFP, Pmyo‐3::mCherry], ynEx236 [Psnb‐1::APL‐1(E371K)::GFP, Pmyo‐3::mCherry], ynEx237 [Psnb‐1::APL‐1(E371K)::GFP, Pmyo‐3::mCherry], ynEx238 [Psnb‐1::APL‐1(E371K)::GFP, Pmyo‐3::mCherry], ynEx242 [Pfln‐1::APL‐1::GFP, Pmyo‐3::mCherry]), ynEx243 [Pfln‐1::APL‐1::GFP, Pmyo‐3::mCherry], ynEx244 [Pfln‐1::APL‐1::GFP, Pmyo‐3::mCherry], ynEx234 [Pcol‐10::APL‐1 (cDNA)::GFP, Pmyo‐3::mCherry], ynEx235 [Pcol‐10::APL‐1 (cDNA)::GFP, Pmyo‐3::mCherry], ynEx239 [Pcol‐10::APL‐1EXT (cDNA), sur‐5::GFP], ynEx240 [Pcol‐10::APL‐1EXT (cDNA), sur‐5::GFP], and ynEx241 [Pcol‐10::APL‐1EXT (cDNA), sur‐5::GFP]. Integrated transgenic lines used were ynIs100 [Papl‐1::APL‐1(yn32), pRF4 rol‐6(su1006gf)], ynIs106 [Papl‐1::APL‐1(yn32 yn5)], ynIs14 [Phsp‐16.2::APL‐1 (cDNA), lin‐15B(+)],Pmyo‐2::GFP), ynIs113 [Pmec‐4::APL‐1 (cDNA)::GFP, Pmyo‐3::mCherry], jsIs1 [Psnb‐1:: SNB‐1::GFP, pRF4 rol‐6(su1006gf)], dvEx371 [Psnb‐1::humanAPP751 (cDNA), Pmtl‐2::GFP], dvEx372 [Psnb‐1::humanAPP751 (cDNA), Pmtl‐2::GFP], zaIs2 [LIN‐14::GFP, rol‐6(su1006)] (Hong et al., 2000; Olsson‐Carter & Slack, 2010); LGI: ynIs109 [Psnb‐1::APL‐1(cDNA)::GFP]; LGII: ynIs105 [Psnb‐1::APL‐1(yn5)(cDNA), sur‐5::GFP]; LGIII: jsIs682 [Prab‐3::GFP::RAB‐3], ynIs12 [Psnb‐1::APL‐1(cDNA), lin‐15B(+)]; LGIV: vsIs13 (lin‐11::pes‐10::GFP, lin‐15(+)), ynIs104 [Prab‐3::APL‐1(cDNA)::GFP, Pmyo‐2::GFP], zIs356 [Pdaf‐16::DAF‐16::GFP, pRF4 rol‐6(su1006gf)]; LGV: ynIs13 [Psnb‐1::APL‐1(cDNA), lin‐15B(+)], ynIs71 [Papl‐1::APL‐1(yn5), sur‐5::GFP], ynIs79 [Papl‐1::APL‐1::GFP], ynIs100 [Papl‐1::APL‐1(yn32)::GFP, pRF4 rol‐6(su1006gf)]; and LGX: dvIs62 (Psnb‐1::humanTDP‐43, Pmtl‐2::GFP), ynIs86 [Papl‐1::APL‐1, sur‐5::GFP], ynIs91 [Prab‐3::APL‐1 (cDNA), pRF4 rol‐6(su1006gf)], ynIs107 [Papl‐1::APL‐1(yn32 D342C/S362C)::GFP, Pmyo‐2::GFP], and ynIs108 [Papl‐1::APL‐1(ΔH, ΔE2)::GFP; sur‐5::GFP].
Generating transgene constructs
To eliminate the heparin sulfate binding domains (H) of APL‐1, the coding region for REEGSPCKWTHSVR, corresponding to amino acids 96–106, was deleted from the pAPL‐1ΔE2::GFP construct (Hornsten et al., 2007), which already had the heparin binding domain within E2 deleted, to generate the Papl‐1::APL‐1(ΔH, ΔE2)::GFP construct with both heparin sulfate binding domains deleted.
RNA interference
RNAi clones were picked from the Ahringer or Vidal libraries. Cultures were grown overnight in LB with 12.5 μg/mL tetracycline and 100 μg/mL ampicillin, diluted to an OD600 of 1, and induced with 1 mM IPTG. This culture was seeded onto NGM agar plates containing tetracycline, ampicillin, and additional IPTG. Empty vector (EV) plasmid pL4440 was used as control.
Lifespan assays
Because our laboratory uses a different growing media (MYOB) than other laboratories, which normally use (NGM), and wild‐type shows a different mean lifespan of 14 days on MYOB vs. 21 days on NGM (Keightley & Caballero, 1997), we performed most experiments on both media as indicated (Tables S1–S4). Lifespan results were similar on MYOB or on NGM. Note that the wild‐type N2 var. Bristol strain has been maintained for many generations in different laboratories and can have mean lifespans ranging from 12 to 17 days at 20°C (Gems & Riddle, 2000). N2 animals from another laboratory (kindly provided by Cathy Savage‐Dunn) showed a similar lifespan as our N2 stock on MYOB plates in four independent trials (198/398 animals). To determine whether the laboratory N2 animals had a longer lifespan on NGM plates, N2 animals were grown on MYOB until the L4 stage and then transferred onto NGM plates for lifespan analysis; these animals showed a mean lifespan of 18.62 ± 0.47 days (60/111 animals). These results indicate that the N2 mean lifespan of 14 days reported here is due to the different composition of the MYOB plates compared to NGM plates.
For all lifespan trials, NGM: L4 animals were picked onto fresh OP50 plates, and then day one adults were placed on either OP50 or RNAi plates containing 50 μm 5‐fluoro‐2′‐deoxyuridine (FUdR), unless otherwise indicated, as described in Ewald et al. (2015). All lifespan trials on MYOB were performed without FUdR. All strains were grown for at least two generations at 20°C, except for daf‐2(e1370) and glp‐1(e2141) animals, which were raised at 15°C. All adult lifespans [except glp‐1(e2141)] were measured starting from the L4 stage at 20°C. Animals carrying the temperature‐sensitive glp‐1(e2141) allele and appropriate control animals were shifted from 15°C to 25°C at the L1 stage and lifespan assays were performed at 25°C.
Pharyngeal pumping assays
The pharyngeal pumping rate was determined by the number of pumps per 20 s; measurements were only taken when worms were on bacteria and pumped at a constant rate without any pauses as described (Ewald et al., 2012b>).
Autofluorescence assays
L4 animals were picked and scored at appropriate times. Animals were placed on an agar pad containing a drop of 10 mm NaN3. Pictures were taken with a Hamamatsu ORCA‐ER digital camera on a Zeiss Axioplan microscope. The area directly behind the last bulb of the pharynx was photographed using Openlab 3.1.3 software (Improvision, Coventry, United Kingdom) with the same settings for all animals and conditions. The autofluorescence intensities were derived by selecting a defined area of the worm in the pictures and computing the ‘mean gray value’ by ImageJ software (NIH).
Western blot analysis
Preparations of animal lysates and Western blots were performed as described (Hornsten et al., 2007). Protein levels were normalized to levels of actin (~40 kDa) for each strain, so that similar amounts of total protein were loaded for each strain. For each blot, the blot was first probed with an actin monoclonal antibody (JLA20 at 1:2000; Developmental Studies Hybridoma Bank, Iowa City, Iowa, United States; secondary antibody anti‐mouse‐goat‐HRP at 1:1000), washed three times with TAE, and then re‐probed with an anti‐APL‐1 antiserum against the extracellular domain of APL‐1 (Hornsten et al., 2007). Relative protein levels were determined by relative intensity to wild‐type (N2) using NIH Image J Gel analyzer.
Quantitative real‐time polymerase chain reaction (qRT‐PCR) assays
Mixed‐stage animals were harvested (>1000 animals; three independent trials; Figs. 5e–f and S3b,c,f). For adulthood qRT‐PCR: one‐day adults were placed on OP50 NGM FUdR plates at 20°C and 8 days later, 200 worms were harvested (Fig. 6d). RNA was isolated with Trizol (TRI REAGENT; Sigma, St. Louis, Missouri, United States), DNase‐treated, and cleaned over a column (RNA Clean & Concentrator™; ZYMO Research, Irvine, California, United States). First‐strand cDNA was synthesized in duplicate from each sample (Invitrogen, Carlsbad, California, United States, SuperScript III). SYBR green was used to perform qRT‐PCR (Applied Biosystems 7900, Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA). For each primer set, a standard curve from genomic DNA accompanied the duplicate cDNA samples (Ewald et al., 2015). mRNA levels relative to N2 control were determined by normalizing to the number of worms and the geometric mean of three reference genes [cdc‐42, pmp‐3, and Y45F10D.4 (Hoogewijs et al., 2008)]. At least two biological replicates were examined for each sample. For statistical analysis, one‐sample t‐test, two‐tailed, hypothetical mean of 1 was used for comparison using Prism 4.0a software (GraphPad, La Jolla, California, United States).
Fat quantification
Fixed Oil Red O (ORO) staining: Worms were synchronized by hypochlorite treatment and day 1 adult worms were fixed. ORO staining of fixed animals and quantification was performed.
Triglyceride quantification: Mixed‐stage worms were collected and prepared as described. Triglyceride levels were measured with the Triglyceride Colorimetric Assay Kit (#10010303; Cayman Chemical, Ann Arbor, MI, USA) and normalized to protein levels using BCA protein assay (Pierce Biotechnology, Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA).
Funding
This work was supported by grants from the Alzheimer's Association, National Institutes of Health (R21AG0339 and R01AG32042), and National Science Foundation (IOS08207) (CL) and a National Institutes of Health RCMI grant (G12‐RR03060‐25) to City College.
Author contributions
CYE and CL designed the experiments, analyzed and interpreted the data. CYE, VM, and CL generated transgenic animals. CYE performed all experiments. CYE and CL wrote the manuscript in consultation with VM.
Conflict of interest
The authors have no competing interests to declare.
Supporting information
Fig. S1 The APL‐1 effects on lifespan correlates with biomarkers of aging.
Fig. S2 The effects on lifespan of APL‐1 overexpressing lines does not correlate with fat content.
Fig. S3 Longevity of Psnb‐1::APL‐1 animals depends on hsf‐1, daf‐16, but not on the unfolded protein response.
Table S1 Hypodermal APL‐1 increases lifespan.
Table S2 APL‐1 mediated lifespan extension requires DAF‐16 and DAF‐12.
Table S3 APL‐1 longevity requires functional APL‐1 and is additive to reduced Insulin/IGF‐1 and reduced germstem cell number mediated longevity.
Table S4 Hypodermal APL‐1 longevity by LIN‐14.
Acknowledgments
We wish to thank Jane Hubbard's group for kindly providing glp‐1 mutants, Cathy Savage‐Dunn's group for kindly providing their N2, Chris Link for the dvEx371, dvEx372, and dvIs62 strains, Erin Cram and Oliver Hobert for providing plasmids, Mahmoud Salem, Monet Bland, and Lana Tolen for confirming lifespan experiments, Sri Devi Narasimhan for help with triglyceride assays, Dan Raps for some microinjections, laboratory members for helpful discussions, Sarah Tichelli for help with statistical analysis, Roy and Gab Ewald, Wolfgang Maier, Cathy Savage‐Dunn, and Piali Sengupta for comments on the manuscript. The apl‐1 genomic representation was made using Exon‐Intron Graphic Maker (http://wormweb.org/exonintron) from Nikhil Bhatla. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
References
- Alzheimer's Association (2015) 2015 Alzheimer's disease facts and figures. Alzheimers Dement. 11, 332–384. [DOI] [PubMed] [Google Scholar]
- Ambros V, Horvitz HR (1984) Heterochronic mutants of the nematode Caenorhabditis elegans . Science 226, 409–416. [DOI] [PubMed] [Google Scholar]
- Ambros V, Horvitz HR (1987) The lin‐14 locus of Caenorhabditis elegans controls the time of expression of specific postembryonic developmental events. Genes Dev. 1, 398–414. [DOI] [PubMed] [Google Scholar]
- Antebi A (2013) Regulation of longevity by the reproductive system. Exp. Gerontol. 48, 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm M, Slack F (2005) A developmental timing microRNA and its target regulate life span in C. elegans . Science 310, 1954–1957. [DOI] [PubMed] [Google Scholar]
- Cabrejo L, Guyant‐Maréchal L, Laquerrière A, Vercelletto M, De la Fournière F, Thomas‐Antérion C, Verny C, Letournel F, Pasquier F, Vital A, Checler F, Frebourg T, Campion D, Hannequin D (2006) Phenotype associated with APP duplication in five families. Brain 129, 2966–2976. [DOI] [PubMed] [Google Scholar]
- Daigle I, Li C (1993) apl‐1, a Caenorhabditis elegans gene encoding a protein related to the human beta‐amyloid protein precursor. Proc. Natl Acad. Sci. USA 90, 12045–12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald CY, Li C (2010) Understanding the molecular basis of Alzheimer's disease using a Caenorhabditis elegans model system. Brain Struct. Funct. 214, 263–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald CY, Li C (2012) Caenorhabditis elegans as a model organism to study APP function. Exp. Brain Res. 217, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald CY, Cheng R, Tolen L, Shah V, Gillani A, Nasrin A, Li C (2012a) Pan‐neuronal expression of APL‐1, an APP‐related protein, disrupts olfactory, gustatory, and touch plasticity in Caenorhabditis elegans . J. Neurosci. 32, 10156–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald CY, Raps DA, Li C (2012b) APL‐1, the Alzheimer's Amyloid precursor protein in Caenorhabditis elegans, modulates multiple metabolic pathways throughout development. Genetics 191, 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald CY, Landis JN, Porter Abate J, Murphy CT, Blackwell TK (2015) Dauer‐independent insulin/IGF‐1‐signalling implicates collagen remodelling in longevity. Nature 519, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gems D, Riddle DL (2000) Defining wild‐type life span in Caenorhabditis elegans . J. Gerontol. A Biol. Sci. Med. Sci. 55, B215–B219. [DOI] [PubMed] [Google Scholar]
- Heestand BN, Shen Y, Liu W, Magner DB, Storm N, Meharg C, Habermann B, Antebi A (2013) Dietary restriction induced longevity is mediated by nuclear receptor NHR‐62 in Caenorhabditis elegans . S. K. Kim, ed. PLoS Genet. 9, e1003651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Gajic V, Hainfellner J, Aguzzi A, Rülicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP (2000) Mice with combined gene knock‐outs reveal essential and partially redundant functions of amyloid precursor protein family members. J. Neurosci. 20, 7951–7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Müller U (2004) Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 23, 4106–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Lee RC, Ambros V (2000) Structure and function analysis of LIN‐14, a temporal regulator of postembryonic developmental events in Caenorhabditis elegans. Mol. Cell. Biol. 20, 2285–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogewijs D, Houthoofd K, Matthijssens F, Vandesompele J, Vanfleteren JR (2008) Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans . BMC Mol. Biol. 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes JT, Liu X, Xu X, Demeler B, Folta‐Stogniew E, Li C, Ha Y (2010) Structural characterization of the E2 domain of APL‐1, a Caenorhabditis elegans homolog of human amyloid precursor protein, and its heparin binding site. J. Biol. Chem. 285, 2165–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornsten A, Lieberthal J, Fadia S, Malins R, Ha L, Xu X, Daigle I, Markowitz M, O'Connor G, Plasterk R, Li C (2007) APL‐1, a Caenorhabditis elegans protein related to the human beta‐amyloid precursor protein, is essential for viability. Proc. Natl Acad. Sci. USA 104, 1971–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova M, Birse D, Hong Y, Ambros V (2005) The Caenorhabditis elegans heterochronic regulator LIN‐14 is a novel transcription factor that controls the developmental timing of transcription from the insulin/insulin‐like growth factor gene ins‐33 by direct DNA binding. Mol. Cell. Biol. 25, 11059–11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D (1995) Age‐related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 15, 1203–1218. [DOI] [PubMed] [Google Scholar]
- Ibáñez‐Ventoso C, Driscoll M (2009) MicroRNAs in C. elegans aging: molecular insurance for robustness? Curr. Genomics 10, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez‐Ventoso C, Yang M, Guo S, Robins H, Padgett RW, Driscoll M (2006) Modulated microRNA expression during adult lifespan in Caenorhabditis elegans . Aging Cell 5, 235–246. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Caballero A (1997) Genomic mutation rates for lifetime reproductive output and lifespan in Caenorhabditis elegans . Proc. Natl Acad. Sci. USA 94, 3823–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ (2010) The genetics of ageing. Nature 464, 504–512. [DOI] [PubMed] [Google Scholar]
- Kirkwood TB, Holliday R (1979) The evolution of ageing and longevity. Proc. R. Soc. Lond. B Biol. Sci. 205, 531–546. [DOI] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, Van Der Ploeg LH, Price DL, Sisodia SS (1997) Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol. Aging 18, 661–669. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Tirosh‐Wagner T, Urban AE, Chen X‐N, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, Sobel EM, Barlow GM, Aylsworth AS, Carpenter NJ, Clark RD, Cohen MY, Doran E, Falik‐Zaccai T, Lewin SO, Lott IT, McGillivray BC, Moeschler JB, Pettenati MJ, Pueschel SM, Rao KW, Shaffer LG, Shohat M, Van Riper AJ, Warburton D, Weissman S, Gerstein MB, Snyder M, Korenberg JR (2009) The genetic architecture of Down syndrome phenotypes revealed by high‐resolution analysis of human segmental trisomies. Proc. Natl Acad. Sci. USA 106, 12031–12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI (2014) Proteostasis and longevity: when does aging really begin? F1000Prime Rep. 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin‐4 encodes small RNAs with antisense complementarity to lin‐14 . Cell 75, 843–854. [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon CJ (2003) Genes that act downstream of DAF‐16 to influence the lifespan of Caenorhabditis elegans . Nature 424, 277–283. [DOI] [PubMed] [Google Scholar]
- Niwa R, Zhou F, Li C, Slack FJ (2008) The expression of the Alzheimer's amyloid precursor protein‐like gene is regulated by developmental timing microRNAs and their targets in Caenorhabditis elegans . Dev. Biol. 315, 418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson‐Carter K, Slack FJ (2010) A developmental timing switch promotes axon outgrowth independent of known guidance receptors. K. Olsson‐Carter & F. J. Slack, eds. PLoS Genet. 6, e1001054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougvie AE, Moss EG (2013) Developmental transitions in C. elegans larval stages. Curr. Top. Dev. Biol. 105, 153–180. [DOI] [PubMed] [Google Scholar]
- Rovelet‐Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D (2006) APP locus duplication causes autosomal dominant early‐onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shariati SAM, De Strooper B (2013) Redundancy and divergence in the amyloid precursor family. FEBS Letters 587, 2036–2045. [DOI] [PubMed] [Google Scholar]
- Shen Y, Wollam J, Magner D, Karalay O, Antebi A (2012) A steroid receptor‐microRNA switch regulates life span in response to signals from the gonad. Science 338, 1472–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del‐Favero J, Cruts M, van Duijn CM, Van Broeckhoven C (2006) APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain 129, 2977–2983. [DOI] [PubMed] [Google Scholar]
- Tanzi RE (2012) The genetics of Alzheimer disease. Cold Spring Harb. Perspect Med. 2, a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL (1987) Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 235, 880–884. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Dillin A (2013) XBP‐1 is a cell‐nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer SW, Klevanski M, Delekate A, Voikar V, Aydin D, Hick M, Filippov M, Drost N, Schaller KL, Saar M, Vogt MA, Gass P, Samanta A, schke AJA, Korte M, Wolfer DP, Caldwell JH, Müller UC (2011) APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J. 30, 2266–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese M, Antebi A, Zheng H (2010) Intracellular trafficking and synaptic function of APL‐1 in Caenorhabditis elegans . C.‐X. Gong, ed. PLoS One 5, e12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 The APL‐1 effects on lifespan correlates with biomarkers of aging.
Fig. S2 The effects on lifespan of APL‐1 overexpressing lines does not correlate with fat content.
Fig. S3 Longevity of Psnb‐1::APL‐1 animals depends on hsf‐1, daf‐16, but not on the unfolded protein response.
Table S1 Hypodermal APL‐1 increases lifespan.
Table S2 APL‐1 mediated lifespan extension requires DAF‐16 and DAF‐12.
Table S3 APL‐1 longevity requires functional APL‐1 and is additive to reduced Insulin/IGF‐1 and reduced germstem cell number mediated longevity.
Table S4 Hypodermal APL‐1 longevity by LIN‐14.