

Abstract
A review is provided on efforts in our laboratory over the last decade to discover anti-HIV agents. The work has focused on computer-aided design and synthesis of non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs) with collaborative efforts on biological assaying and protein crystallography. Numerous design issues were successfully addressed including the need for potency against a wide range of viral variants, good aqueous solubility, and avoidance of electrophilic substructures. Computational methods including docking, de novo design, and free-energy perturbation (FEP) calculations made essential contributions. The result is novel NNRTIs with picomolar and low-nanomolar activities against wild-type HIV-1 and key variants that also show much improved solubility and lower cytotoxicity than recently approved drugs in the class.
Keywords: Structure-based design, Free energy perturbation, Anti-HIV drugs, NNRTIs
1. Introduction
Recognition of the on-going AIDS epidemic occurred in the early 1980s. Drug development proceeded rapidly leading to FDA-approval of AZT in 1987, capitalizing on earlier work on the use of nucleic acid analogues to inhibit the reverse transcriptase (RT) enzyme found in retroviruses.1 The search for additional anti-HIV drugs and targets soon led to other nucleoside inhibitors (NRTIs), HIV protease inhibitors (PIs), and the non-nucleoside class of HIV-RT inhibitors (NNRTIs). Application of structure-based drug design (SBDD), which was in its infancy in the 1980s, became possible for anti-HIV agents with the first reports of crystal structures for HIV protease2 and reverse transcriptase.3,4 In fact, discovery of PIs guided by protein crystallography is considered among the earliest successes of SBDD.5,6 In view of the continuing need to develop new anti-HIV agents with improved therapeutic spectrum, safety, and pharmacological properties, we became involved in collaborative efforts at Yale that had roots in the development of stavudine (Zerit) and the pioneering crystallography for HIV-RT in the Prusoff7 and Steitz laboratories.3,4 Our initial, purely computational work evolved into joint experimental and computational discovery of next-generation NNRTIs with striking potency and improved solubility and safety. On this journey, much has been learned about the challenges of drug discovery and about the utility of computational methods in addressing them.
2. Background and early computational studies
Since the late 1970s our laboratory has been engaged in fluid simulations using Monte Carlo statistical mechanics (MC) and molecular dynamics (MD). Our first MD simulations for proteins in water were in the mid-1980s,8,9 at which time we were also actively pursuing free-energy calculations with many applications including study of solvent-effects on reaction kinetics and computation of relative free-energies of binding for organic host–guest systems.10 Our preferred method for computing free-energy changes was and remains free-energy perturbation (FEP) calculations using the procedures that we introduced in 1985.11,12 However, we had also explored ‘linear response’ (LR) methods,13 which attempt to estimate binding energies from changes in energy components and surface areas from MC or MD simulations of bound and unbound complexes.14 A combination of insufficient software and computer resources kept us from routinely applying FEP calculations to protein–ligand binding until 1996. By this time, we had developed the MCPRO program15 for this purpose and had started using PC clusters.16 The first applications of our MC/FEP methodology were then to cyclosporine/cyclophilin and trypsin/benzamidine complexes;17,18 the accord with experimental binding data confirmed the potential of the approach.
In collaboration with Marilyn Kroeger Smith and co-workers at the National Cancer Institute, our initial simulations of complexes of HIV-RT were with TIBO analogues,19,20 which were discovered at the Rega Institute21 and along with HEPT derivatives were the first known NNRTIs.22 Though the lead HEPT (emivirine) and TIBO (tivirapine) compounds did not become drugs, by 1998 three NRRTIs had been approved by the FDA: nevirapine (1996), delavirdine (1997), and efavirenz (1998). Subsequently, etravirine (TMC125) and rilpivirine were FDA approved in 2008 and 2011 (Fig. 1). A key issue with the development of NNRTIs has always been their efficacy against wild-type HIV-1 and variant forms that are induced by drug treatment.23,24 Common clinically-observed mutations to HIV-RT that can cause resistance to NNRTIs include L100I, K101P, K103N, V106M, Y181C, Y188L, G190S, and M230L, with K103N, V106M and Y181C being the most common and the double variant K103N/Y181C being especially challenging. The first-generation NNRTIs, nevirapine and delavirdine, are not effective against viral strains containing these mutations, while efavirenz retains potency against the Y181C variant, but it is ineffective against virus bearing K103N or V106M mutations. The third-generation compounds etravirine and rilpivirine show significantly improved resistance profiles, though resistance occurs for some currently less common mutations such as K101P, E138K, and Y181V.24 Today, efavirenz and rilpivirine are the most used in front-line therapies as they are the NNRTI component of the triple-combination medications Atripla and Complera, which also contain the NRTIs tenofovir and emtricitabine.22
Figure 1.

Structures of FDA-approved NNRTIs.
In view of the importance of resistance, we tested the utility of MC/LR and MC/FEP calculations in estimating the changes in activity from L100I, V106A, and Y181C mutations for several NNRTIs including two TIBOs, nevirapine, emivirine, and efavirenz.20,25,26 The FEP calculations are performed for the given mutation with apo RT and in the presence of the inhibitor, the difference giving the predicted change in free energy of binding. At the time, a crystal structure had not yet been reported for the complex of HIV-RT with efavirenz. The good accord between the FEP results and experimental activity results (fold changes) provided support for our structural model of the complex that came from docking calculations, 25 and which was then fully confirmed by a subsequent crystal structure.27 A similar prediction for the structure of the etravirine/RT complex was based on computation of fold changes for etravirine compared to nevirapine and efavirenz,28 and again was eventually confirmed by crystallography.29 We were also able to use FEP calculations to sort out the significant discrepancies between two crystal structures that had been reported for the complex of efavirenz with K103N RT.30 Binding results for efavirenz versus two analogues were computed using both crystal structures. The results were in much better agreement with experimental data for one of the crystal structures than the other; the differences were attributed to the use of low-pH conditions for growth of the crystals that gave the inconsistent results.
Through these studies and work with other proteins, confidence had been gained that at least qualitatively correct predictions could be made on the effects of changes in protein residues or in inhibitors on free energies of binding using MC/FEP calculations. The stage was set for prospective studies in which FEP calculations could guide lead optimization to enhance the efficiency of drug discovery by focusing on synthesis and assaying of the most promising compounds. The utility of the FEP approach had not been established by 2004.31 If successful, it would also be an important statement on the practical value of the widespread activities in computer simulations of biomolecular systems over the prior twenty-five years. For NNRTIs, the goal was to discover new compounds with improved spectrum, safety margins, and properties, especially solubility.
3. Initial design of NNRTIs
Of course, it is not common for a computational group to take-on experimental studies. For lead optimization work under any circumstances, it is expected that a hundred or more compounds may need preparation for each chemical series that is investigated, and there is no practical way to do that except in one’s own laboratory. Fortunately, I had had constant exposure to and interest in organic synthesis starting in graduate school, so I felt comfortable overseeing that activity. My past experience was also helpful in giving me a good sense of the difficulty of synthesis of a proposed compound, which is an important design consideration. In addition, my colleague Andrew Hamilton kindly provided further guidance and initial lab space. The first synthetic postdoctoral fellow, Juliana Ruiz-Caro, was hired in 2004, followed by Vinay Thakur and Joseph Kim in 2005. Critically, an expert on HIV biology, Karen Anderson, in the Yale School of Medicine was willing to have the biological testing performed in her laboratory. The key assays in the field use human T-cells infected with live HIV-1 virus, which requires a BL3 facility; they yield an EC50 for cytopathic protection and a CC50 for the cytotoxicity towards the T-cells.32,33
In our work, the initial lead compounds are obtained by virtual screening (docking) or by de novo design.34 For the latter, starting around 2000, I wrote a program called BOMB (Biochemical and Organic Model Builder), which builds and scores libraries of ligands, starting from a small core like benzene that is placed in a binding site of a biomolecule.34 In a BOMB run, up to four substituents, linking groups, or ring systems from a library of more than 700 options can be added to the core. Our first NNRTIs arose from BOMB designs followed by FEP-guided optimization. The initial reports were made in a series of four communications in 2006.35–38 Based on analyses of the NNRTI binding site and the structures of known inhibitors, thousands of molecules were built in U-Het-NH-PhX and Het-NH-PhX-U motifs, where U is an unsaturated hydrophobic group and Het is an aromatic heterocycle. To steer away from compounds similar to etravirine, the focus became the latter motif. As illustrated in Figure 2 for a derivative where Het is pyrimidine and U is dimethylallyloxy, the design features a hydrogen bond with the carbonyl group of Lys101 and the anilinyl NH of the inhibitor, and placement of the U group in the π-box formed by Tyr181, Tyr181, Phe227, and Trp229. MC/FEP calculations were used to optimize the choices for Het and the substituents other than U on the phenyl ring. This led to focus on 2-thiazole, 2-pyrimidine, and 1,3,5-triazine for Het and a 4-chloro or 4-cyano substituent. For U, numerous viable options were indicated by BOMB of which 20 were eventually tested.36,37 Dimethylallyloxy (ODMA) emerged as best, followed by thiophenyl. When assayed, the parent thiazole 1 (X = H) and pyrimidine 2 (X = Y = H) were modest NNRTIs with EC50 values towards wild-type HIV-1 of 10 and 30 μM; however, addition of X = Cl brought the EC50s down to 0.3 and 0.2 μM, respectively (Table 1). Further headway was made, as predicted, with the cyano analogue 1 (X = CN) with an EC50 of 0.2 μM, but with an unexpected drop in the CC50 from 26 μMto 0.5 μM. The pyrimidine analogue 2 (X = CN, Y = H) showed an even more dramatic increase in potency at 17 nM (0.017 μM); however, it was remarkably cytotoxic with a CC50 of 36 nM.
Figure 2.

Illustration from the crystal structure of the NNRTI 2 (X = CN, Y = OMe) bound to HIV- RT (PDB ID: 4KO0). Carbon atoms of the inhibitor are in yellow; the hydrogen bond with Lys101 is dashed; the side chain of Lys101 has been removed for clarity.
Table 1.
Anti-HIV activity (EC50) and cytotoxicity (CC50) of thiazoles and azines
| Compd | X | Y | EC50 (μM) | CC50 (μM) |
|---|---|---|---|---|
| 1 | H | — | 10 | 23 |
| 1 | Cl | — | 0.30 | 26 |
| 1 | CN | — | 0.21 | 0.5 |
| 2 | H | H | 30 | >100 |
| 2 | Cl | H | 0.20 | 2.5 |
| 2 | CN | H | 0.017 | 0.036 |
| 2 | Cl | OMe | 0.010 | 9.0 |
| 2 | Cl | SMe | 0.018 | 2.8 |
| 2 | CN | OMe | 0.002 | 0.230 |
| 3 | CN | OMe | 0.011 | 23 |
| 3 | CN | SMe | 0.005 | 8.4 |
| 4 | Cl | — | 0.005 | 17 |
| 5 | Cl | — | 0.130 | 17 |
| 6 | Cl | — | 0.019 | 20 |
| 7 | Cl | — | 0.900 | 9.2 |
| 8 | CH3 | — | NA | 61 |
| 9 | Cl | — | 0.820 | 20 |
| 10 | Cl | — | 0.310 | >100 |
| 11 | CN | — | 0.013 | 7.4 |
| Nevirapine | 0.11 | >100 | ||
| Efavirenz | 0.002 | 15 | ||
| Etravirine | 0.001 | 11 | ||
| Rilpivirine | 0.00067 | 8 |
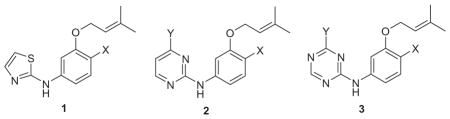
MC/FEP calculations were also used to select a substituent Y for the pyrimidine ring in 2 and to establish its favored orientation as either pointing in towards Phe227 or flipped to point outwards towards Lys103 (Fig. 2). Inward was strongly favored and the compounds with Y = methyl, ethyl, methoxy, and thiomethyl were all predicted and found to be much more potent than the Y = H reference. With X = Cl, the Y = methoxy and thiomethyl compounds were the most potent with EC50 values of 10 and 18 nM and with CC50 values of 9 and 3 μM. To see if the addition of the methoxy group might reduce the cytotoxicity for the X = CN cases, several analogues were prepared and 2 (X = CN, Y = OMe) did show an EC50 of 2 nM and CC50 of 0.23 μM. This compound, JLJ135, was the 135th compound that had been synthesized. The 2-nM potency was a substantial success for the design strategy, particularly since many of the first 80 or so compounds showed little or no activity, and compound JLJ047 (1 (X = Cl)) was the first one with an EC50 below 1 μM. In retrospect, many of the early compounds were simply too small, and quite a bit of effort had been spent with alternatives for the ODMA group. For example, the analogues of 1 (X = Cl) with the ODMA replaced by 3-thienylmethoxy or 1-cyclopentenyl-methoxy groups gave EC50 values of 5–6 μM.36 However, the cytotoxicity of 0.23 μM for JLJ135 remained unacceptable.
As an alternative, the synthesis of triazenes 3 was pursued as little change in activity was predicted by MC/FEP calculations in comparison to the pyrimidines 2. Fortunately, it turned out that the triazenes were much less cytotoxic such that X = CN analogues of 3 with Y = OMe and SMe had EC50 values of 11 and 5 nM and CC50 values of 42 and 8 μM (Table 1).37 There is no rationale as there is no understanding of the mechanisms of cytotoxicity for these or any other NNRTIs that we have worked with. This is a good illustration of the unexpected downs and ups that are regularly encountered during lead optimization, and why anyone engaged in it needs to be prepared to synthesize hundreds of compounds.
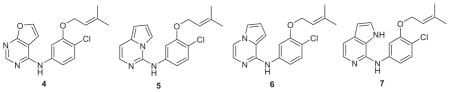
At this point, given the potency of the Y = OMe compounds and, though we did not have a crystal structure of a complex for any of our compounds with RT, the expectation that the methoxy group was oriented as in Figure 2, it seemed possible to cyclize the substituent into the pyrimidine ring and consider 6:5-bicyclic analogues like 4–7. The potential advantages were increased activity and novelty. In all, eight alternative bicyclic heterocycles were considered and MC/FEP calculations were carried out to rank them. Seven were then synthesized and the observed activities were in almost the exact order as the predicted ones. 4 and 7 were predicted and found to be the most and least active (Table 1), and the correct order was also predicted for the interesting isomeric pair 5 and 6.38 With an EC50 of 5 nM, 4 was our most potent NNRTI that did not contain a cyano group. This example well illustrates the power of the FEP approach as the ordering of the activities, even though the inhibitors are isosteric, was far from obvious owing to uncertain differences in electrostatic interactions and desolvation. Thus, in 2006 the joint efforts at Yale had yielded several NNRTIs including the particularly novel 4 that have similar activities and cytotoxicities as efavirenz and etravirine, and more than 20-fold greater potency than nevirapine (Table 1).
In collaboration with the group of Eddy Arnold at Rutgers, efforts were also underway to obtain crystals structures for some of the more potent compounds in complex with HIV-RT. This did result in a 1.95-Å structure for JLJ135 (2 (X = CN, Y = OMe)) in mid-2006, which was too late to include in the initial publications.35–38 The structure (Fig. 1) was eventually published in 2013 in the context of seeking more soluble NNRTIs, as discussed below.39 The crystal structure confirmed the correctness of the modeling, which had also been supported by the structure–activity data and numerous correct predictions during the evolution of 1–7. Another crystal structure for any complexes with our compounds was not obtained until 2012, so key design decisions were all being made based on computational modeling.
4. A docking adventure
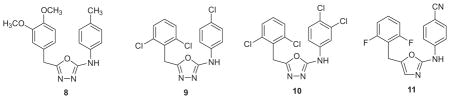
New lead series were also pursued with a combined similarity search and docking effort that used Glide 3.5 and the Maybridge collection of ca. 70,000 compounds.40 The Maybridge compounds were augmented with 26 known NNRTIs including several of ours, and the top 100 compounds from the docking were post-scored with an MM-GB/SA method. The procedure was applied to both a wild-type crystal structure and a K103N variant. Nine or ten of the known NNRTIs scored in the top-10 ranked compounds for both cases. Sixteen of the top ranking library compounds were then purchased and assayed, but none were found to be active in the MT-2 cell assay. Our expectation was that there were probably several near-misses associated with the compounds being a little too big or having a misplaced substituent on an otherwise viable core. The prior work had clearly demonstrated the sensitivity of activity to structure for NNRTIs, e.g., the 30–150 fold effect of just adding a chlorine to the parent thiazole 1 or pyrimidine 2 (Table 1).
This notion was pursued by examining the best-ranked library compound S10087 (8).41,42 The compound fits the U-Het-NH-PhX motif, though the 5-membered central ring was unprecedented for NNRTIs. The substituent pattern was analyzed by removing the methyl and methoxy groups of 8 and running a ‘chlorine scan’ with MC/FEP calculations, in which the impact of adding a chlorine to each position on the core is evaluated. This predicted that the best sites for substitution were the 3- and 4-positions in the anilinyl ring and at the 2- and 6-positions of the benzyl ring. Several compounds with one or two chlorines were synthesized, but were not active. However, with three (9) or four chlorines (10) active compounds were finally obtained with EC50 values of 0.82 and 0.31 μM (Table 1).41
The core structure then received an exhaustive FEP analysis for the substituents, 5-ring heterocycle, and benzyl linker.42 Cyano was still favored in the 4-position of the anilinyl ring, while CH2, CHCH3, and NCH3 were predicted to be viable for the linker, and 2,5-oxazole was the only heterocycle preferable to 2,5-oxadiazole. These predictions were confirmed by synthesis and assaying of multiple analogues, and the oxazole 11 emerged as the most potent with an EC50 of 13 nM (Table 1).42 Evolution of the false-positive 8 from docking to highly active 11 including the testing of alternative linkers and heterocycles required the synthesis of ca. 20 compounds. This provides a good illustration of the efficiencies that are possible with the FEP guidance and of how close an inactive library compound can be to a potent inhibitor.
5. Trouble with Y181C
So far, only wild-type (WT) activities had been assessed. At the beginning of 2008, the assays were extended to include viral strains incorporating the important Y181C and Y181C/K103N mutations. Results for our best compounds such as the triazene 3 (X = CN, Y = OMe) and 11 were disappointing (Table 2). The triazene yielded an EC50 of 12.5 μM for the Y181C variant and was inactive towards the double mutant. The oxazole 11 showed no activity towards either strain, though it should be noted that an EC50 cannot be obtained above the CC50, so a low CC50 can mask any anti-viral activity. The results seemed surprising since it was thought that smaller, flexible inhibitors should perform better towards the variants than larger more rigid molecules like nevirapine and TIBOs.26,43 Both the analogue of 3 and oxazole 11 are smaller than etravirine and have a similar number of rotatable bonds, but etravirine maintains strong activity against the variant viral strains.26 Further analysis suggested that the problem is more associated with greater contact and contribution to binding for our compounds with Tyr181. Thus, modifications were sought that would diminish the contact with Tyr181 and improve contacts elsewhere to maintain low-nM activities.
Table 2.
Anti-HIV activity (EC50) and cytotoxicity (CC50) in μMa
| Compd | WT EC50 | Y181C EC50 | K103N/Y181C EC50 | CC50 |
|---|---|---|---|---|
| 3b | 0.011 | 12.5 | NA | 23 |
| 11 | 0.013 | NA | NA | 7.4 |
| 12 | 0.006 | 0.42 | NA | 11 |
| 13 | 0.011 | NA | NA | 2.2 |
| 14 | 0.031 | 3.2 | 4.5 | 16 |
| 15 | 0.001 | 0.0069 | 0.21 | 4.7 |
| 16 | 2.5 | ND | ND | 38 |
| 17 | 0.048 | 0.25 | ND | 1.2 |
| 18 | 0.0017 | 0.015 | NA | 1.8 |
| 19 | 4.8 | ND | ND | 72 |
| 20 | 0.017 | 0.24 | 0.57 | 21 |
| 21 | 0.00032 | 0.016 | 0.085 | 45 |
| 25 | 0.010 | NA | 0.80 | 1.2 |
| 26 | 0.019 | 1.9 | 0.26 | >100 |
| 27 | 0.00038 | 0.31 | 0.011 | >100 |
| 28 | 0.0011 | 0.008 | 0.006 | >100 |
| 29 | 0.00052 | 0.0071 | 0.032 | 16 |
| 30 | 0.0011 | 0.0013 | 0.007 | 9.5 |
| 33 | 0.0012 | 0.012 | 0.0013 | 4.5 |
| 34 | 0.00019 | 0.350 | 0.070 | 10 |
| Nevirapine | 0.11 | NA | NA | >100 |
| Efavirenz | 0.002 | 0.010 | 0.030 | 15 |
| Etravirine | 0.001 | 0.008 | 0.005 | 11 |
| Rilpivirine | 0.00067 | 0.00065 | 0.002 | 8 |
NA = not active. ND = not determined.
X = CN, Y = OMe analogue.
One idea was to extend the inhibitors to the lower-right in Figure 2 into a channel between Phe227, Val106, and Pro236. For the azoles, replacement of the cyano group in 11 was sought with various linker-heterocycle combinations as ‘eastern extensions’ that were built with BOMB.44 The intention was to improve the baseline activity such that mutations could be better tolerated, i.e., the rising tide floats all boats approach. First, addition of a chlorine adjacent to the cyano group in 11 to give 12 did boost the wild-type activity to 6 nM and yield a measurable activity of 0.42 μM towards the Y181C variant. Then, linkers with 0–3 heavy atoms, mostly terminated in pyridines, were considered. The best compounds had methoxy or oxy linkers as in 13 and 14. Though these embodied novel replacements for a cyano group, they did not improve upon the activities of 12 (Table 2).44 Though our focus turned to other series, the oxazoles were revisited three years later, after it was decided to explore additions to the 4-position of the benzyl ring.45 Such modification had previously been avoided because there did not appear to be room for a group much larger than methyl without steric clashes for the WT protein. However, a very revealing set of FEP calculations considered eleven replacements for the 4-hydrogen in both the WT and Y181C variants.45 The predictions were that the WT potency would be optimal with ethyl, and isopropyl would be optimal for the Y181C form. Ten analogues were synthesized, and remarkably ethyl (15) did turn out to be optimal for the WT strain (1.3 nM), while isopropyl was 5 nM, and ethyl and isopropyl tied at 7 nM for the Y181C variant. The EC50 values also improved for the double variant to 210 nM for 15 and 120 nM for the isopropyl analogue. The relatively large improvement in going from hydrogen (12) to ethyl (15) or isopropyl for the Y181C strain was expected from better filling of the space vacated by the change from tyrosine to cysteine (Fig. 3). However further size increases were detrimental for addressing the WT virus such as for cyclopropylmethyl (68 nM) or sec-butyl (120 nM).45 Thus, with a careful size balance, it was possible to obtain sub-10 nM potency towards both the WT and Y181C variants with 15 and the isopropyl analogue. The advancement from the inactive 8 is notable.
Figure 3.

Snapshot of 15 bound to Y181C HIV-RT from an MC/FEP simulation. Some residues and all water molecules have been deleted for clarity. Carbon atoms of the inhibitor are in yellow.
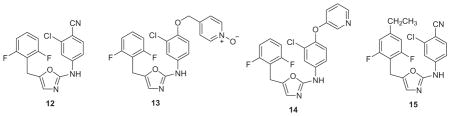
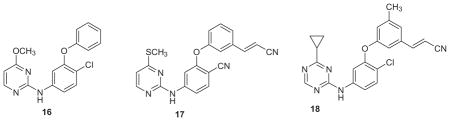
Returning to the azine series 2 and 3, an idea was to diminish the contact of the ODMA group with Tyr181 and extend the compounds towards the back in Fig. 2 in the channel below Trp229. Modeling indicated that replacement of the ODMA group by phenoxy or thiophenyl would decrease the interaction with Tyr181 and place more emphasis on aryl-aryl interaction with Tyr188. Though the net result was a large loss in potency in the WT assay for 16 (EC50 = 2.5 μM) versus 2 (X = Cl, Y = OMe) at 10 nM,37 perhaps judicious substitution of the phenoxy group could compensate. Chlorine and methyl scans with MC/FEP calculations revealed that substitution at the 3- and 5- positions of the phenoxy ring were much favored and that several conformers could be populated that would direct a thin substituent such as chloro, cyano, or cyanovinyl either between Tyr181 and Tyr188 or towards the channel below Trp229 (Fig. 4).46 Such compounds were synthesized including 17 (EC50 = 48 nM), and eventually the triazine 18 (1.7 nM).46 FEP calculations made another important contribution in predicting that a cyclopropyl, isopropyl, or thiomethyl substituent in the triazine or pyrimidine ring would provide significant activity boosts, which indeed turned out to be five-fold over methoxy. Furthermore, as expected, the elaborated compounds did gain significant activity towards the Y181C–containing viral strain. For example, 18 provided an EC50 of 15 nM (Table 2), though it was not active against the double variant. This study again had some toxicity twists. For example, the analogue of 18 with the methyl group replaced by chlorine has great potency with EC50 values of 2.5 and 4.9 nM towards the WT and Y181C variants; however, its CC50 is 73 nM. In addition, thiophenyl analogues were intrinsically more active than their phenoxy counterparts; however, they were not pursued owing to their ca. 10-fold greater cytotoxicity.
Figure 4.

Computed structure for an analogue of 17 bound to WT HIV-RT in the conformation with the cyanovinyl group pointing into the channel below Trp229.
6. Catechol diethers
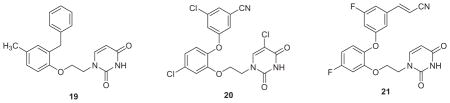
In parallel with the lead optimization work, new core structures were being sought through a more elaborate docking effort that emphasized potential Y181C activity. This time consensus hits were sought by docking the ZINC library of more than two million compounds using a conventional WT crystal structure, one with an alternative uncommon “down” structure for Tyr181, and one for the Y181C variant.47 Though only nine compounds were purchased, three were found to show low-micromolar activity towards the WT virus, the Y181C variant, or both. All three have been pursued, though the predominant focus was placed on hit 19, which had 4.8 μM potency towards the WT virus and a CC50 of 72 μM. Its predicted solubility with QikProp was also good,48 the structure including the uracilylethoxy substituent was novel, and it appeared to fit the Tyr181-down structure well. The apo protein has both Tyr181 and Tyr188 down, collapsed into the space where the inhibitors reside in Figs. 2 and 4. So, the thoughts were that the reorganization penalty for inhibitor binding might be less with Tyr181 down and that interactions between inhibitors and Tyr181 might be diminished along with impact of the Y181C mutation.
By this point, our approach should be evident. Substitution patterns, linkers, and the heterocycle are going to be scrutinized by MC/FEP calculations. All possible placements of one or two chlorines on the terminal phenyl ring were considered first. The prediction was that 2,4-, 2,5-, 2,6-, and 3,5-disubstitution were all viable. This immediately paid off as the four corresponding dichlorides of the analogue of 19 with the methyl group replaced by chlorine were synthesized and yielded WT activities of 2.9, 0.38, 0.31, and 1.3 μM.49 The methylene and ethoxy linkers were then analyzed for placement of oxygen atoms at any position. The FEP result was that the gain should be striking for conversion of the methylene linker to an oxygen atom, yielding what can be referred to as a catechol diether. This appeared to have a large conformational component as diphenyl ethers prefer a perpendicular conformation that is ideal for the desired complexation (Fig. 5), while diphenylmethanes prefer an open clam-shell geometry.49 The first compound of this type that was synthesized had aWT EC50 of 0.14 μM, and quick progress was made to compounds such as 20, which has a WT potency of 17 nM and shows mid-nanomolar activity for both mutant viral strains (Table 2). Synthesis of a few more analogues led to some excellent potencies, for example, the difluoro compound 21 (JLJ506) has EC50 values of 0.32, 16, and 85 nM towards the WT, Y181C, and K103 N/Y181C variants. Its dichloro analogue (JLJ494) has a stunning EC50 of 0.055 nM (55 picomolar) towards wild-type virus, and it is likely the most potent known anti-HIV agent in the standard T-cell assay. Alternatives for the uracilylethoxy substructure were also sought, but none have yet to emerge as superior.
Figure 5.

Computed structure for JLJ494, the dichloro analogue of 21, bound to WT HIV-RT. Note the expected ‘down’ orientation of Tyr181 and hydrogen bonds between the uracilyl OCNH fragment and the backbone NH and CO of Lys103.
The progress was made without the benefit of a crystal structure for any of our compounds since JLJ135 bound to RT (Fig. 2). However, the good accord between the FEP predictions and structure–activity data provided confidence in our modeled structures (Fig. 5). The situation finally changed when Kathleen Frey joined the Anderson laboratory in 2012 and was rapidly able to obtain 2.9-Å crystal structures for both JLJ494 and JLJ506 bound to WT RT (PDB IDs: 4H4M and 4H4O).50 The findings were in essentially exact accord with what had been published a year earlier (Fig. 5) including the down-orientation of Tyr181, the hydrogen bonding with Lys103, the conformation of the ethoxy linker, and the positioning of the cyanovinylphenyl group.49 The modeling also raised the possibility that the extreme potency of JLJ494 might benefit from a halogen bond between the chlorine on the terminal ring and the oxygen atom of Pro95. Though the chlorine fills the space well, the crystallography showed that the interaction with an O-Cl separation of 4.72 Å is ca. 1 Å longer than optimal. Additional crystal structures for analogues of 21 were subsequently reported along with detailed analyses of the origin of the effects on activity for variation of the two halogens.51
7. Replacement of the cyanovinyl group
Medicinal chemists have become sensitized to avoid what are perceived as undesirable features in potential drug molecules. The features are associated with false positives in assays, toxicities, and unwanted covalent modification of proteins or nucleic acids.52,53 Though exceptions to the rules are common,54 a conservative approach is wise when one considers the costs of human clinical trials. In viewing rilpivirine (Fig. 1), 18, or 21, medicinal chemists are immediately drawn to the cyanovinyl group as a potential liability through action as a Michael acceptor leading to off-target covalent modifications. Though rilpivirine is an FDA-approved drug, search of a database of ~ 1900 approved drugs only found four other examples with a cyanovinyl substructure.55 While we were pursuing replacements for the cyanovinyl group in the catechol diethers, confirmation of the concern came from the failure of the NNRTI fosdevirine (GSK-2248761, 22) in phase IIb clinical trials.56 After four weeks of treatment, 5 of 20 subjects experienced seizures. Two prominent metabolites were found to arise from cysteine addition of glutathione to the vinyl group of 22, and one was implicated as the source of the neurotoxicity.56
Our idea was to replace the cyanovinylphenyl group in the catechol diethers with a 6:5 bicyclic heterocycle as in 23 or 24. For 23, R would likely be cyano, while in 24 the cyano group is replaced by an azole nitrogen. From MC simulations of complexes as in Fig. 5, it was apparent that there was a water molecule hydrogen bonded to the nitrile nitrogen atom and perhaps the water molecule could drift farther into the channel and hydrogen-bond with the azole nitrogen atom in 24. The big problem was to decide among the numerous possibilities for W, X, Y, and Z in 23 and 24. Synthesis of many alternatives would be challenging since the specific substitution patterns for the bicyclic heterocycles in 23 and 24 were required. It was also very unclear which heterocycles would have the most favorable interactions with Trp229 (Fig. 5) and if competitive activity with the cyanovinyl-containing compounds like 21 could be achieved.
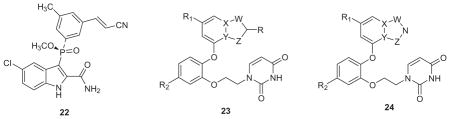
Of course, this was a perfect opportunity for a heterocycle scan as done for 4–7.38 So, MC/FEP calculations were carried out for 18 alternatives of 23 and 24 with R1 = H and R2 = Cl. The resultant rankings strongly favored 23 with the indole, benzofuran, and indolizine options that are reflected in 25–27. All three structure types were synthesized and gave potent NNRTIs (Table 2).55 The indolizines were the most potent including 27 and its R1 = R2 = F analogue, which both have a WT EC50 of 0.4 nM. 27 (JLJ555) also showed no cytotoxicity and strong activity towards the K101N/Y181C variant (EC50 = 11 nM); however, it was oddly less potent towards the Y181C strain (EC50 = 310 nM), though this is reconsidered below. In addition, examples of the isomeric indoles, benzofurans, and indolizines were synthesized. As predicted by the FEP calculations, they were less active than the illustrated isomers. A 2.9-Å crystal structure was also reported for 27 bound to WT RT (PDB ID: 4MFB).55
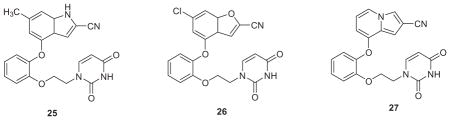
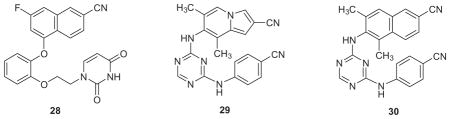
With recognition of the viability of replacing the cyanovinylphenyl group with a cyano-substituted bicyclic heterocycle, two additional investigations were carried out. First, structure building with BOMB indicated that a 6-cyanonaphthyl group might also fit in place of the bicyclic heterocycle in the catechol diethers. This led to the synthesis of thirteen examples including 28.57 The results were gratifying as several of the analogues showed sub-20 nM activity against all three viral strains. For example, the EC50 values for 28 are 1.1, 8.0, and 6.0 nM towards the WT, Y181C, and K103N/Y181C strains (Table 2), while the results for the desfluoro analogue are 0.53, 19, and 15 nM; both compounds also have CC50s >100 μM.57 Secondly, our cyanovinylphenyl mimics were then merged with the diaminopyrimidine substructure of rilpivirine or the corresponding triazine to give 22 hybrid NNRTIs including 29 and 30.58 The activities of these compounds are excellent, and as also intended, there was a dramatic boost in the aqueous solubilities (vide infra). Crystal structures were also reported for desfluoro-28, 29, and 30 bound to WT HIV-RT.57,58
8. Aqueous solubility
In view of the hydrophobic nature of the NNRTI binding site (Fig. 1), it is not surprising that poor aqueous solubility has been a common feature for NNRTIs. Poor solubility is often associated with irregularities in assays, aggregation phenomena, low bioavailability, and difficulties in formulation.48,59 The problem is well known for the TMC compounds etravirine, rilpivirine, and their predecessor dapivirine (TMC120), which is identical to rilpivirine with replacement of the cyanovinyl group by a methyl group. Oral drugs typically have aqueous solubilities S of 10 μM–10 mM, corresponding to 4–4000 μg/mL for a drug with a molecular weight of 400 using the common solubility units.48 However, the three TMC compounds have aqueous solubilities well below 1 μg/mL, which has led to formulation difficulties, and evaluation of dapivirine as a topical microbicide.61,62
In order to improve the aqueous solubility of a compound, analogues can be explored that decrease the stability of the crystalline state (lower the melting point and heat of sublimation) and/or make the free energy of hydration more favorable. The common choices are to increase polarity, increase torsional flexibility, and decrease planarity.63 Thus, a common strategy is to introduce flexible side chains with polar functionality such as polyethers and often successful solubilizing groups such as morpholine.64 However, there are no guarantees because the addition of polar functional groups may also enhance polar intermolecular interactions, especially hydrogen-bonding in the crystals. Thus, there is still considerable empiricism in searching for solubility-enhancing modifications that do not significantly diminish the biological activity of the compounds. In this regard, the best choice is to attach the solubilizing substituent at a site in an inhibitor that is known to be solvent exposed in the protein-inhibitor complex. It is also possible that the situation cannot be rectified and the only option is to seek a less problematic chemical series.
As a challenge, we decided to attempt to improve the solubility of the TMC compounds and our azines 2 and 3 by making attachments that would probe a largely unexplored region of the NNRTI binding site, namely the ‘entrance channel’, which is in front of Lys103 in Figure 2. Based on our experience, modifications elsewhere would be likely to have more negative effects on the antiviral activity. Thus, a variety of attachments to C6 of the azine ring were modeled such as the morpholinylethoxy appendage in 31 and ca. 25 compounds were synthesized.39,65 It was found that many groups could be tolerated, though with some loss in potency. For example, the WT EC50 for the mono-methoxy 3 (X = CN, Y = OMe) is 11 nM, while it is 22 nM for the 4,6-dimethoxy analogue, and it is 92 nM for 31. A 2.9-Å crystal structure was also obtained for 31 complexed with WT RT.39 In Figure 6, the morpholinyl group is seen to emerge into a broad opening on the surface of RT in the vicinity of Glu28B and Lys32B. This indicates that larger terminal attachments are possible. Indeed, dimeric NNRTIs including 32 were prepared and found to show significant activity, similar to that of nevirapine, e.g., the WT EC50 for 32 is 170 nM.65 Though such constructs may seem somewhat whimsical, one could envision chimeric inhibitors in an NNRTI1-linker-NNRTI2 format, where the two NNRTIs have different resistance profiles. More elaborate possibilities could have several different inhibitors attached to a core, like a key ring with keys for different locks. Bifunctional inhibitors NNRTI-linker-Inh, where Inh is a member of a different class of anti-HIV agent, have also been explored.66,67
Figure 6.

Rendering from the crystal structure of 31 bound to WT HIV-1 RT (PDB ID: 4KKO). The morpholinylethoxy side chain is seen to emerge into a large opening on the protein’s surface. No residues have been omitted.
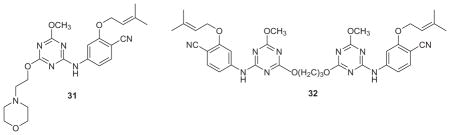
Aqueous solubilities are measured in our laboratory with a well-established shake-flask procedure at a pH of 6.5.68 3 (X = CN, Y = OMe) is indeed poorly soluble like the TMC compounds with S = 0.1 μg/mL; however, addition of the morpholinylethoxy group has a profound effect yielding a viable solubility of 42.2 μg/mL for 31, and the propoxy homolog has S = 52.3 μg/mL.39 For comparison, the solubilities of dapivirine, efavirenz, and nevirapine are 0.15, 68.0, and 167 μg/mL, as summarized in Table 3.
Table 3.
Aqueous solubility at pH 6.5 (S, μg/mL)
The strategy was then applied to the TMC scaffold with good success.69 The best NNRTI that emerged from this study was 33, which may be viewed as the triazine relative of rilpivirine with the added morpholinylpropoxy solubilizing group. This compound retains excellent potency with EC50 values of 1.2, 12, and 1.3 nM towards the WT virus and the Y181C and K103N/Y181C strains (Table 2) and its solubility at 14.2 μg/mL shows a 700-fold improvement over rilpivirine (Table 3). The analogue with the cyanovinyl group replaced by a methyl group has similar solubility (15.3 μg/mL); the crystal structure for its complex with WT RT was subsequently determined and showed it was shifted 3-Å deeper into the NNRTI binding site than its analogue lacking the morpholinylpropoxy group.70 The 2,6-difluoro-4-methyl analogue 34 is also interesting and more soluble at 22.9 μg/mL; it is extraordinarily potent towards the WT virus with an EC50 of 0.190 nM (190 picomolar), while it shows 70 nM potency towards the double variant, but it oddly, like 33, is significantly less potent at 350 nM towards the Y181C variant. Typically, the activity of an NNRTI is less towards the K103N/Y181C strain than it is towards either constituent single variant. For example, dapivirine is reported to have EC50 values of 1.2, 7, and 54 nM in MT-4 cell assays with WT, Y181C, and K103N/Y181C virus,60 while our values are 0.7, 39, and 39 nM with MT-2 cells.69 In this and other instances, it seems that our Y181C–containing strain is particularly challenging.
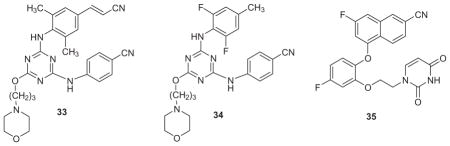
In addition, the measured solubilities of other key compounds are provided in Table 3. The catechol diethers (21, 27, 28) and modified azines (29, 30) generally have aqueous solubilities of 10–40 μg/mL, which is a large improvement over the TMC compounds and falls into the normal oral-drug range.48 Some unusual cases have been observed. For example, the parent 1-naphthyl catechol diether, desfluoro-28, has a solubility of 4.3 μg/mL, the monofluoro 28 is 9.1 μg/mL, and then there is a significant boost to 82.9 μg/mL for adding the second fluorine in 35. Normally replacing an aromatic hydrogen by fluorine increases the octanol water partition coefficient, log Po/w, and decreases aqueous solubility. 48 The exceptions most likely reflect structural subtleties in the crystalline state.
9. Final results and summary
There has also been external testing of some of our compounds. Several catechol diethers were tested in single-round infectivity assays using CD4+ T cells from blood donors in the laboratory of Robert Siliciano at Johns-Hopkins University School of Medicine. After addition of the test compounds and infection with viral strains, the cells are incubated for three days at 37 °C, and then the extent of infectivity is quantified by flow cytometry.72 The evaluations were carried out with several HIV-1 strains including WT and ones bearing the Y181C, K103N, and K101P mutations in RT. We were particularly interested in the latter mutation as multiple variations of Lys101 (K101E, K101H, K101P) are known to cause resistance to all FDA-approved NNRTIs.23,24 With continued use of the combination therapies, Atripla and Complera, increased prominence of K101 variants can be expected.
Results are summarized in Table 4 for the difluorocyanovinyl-containing 21, the indolizine 27, efavirenz (efv) and rilpivirine (rpv).72 The results are clearly striking, especially for 27, which shows the greatest potency among the four NNRTIs for all viral strains, no cytotoxicity towards T-cells, and good solubility. Both 21 and 27 show excellent activity towards the WT and Y181C—containing strains with no loss of potency for the K101P variant. Efavirenz and rilpivirine are not effective against the K101P strain, showing fold changes of 58 and 88, and as usual, efavirenz is not effective against K103N variants. Crystal structures were also compared for efavirenz, rilpivirine, and 27 with WT RT, and a new structure for 27 with K101P RT was reported.72 Since the catechol diethers do not have the hydrogen bond with the backbone carbonyl of Lys101 that is characteristic of almost all NNRTIs including 2 (Fig. 2), efavirenz, and the TMC compounds, the catechol diethers are positioned farther back in the NNRTI binding site and do not contact residue 101. Consequently, they are not affected by changes to Lys101, and the binding sites for the complexes of 27 with WT and K101P RT superimpose with an rmsd of only 0.4 Å.72 Conversely, NNRTIs that hydrogen bond with Lys101 are sensitive to changes at that position; K101P can be expected to be particularly damaging since it changes the backbone conformation and it also removes the NH that is generally forming a second hydrogen bond with the NNRTI, e.g., with the carbonyl oxygen of efavirenz and a pyrimidine nitrogen of rilpivirine. Further antiviral testing has been carried out for some of the catechol diethers on E138K-containing strains in the MT-2 cell assays. The performance of 27 remains superb with EC50 values of 0.9 and 0.75 nM for HIV-1 strains containing E138 K and E138K/M184V.72 Thus, disruption of the commonly observed Lys101-Glu138 salt bridge also does not affect the catechol diethers.
Table 4.
Results of single-round infectivity assays (nM), CC50, and aqueous solubility
| efv | rpv | 21 | 27 | |
|---|---|---|---|---|
| WT EC50 (nM) | 15 | 13 | 2 | 1 |
| Y181C EC50 (nM) | 41 | 51 | 8 | 2 |
| K103N EC50 (nM) | 806 | 13 | 89 | 3 |
| K101P EC50 (nM) | 870 | 1142 | 2 | 1 |
| CC50 (μM) | 15 | 8 | 45 | >100 |
| Solubility (μg/mL) | 68 | 0.02 | 10.8 | 37.9 |
Additional testing has included typical preclinical studies for off-target activity carried out by Eurofins Panlabs using their HitProfiling+CYP450 Screen. The compounds are tested at a fixed concentration of 10 μM for inhibition of five cytochrome P450s and thirty ion channels, receptors, and transporters including hERG. In these assays, 21 gives no inhibition values above 50% and 27 gives only one for CYP 2C19. It is highly unusual to not have several hits in these screens. We also had efavirenz tested; it yields three hits, a calcium channel, a sodium channel, and serotonin receptor 5-HT2B. Rilpivirine was not tested since it is not soluble at 10 μM.
In summary, our efforts to contribute to the improvement of HIV/AIDS therapies have come a long way since our initial modeling of effects of mutations in RT on binding to our discovery and optimization of anti-HIV agents yielding the results in Tables 2–4. Along the way numerous challenges of general importance for SBDD have been addressed including what to do when virtual screening fails, the utility of FEP calculations for guiding lead optimization, how to obtain activity towards multiple targets (variants), replacements of undesirable functional groups with heterocyclic alternatives, and ways to improve solubility. The work has required a close-knit team effort for computation, synthesis, biological assaying, and protein crystallography. Though the details of the synthetic chemistry can be found in the cited publications, it should be recognized that synthesis is the most labor intensive aspect of SBDD work, and those who carry it out deserve special recognition. They have remarkable perseverance that enables them to try one more route to overcome the regular failure of well-planned reaction schemes.
Acknowledgments
Gratitude is expressed to the National Institute of General Medical Sciences (GM032136) and the National Institute of Allergy and Infectious Diseases (AI44616) for providing the funds that have enabled the computational and synthetic work in the Jorgensen laboratory. Deep gratitude is also expressed to the numerous co-workers who have contributed to the anti-HIV projects in the Jorgensen and Anderson laboratories. Special thanks go to Prof. Karen S. Anderson who has been the perfect colleague and whose great skills with the biological assaying and protein crystallography have been essential to the success of the described work. My long-time associate Dr. Julian Tirado-Rives is also given sincerest thanks for being key to our computational efforts and advances.
References and notes
- 1.Reynolds C, de Konig CB, Pelly SC, van Otterlo WAL, Bode ML. Chem Soc Rev. 2012;41:4657. doi: 10.1039/c2cs35058k. [DOI] [PubMed] [Google Scholar]
- 2.Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, Herber WK, Sigal IS, Darke PL, Springer JP. Nature. 1989;337:615. doi: 10.1038/337615a0. [DOI] [PubMed] [Google Scholar]
- 3.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Science. 1992;256:1783. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 4.Smerdon SJ, Jäger J, Wang J, Kohlstaedt LA, Chirino AJ, Friedman JM, Rice PA, Steitz TA. Proc Natl Acad Sci USA. 1994;91:3911. doi: 10.1073/pnas.91.9.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vacca JP, Condra JH. Drug Discovery Today. 1997;2:261. [Google Scholar]
- 6.Gohlke H, Klebe G. Angew Chem, Int Ed. 2002;41:2644. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 7.Lin TS, Schinazi RF, Pusoff WH. Biochem Pharmacol. 1987;36:2713. doi: 10.1016/0006-2952(87)90253-x. [DOI] [PubMed] [Google Scholar]
- 8.Jorgensen WL, Tirado-Rives J. J Am Chem Soc. 1988;110:1657. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 9.Jorgensen WL, Tirado-Rives J. Chem Scripta. 1989;29A:191. [Google Scholar]
- 10.Jorgensen WL. Acc Chem Res. 1989;22:184. [Google Scholar]
- 11.Jorgensen WL, Ravimohan C. J Chem Phys. 1985;83:3050. [Google Scholar]
- 12.For a review, see: Jorgensen WL, Thomas LT. J Chem Theory Comput. 2008;4:869. doi: 10.1021/ct800011m.
- 13.Åqvist J, Medina C, Sammuelsson JE. Protein Eng. 1994;7:385. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 14.Lamb ML, Jorgensen WL. Curr Opin Chem Biol. 1997;1:449. doi: 10.1016/s1367-5931(97)80038-5. [DOI] [PubMed] [Google Scholar]
- 15.Jorgensen WL, Tirado-Rives J. J Comput Chem. 2005;26:1689. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 16.Tirado-Rives J, Jorgensen WL. J Comput Chem. 1996;17:1385. doi: 10.1002/(SICI)1096-987X(199608)17:11<1385::AID-JCC11>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 17.Pierce AC, Jorgensen WL. Angew Chem, Int Ed. 1997;36:1466. [Google Scholar]
- 18.Essex JW, Severance DL, Tirado-Rives J, Jorgensen WL. J Phys Chem. 1997;101:9663. [Google Scholar]
- 19.Smith RH, Jr, Jorgensen WL, Tirado-Rives J, Lamb ML, Janssen PAJ, Michejda CJ, Kroeger Smith MB. J Med Chem. 1998;41:5272. doi: 10.1021/jm9804174. [DOI] [PubMed] [Google Scholar]
- 20.Kroeger Smith MB, Lamb ML, Tirado-Rives J, Jorgensen WL, Michejda CJ, Ruby SK, Smith RH., Jr Protein Eng. 2000;13:413. doi: 10.1093/protein/13.6.413. [DOI] [PubMed] [Google Scholar]
- 21.Pauwels R, Andries K, Desmyter J, Schols D, Kukla MJ, Breslin HJ, Raeymaeckers A, Van Gelder J, Woestenborghs R, Heykants J, Schellekens K, Janssen MAC, De Clercq E, Janssen PAJ. Nature. 1990;343:470. doi: 10.1038/343470a0. [DOI] [PubMed] [Google Scholar]
- 22.De Clercq E. Adv Pharmacol. 2013;67:317. doi: 10.1016/B978-0-12-405880-4.00009-3. [DOI] [PubMed] [Google Scholar]
- 23.Iyidogan P, Anderson KS. Viruses. 2014;6:4095. doi: 10.3390/v6104095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basson AE, Rhee SY, Parry CM, El-Khatib Z, Charalambous S, De Oliveira T, Pillay D, Hoffmann C, Katzenstein D, Shafer RW, Morris L. Antimicrob Agents Chemother. 2015;59:960. doi: 10.1128/AAC.04215-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rizzo RC, Wang DP, Tirado-Rives J, Jorgensen WL. J Am Chem Soc. 2000;122:12898. [Google Scholar]
- 26.Wang DP, Rizzo RC, Tirado-Rives J, Jorgensen WL. Bioorg Med Chem Lett. 2001;11:2799. doi: 10.1016/s0960-894x(01)00510-8. [DOI] [PubMed] [Google Scholar]
- 27.Ren J, Milton J, Weaver KL, short SA, Stuart DI, Stammers DK. Struct Fold Des. 2000;8:1089. doi: 10.1016/s0969-2126(00)00513-x. [DOI] [PubMed] [Google Scholar]
- 28.Blagović MU, Tirado-Rives J, Jorgensen WL. J Am Chem Soc. 2003;125:6016. doi: 10.1021/ja034308c. [DOI] [PubMed] [Google Scholar]
- 29.Kertesz DJ, Brotherton-Pleiss C, Yang M, Wang Z, Lin X, Qiu Z, Hirschfeld DR, Gleason S, Mirzadegan T, Dunten PW, Harris SF, Villasenor AG, Hang JQ, Heilek GM, Klumpp K. Bioorg Med Chem Lett. 2010;20:4215. doi: 10.1016/j.bmcl.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 30.Blagović MU, Tirado-Rives J, Jorgensen WL. J Med Chem. 2004;47:2389. doi: 10.1021/jm0303507. [DOI] [PubMed] [Google Scholar]
- 31.Jorgensen WL. Science. 2004;303:1813. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 32.Lin TS, Luo MZ, Liu MC, Pai SB, Dutschman GE, Cheng YC. Biochem Pharmacol. 1994;47:171. doi: 10.1016/0006-2952(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 33.Ray AS, Yang Z, Chu CK, Anderson KS. Antimicrob Agents Chemother. 2002;46:887. doi: 10.1128/AAC.46.3.887-891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jorgensen WL. Acc Chem Res. 2009;42:724. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jorgensen WL, Ruiz-Caro J, Tirado-Rives J, Basavapathruni A, Anderson KS, Hamilton AD. Bioorg Med Chem Lett. 2006;16:663. doi: 10.1016/j.bmcl.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 36.Ruiz-Caro J, Basavapathruni A, Kim JT, Wang L, Bailey CM, Anderson KS, Hamilton AD, Jorgensen WL. Bioorg Med Chem Lett. 2006;16:668. doi: 10.1016/j.bmcl.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 37.Thakur V, Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2006;16:5664. doi: 10.1016/j.bmcl.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 38.Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. J Am Chem Soc. 2006;128:15372. doi: 10.1021/ja066472g. [DOI] [PubMed] [Google Scholar]
- 39.Bollini M, Frey KM, Cisneros JA, Spasov KA, Das K, Bauman JD, Arnold E, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2013;23:5209. doi: 10.1016/j.bmcl.2013.06.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barreiro G, Guimaraes CRW, Tubert-Brohman I, Lyons TM, Tirado-Rives J, Jorgensen WL. J Chem Inf Model. 2007;47:2416. doi: 10.1021/ci700271z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barreiro G, Kim JT, Guimaraes CRW, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. J Med Chem. 2007;50:5324. doi: 10.1021/jm070683u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeevaart JG, Wang L, Thakur VV, Leung CS, Tirado-Rives J, Bailey CM, Domaoal RA, Anderson KS, Jorgensen WL. J Am Chem Soc. 2008;130:9492. doi: 10.1021/ja8019214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das K, et al. J Med Chem. 2004;47:2550. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 44.Leung CS, Zeevaart JG, Domaoal RA, Bollini M, Thakur VV, Spasov K, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2010;20:2485. doi: 10.1016/j.bmcl.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bollini M, Gallardo-Macias R, Spasov KA, Tirado-Rives J, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2013;23:1110. doi: 10.1016/j.bmcl.2012.11.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jorgensen WL, Bollini M, Thakur VV, Domaoal RA, Spasov KA, Anderson KA. J Am Chem Soc. 2011;133:15686. doi: 10.1021/ja2058583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nichols SE, Domaoal RA, Thakur VV, Bailey CM, Wang L, Tirado-Rives J, Anderson KS, Jorgensen WL. J Chem Inf Model. 2009;49:1272. doi: 10.1021/ci900068k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jorgensen WL, Duffy EM. Adv Drug Deliv Rev. 2002;54:355. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 49.Bollini M, Domaoal RA, Thakur VV, Gallardo-Macias R, Spasov KA, Anderson KS, Jorgensen WL. J Med Chem. 2011;54:8582. doi: 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frey KM, Bollini M, Mislak AC, Cisneros JA, Gallardo-Macias R, Jorgensen WL, Anderson KS. J Am Chem Soc. 2012;134:19501. doi: 10.1021/ja3092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frey KM, Gray WT, Spasov KA, Bollini M, Gallardo-Macias R, Jorgensen WL, Anderson KS. Chem Biol Drug Des. 2014;83:541. doi: 10.1111/cbdd.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rishton GM. Drug Discovery Today. 2003;8:86. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- 53.Baell JB, Holloway GA. J Med Chem. 2010;53:2719. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 54.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Chem Res Toxicol. 2011;24:1345. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 55.Lee WG, Gallardo-Macias R, Frey KM, Spasov KA, Bollini M, Anderson KS, Jorgensen WL. J Am Chem Soc. 2013;135:16705. doi: 10.1021/ja408917n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Castellino S, Groseclose MR, Sigafoos J, Wagner D, de Serres M, Polli JW, Romach E, Myer J, Hamilton B. Chem Res Toxicol. 2013;26:241. doi: 10.1021/tx3004196. [DOI] [PubMed] [Google Scholar]
- 57.Lee WG, Frey KM, Gallardo-Macias R, Spasov KA, Bollini M, Anderson KS, Jorgensen WL. ACS Med Chem Lett. 2014;5:1259. doi: 10.1021/ml5003713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee WG, Frey KM, Gallardo-Macias R, Spasov KA, Chan AH, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2015;25:4824. doi: 10.1016/j.bmcl.2015.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 60.Janssen PAJ, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Béthune M-P, Pauwels R, Das K, Clark AD, Jr, Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. J Med Chem. 2005;48:1901. doi: 10.1021/jm049534r. [DOI] [PubMed] [Google Scholar]
- 61.Weuts I, Van Dycke F, Voorspoels J, de Cort S, Stokbroekx S, Leemans R, Brewster ME, Xu D, Segmuller B, Turner YTA, Roberts CJ, Davies MC, Qi S, Craig DQM, Reading M. J Pharm Sci. 2011;100:260. doi: 10.1002/jps.22242. [DOI] [PubMed] [Google Scholar]
- 62.Baert L, et al. Eur J Pharma Biopharma. 2009;72:502. doi: 10.1016/j.ejpb.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 63.Ishikawa M, Hashimoto Y. J Med Chem. 2011;54:1539. doi: 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- 64.Roughley SD, Jordan AM. J Med Chem. 2011;54:3451. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- 65.Ekkati AR, Bollini M, Domaoal RA, Spasov KA, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2012;22:1565. doi: 10.1016/j.bmcl.2011.12.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bailey C, Sullivan T, Iyidogan P, Tirado-Rives J, Chung R, Ruiz-Caro J, Mohamed E, Jorgensen WL, Hunter R, Anderson KA. J Med Chem. 2013;56:3959. doi: 10.1021/jm400160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhan P, Liu X. Curr Med Chem. 2013;20:1743. doi: 10.2174/0929867311320130011. [DOI] [PubMed] [Google Scholar]
- 68.Baka E, Comer JEA, Takács-Novák K. J Pharm Biomed Anal. 2008;46:335. doi: 10.1016/j.jpba.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 69.Bollini M, Cisneros JA, Spasov KA, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2013;23:5213. doi: 10.1016/j.bmcl.2013.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mislak AC, Frey KM, Bollini M, Jorgensen WL, Anderson KA. Biochim Biophys Acta. 2014;1840:2203. doi: 10.1016/j.bbagen.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morelock MM, Choi LL, Bell GL, Wright JL. J Pharm Sci. 1994;83:948. doi: 10.1002/jps.2600830706. [DOI] [PubMed] [Google Scholar]
- 72.Gray WT, Frey KM, Laskey SB, Mislak AC, Spasov KA, Lee WG, Bollini M, Siliciano RF, Jorgensen WL, Anderson KS. ACS Med Chem Lett. 2015;6:1075. doi: 10.1021/acsmedchemlett.5b00254. [DOI] [PMC free article] [PubMed] [Google Scholar]