

Abstract
RATIONALE
The metabolite profiling of a NIST plasma Standard Reference Material (SRM 1950) on different LC-MS platforms showed significant differences. Although these findings suggest caution when interpreting metabolomics results, the degree of overlap of both profiles allowed us to use tandem mass spectral libraries of recurrent spectra to evaluate to what extent these results are transferable across platforms and to develop cross-platform chemical signatures.
METHODS
Non-targeted global metabolite profiles of SRM 1950 were obtained on different LC-MS platforms using reversed phase chromatography and different chromatographic scales (nano, conventional and UHPLC). The data processing and the metabolite differential analysis were carried out using publically available (XCMS), proprietary (Mass Profiler Professional) and in-house software (NIST pipeline).
RESULTS
Repeatability and intermediate precision showed that the non-targeted SRM 1950 profiling was highly reproducible when working on the same platform (RSD < 2%); however, substantial differences were found in the LC-MS patterns originating on different platforms or even using different chromatographic scales (conventional HPLC, UHPLC and nanoLC) on the same platform. A substantial degree of overlap (common molecular features) was also found. A procedure to generate consistent chemical signatures using tandem mass spectral libraries of recurrent spectra is proposed.
CONLUSIONS
Different platforms rendered significantly different metabolite profiles, but the results were highly reproducible when working within one platform. Tandem mass spectral libraries of recurrent spectra are proposed to evaluate the degree of transferability of chemical signatures generated on different platforms. Chemical signatures based on our procedure are most likely cross-platform transferable.
Keywords: Human Plasma Standard Reference Material, Non-targeted Metabolomics, Global Metabolite Profiling, Liquid Chromatography-Electrospray Mass Spectrometry, Chemical Signatures
INTRODUCTION
Metabolomics has emerged as a field of great significance for biomedical research. It is generally recognized that DNA sequencing or protein profiling are not sufficient for diagnostic or therapeutic purposes[1]. In contrast, metabolic profiles reflect both endogenous and exogenous influences and can provide a functional readout of the cellular state. These profiles are easier to correlate with phenotype than genomic or proteomic information because metabolites are not subject to epigenetic regulation or post-translational modifications as are genes and proteins, respectively.
Metabolomics experiments can either be targeted or non-targeted. Targeted profiling entails searching for known chemical compounds. In non-targeted profiling, the goal is to identify as many metabolites as possible in each measurement. The majority of metabolomic applications in the literature are targeted consequently there exists a wealth of information on analyzing different classes of metabolites. Non-targeted profiling is a more recent development[1, 2]. Liquid chromatography mass spectrometry (LC-MS) is widely used because of its high sensitivity and large dynamic range, which are necessary to cover analyte concentrations of nine orders of magnitude or more[3]. Furthermore, LC-MS is useful when the metabolites are non-volatile or thermally unstable[4] giving the method a good combination of “versatility and robustness”[5] even when working with different platforms[6].
Non-targeted profiling has been performed on a variety of human biological fluids including plasma/serum, urine and cerebrospinal fluid. The global profiling of small molecules of the human plasma metabolome has been discussed in several publications[3, 7, 8]. Plasma has been profiled for the discovery of biomarkers in cancers. A few examples include hepatocellular carcinoma[9], pancreatic[10], epithelial ovarian[11], prostate[12], and lung[13] cancers. In these types of experiments metabolites specific to the disease of interest are sought by comparison of results between control and case samples. The analytical variability is a serious impediment to this determination.
Several papers have focused on the biological variability in metabolomics experiments. Cai et al. found that the highly polar metabolites in human plasma could be used to discriminate samples by sex and age[14]. Lawton et al. found that metabolites that display differences in age, sex and race can be found in human plasma. The most differences were associated with age, followed by sex and then race[8]. Studies of analytical variability are scarce and mostly focused on differences resulting from different sample preparation methods[15–17]. However, in the course of expanding the NIST Standard Reference Material and Data (SRM/D) online database project[18], significant differences in the chromatographic patterns of a human plasma Standard Reference Material (SRM 1950) were found on different LC-MS platforms (where platform refers to a particular LC coupled to a specific mass spectrometer), even when following the same sample preparation protocol. To date, several studies on SRM 1950 have been completed at NIST, NIH and other laboratories using GC-MS, LC-MS, NMR and clinical tests[18–25]. It is worth mentioning that comparisons of the performance of different LC-MS platforms for the field of proteomics have been previously reported[26].
In this work, we analyzed SRM 1950 across several LC-MS platforms - following the same sample preparation procedures. These included different liquid chromatography separations (conventional HPLC, UHPLC and nanoLC) and different mass spectrometers (Q-TOF and Orbitrap). XCMS [27–29], the NIST pipeline (see supplemental info and http://peptide.nist.gov) and Agilent Mass Profiler Professional (MPP) were used as pattern recognition tools to find and report differences in the chromatographic patterns. The NIST pipeline has proven very effective for providing performance metrics for LC-MS in proteomic analyses[30]. Recently it has been modified to work with small molecules other than peptides. This paper represents a continuation of previous work[18] and our objectives are to 1) report the variability of plasma analysis on different LC-MS platforms and compare the chromatographic patterns, 2) establish a precision limit where possible and 3) provide a workflow and tools for systematically analyzing variability in metabolomics studies and bringing consistency to cross-platform analysis with the use of tandem mass spectral libraries of recurrent spectra.
Three aspects distinguish this work from prior comparison studies in the metabolomics field. The first is the use of a Standard Reference Material to facilitate the study of variability arising from experimental conditions excluding the potential for biological variability. The reference material used is publically available enabling others to reproduce our results. The second distinguishing aspect is the use of in-house developed software in conjunction with publically available software to provide LC-MS performance metrics in metabolomics analyses. The third aspect is the fact that most comparisons are based on tandem mass spectral library metabolite identifications versus unidentified extracted molecular features. By working only with recurrent features (identified by their MS/MS spectra) we ensure that we always extract the same feature from run to run or batch to batch.
The Results and Discussion section covers four main themes. First, we discuss the procedure to generate material-oriented tandem mass spectral libraries, which are necessary to follow the methodology we are proposing for cross-platform comparisons. These spectral libraries include all recurrent spectra of identified and unidentified ions in a particular material. Next we discuss the differences observed when comparing different chromatographic separations (conventional HPLC, UHPLC and nanoLC). Following that, we analyze differences when using different mass spectrometers. Finally, we give a brief account of complicating factors that can account for the observed differences.
EXPERIMENTAL METHODS1
Reagents
All reagents were HPLC grade. Water and acetonitrile were manufactured by J.T. Baker and obtained from VWR (Radnor, PA, USA). All remaining reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Sample Preparation
SRM 1950 was handled at Biosafety Level (BSL) 2 as recommended in the Certificate of Analysis. The material was disinfected with ethanol which also resulted in protein precipitation. Ethanol is a potent bactericidal and virucidal agent at concentrations of 60%–80%[31]. An ethanol concentration of 70% (v/v) for 30 min[32] at 0 °C was utilized. Following disinfection, the material was considered safe for handling at BSL 1. Briefly, SRM 1950 was allowed to thaw at room temperature. Ice-cold ethanol (200 proof) was added to a concentration of 70% (v/v). The samples were vortexed for 15 s then allowed to sit on ice for 30 min. The percent of ethanol was then adjusted to 80% (v/v) and samples were vortexed and stored at −20 °C overnight. The next day, the samples were centrifuged at 4 °C for 15 min at 14,000×g and the supernatant was evaporated to dryness in a speedvac (Labconco, Kansas City, MO, USA). The metabolites were reconstituted in 1% (v/v) acetonitrile with 0.1% (v/v) formic acid and centrifuged at 4 °C for 15 min at 14,000 × g to remove any insoluble material prior to analysis. The supernatant, excluding a yellow hydrophobic material on the top, was stored at −20 °C until analysis.
LC-MS/MS Analysis
The metabolites were separated by three LC methods, namely using conventional HPLC, UHPLC and nanoLC. Each type of separation was coupled to either a 6530 Accurate-Mass Q-TOF mass spectrometer (Agilent Technologies, Inc., Santa Clara, CA) with a Jet Stream ESI interface or an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Waltham, MA) giving six possible combinations. The data was collected in positive ion mode with data-dependent MS/MS acquisition, exept for the semi-quantitative study which was acquired as MS only. The details of the MS settings for the Q-TOF instrument can be found in the supporting information, Table SI_1, while those for the Orbitrap instrument can be found in Table SI_2.
Q-TOF Analysis
Conventional HPLC and UHPLC separations were done on an Agilent 1290 Infinity HPLC while nanoLC separation used an Agilent 1200 Series HPLC with a nano and cap flow splitter. Two different conventional HPLC columns were used with similar results. These columns were a Waters SunFire C18 (1.0 × 150 mm, 100 Å, 3.5 µm) and an Agilent ZORBAX Eclipse Plus RR C18 (2.1 × 150 mm, 95 Å, 3.5 µm). The UHPLC column was a ZORBAX Eclipse Plus RRHD C18 (2.1 × 150 mm, 95 Å, 1.8 µm) and was used with a guard column. For the nanoLC, a Polaris-HR-Chip 3C18 (150 mm, 180 Å, 3 µm C18 chip with 360 nL trap column) was used with the HPLC-Chip Cube MS Interface. HPLC mobile phases A and B consisted of 0.1% (v/v) formic acid in water and acetonitrile, respectively. The HPLC flow rates were 75 µL/min and 210 µL/min for the SunFire and ZORBAX Eclipse Plus columns, respectively. Approximately 565 µg or 1150 µg of metabolites were injected for the SunFire column and ZORBAX Eclipse Plus columns, respectively. The gradient (Gradient 1) in (v/v %) started at 1% B, increased linearly to 20% B in 30 min, to 90% B in 10 min, was maintained at 90% B for 5 min, returned to 1% B in 2 min and then was maintained at 1% B for 20 min. Approximately 7–8 µg of metabolites was analyzed for nanoLC, and Gradient 1 was used with a flow rate of 2.5 µL/min and 0.4 µL/min for the trap and analytical columns, respectively. The flow rate and gradient used for UHPLC was scaled to the ZORBAX Eclipse Plus RR column to maintain the same separation using the HPLC Calculator Version 3.0[33, 34] and the same amount of sample was analyzed. The UHPLC flow rate was 408 µL/min and the gradient (Gradient 2) started at 1% B, increased linearly to 20% B in 15.43 min, to 90% B in 5.14 min, was maintained at 90% B for 2.57 min, returned to 1% B in 1.03 min and then was maintained at 1% B for 10.29 min. Blanks using the same method were run before the first and between each sample injection. In addition, for nanoLC, the trap and analytical columns were washed separately at 90% B for one hour after each sample run.
Orbitrap Analysis
The same columns and HPLC system as above for the HPLC separation were used with Gradient 1 for the analysis. Approximately 1100 µg were injected, depending on the run. For the nanoLC separation, the HPLC system was an Eksigent 2D Classic nanoLC. Gradient 1 was used at a flow rate of 300 nL/min. The column was an Agilent ZORBAX 300StableBond RR C18 (75 µm × 150 mm, 300 Å, 3.5 µm). Approximately 221 ng of metabolites were analyzed. Blanks using the same method were run before the first and between each sample injection.
Data Analysis
All data were analyzed using XCMS and the NIST pipeline. The data from each platform was searched against a metabolite library with the NIST MSPepSearch software[35–37] to provide tandem mass spectral library identifications. All hits were manually curated because of the many complicating factors (see discussion) with library identifications. The identifications were manually checked and a quality index with four levels (E: excellent, G: good, A: acceptable and P: poor) was developed. Poor matches and duplicated identifications were removed. The entire Q-TOF data set was also processed using Agilent’s Mass Profiler Professional Software.
RESULTS AND DISCUSSION
Below we analyze the relative performance of individual mass spectrometers and different chromatographic separations and make comparisons between them using open, proprietary and in-house software. It is worth remarking that the current study purposely uses the same standard reference material and LC instrument on two different mass spectrometers. Therefore, some differences were expected and were considered only due to differences in the platform performances regarding background ions, artifacts, ionization efficiency, data processing, etc.
Material-oriented Tandem Mass Spectral Libraries
In order to generate chemical signatures that could be validated across different separations (and also across different LC-MS platforms), we have added another step to the workflow. We have built tandem mass spectral libraries of all recurrent spectra[38]for the materials of interest, so it makes it possible to identify common molecular features in different runs through library match. To build the tandem mass spectral libraries, in-house software and data from more than 300 runs of human plasma and urine standard reference materials on different platforms were used. In most LC-MS/MS runs, between two and three thousand tandem mass spectra were acquired, with at least three replicate runs of each sample. NIST-developed software was used along with available commercial software to align chromatograms, derive chromatographic peak shapes, intensities and retention times and to find and cluster recurrent spectra. High quality spectra of these recurrent, but unidentified, species were used to build tandem MS libraries for each material. These libraries can be accessed by the library search software as an independent library or as part of the NIST MS/MS library through a labeling system (plasma, urine, etc.). NIST spectral data uses an index file structure (NIST#, CAS, etc.) to specify certain attributes of the data; the access structure is expressed with these attributes and logical operations over the attributes. 695 components of the plasma samples have been identified previously using the NIST tandem mass spectral library (version 11)[18]. To build the libraries of recurrent spectra, spectra were first extracted from original data files by the NIST pipeline using settings designed to minimize spurious spectra, followed by searching the NIST tandem mass library with all spectra. The spectra that could not be identified were then filtered to remove poorly deconvoluted data and clustered. The results were assumed to be unidentified components. It was required each unidentified spectrum to be found in multiple replicates. Over the past two years the NIST Mass Spectrometry Data Center has built several of these material-oriented libraries for common materials, such as human plasma and urine, cell extracts, etc., for both LC-MS and GC-MS applications. At this point, the human serum tandem mass library of recurrent spectra contains 14,481 high quality spectra derived from the analysis of SRM 1950. Spectra have been recorded on different mass spectrometers (Waters QQQ, Agilent Q-TOF and Thermo Orbitrap) and different chromatographic scales (conventional HPLC, UHPLC and nanoLC). All spectra in the library were labeled as identified, recurrent, contaminants, or artifacts, so we can assign a prior probability to each spectrum (compound) regarding its potential biological origin.
Chromatography Differences between conventional HPLC, UHPLC and nanoLC
Data from different chromatographic separations on the Agilent HPLC/Q-TOF system (conventional HPLC, UHPLC and nanoLC), consisting of five technical replicates and three different batches, was analyzed using XCMS, the NIST pipeline and MPP. On average, the numbers of extracted molecular features using XCMS were 6455, 4844, and 1012, respectively. 4259 common features were found between conventional and UHPLC analysis, and only 424 between nanoLC and conventional (336 between nanoLC and UHPLC). These numbers do not show much variation across batches and regularly the same molecular features were consistently found across batches. It is worth mentioning that an optimized set of parameters and a deisotoping algorithm were used for the molecular feature extraction. The repeatability and intermediate precision of the conventional and UHPLC analyses, based on the variations of the relative abundances of the molecular features, were acceptable to excellent (RSD < 2%), as demonstrated in Figure 1. It shows three (not aligned or normalized) chromatograms from the conventional HPLC/Q-TOF analysis of SRM 1950. For the sake of simplicity, it shows only 50 extracted molecular features confidently identified with tandem mass spectral library searching for three (out of five) replicates.
Figure 1.
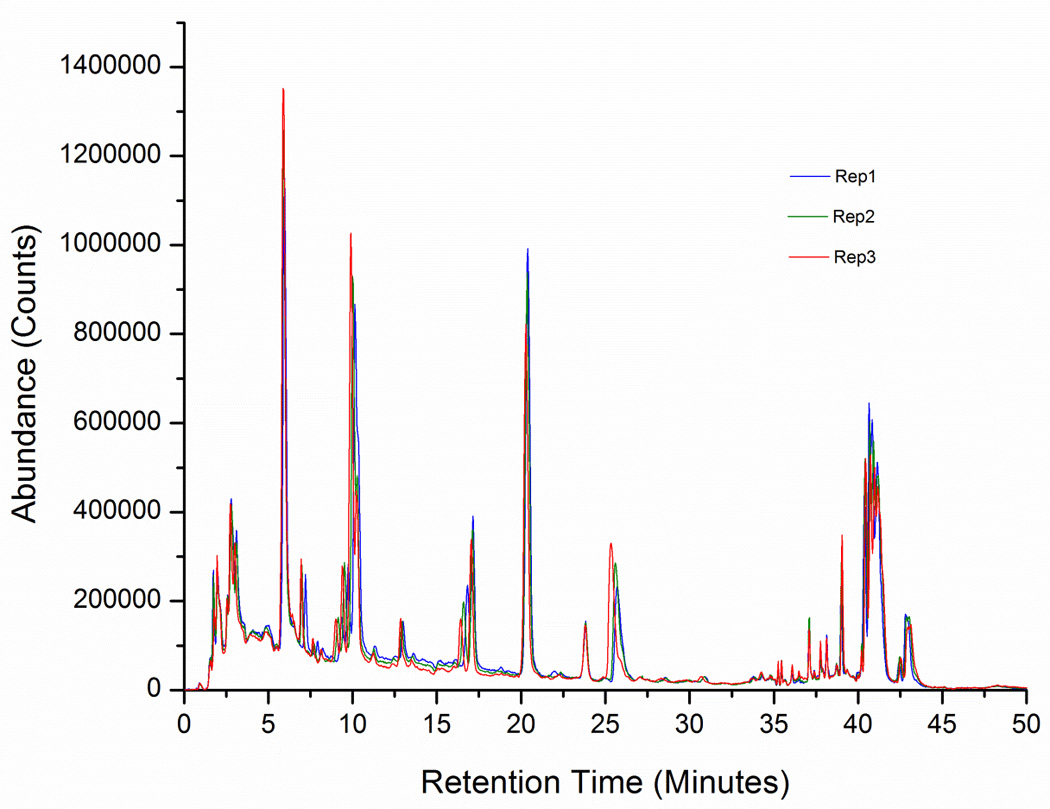
The degree of overlap between the conventional and UHPLC analyses was relatively high. Surprisingly, the number of extracted molecular features using the optimized XCMS settings was higher for the conventional analysis despite the fact that the UHPLC chromatography performance (discussed later on) was actually better than the conventional analysis. It might be thought that this is a consequence of the conventional analysis being less robust than the UHPLC analysis, so many of the additional molecular features found in the conventional analysis are borderline background or low intensity features. However, this is not always the case and we will see later that a significant number of identified features are unique to each separation. It raises some concerns about the indiscriminate use of feature finding algorithms in untargeted, unsupervised approaches, to generate chemical signatures correlated to a biological or other effect based only on retention time alignment and accurate mass. In addition, when comparing separations at different scales (e.g., conventional vs. UHPLC) there is no trivial rescaling factor for the LC retention time[39].
When the plasma tandem mass spectral library of recurrent spectra was used to compare the conventional and the UHPLC analyses on the Q-TOF mass spectrometer, the number of common features was significantly reduced from 4259 to 637 with the library match step (see Figure 2a). However, a significant portion of this reduction could be due to the fact that many common extracted features were not found in the library, either because the spectra were not of high enough quality or because they were not sampled for MS/MS. Currently, new experiments targeting these compounds are in progress.
Figure 2.
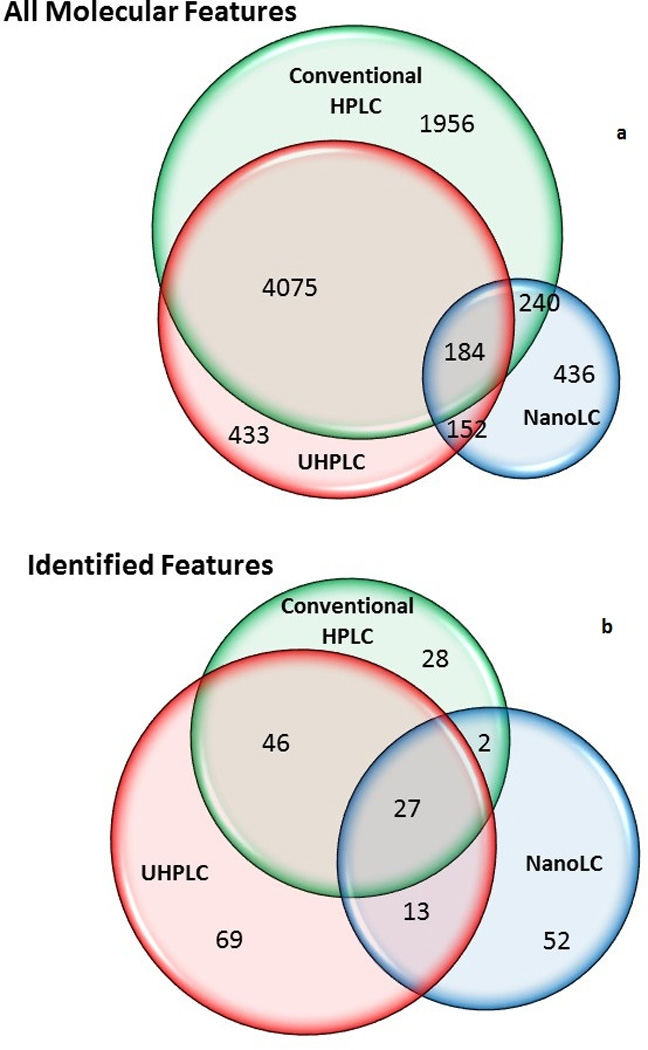
The overlap is further reduced if only identified features are included in the analysis. Specifically, this means molecular features that found a match in the NIST tandem mass spectral library of pure compounds (not the library of recurrent spectra). For example, a total of 237 different compounds were identified from a single replicate of each type of analysis using the Q-TOF mass spectrometer, but results differ significantly across different chromatographic separations (conventional HPLC, UHPLC or nanoLC). Table I shows the chemical name, Human Metabolome Database (HMDB) index, parent ion formula, relative abundance (as percent of the most abundant peak in each chromatogram) and the percent of the corresponding relative standard deviation (RSD) for the 50 extracted molecular features shown in Figure 1. The ion formulas (MS1) were automatically generated based on how well they match the experimental mass, abundance pattern of the isotopic cluster, and m/z spacing between the monoisotopic ion and the A+1 and A+2 ions. The library precursor ion m/z values are given at the level of precision of the matching library spectrum. The NIST tandem mass library currently contains more than 10,000 compounds of biological and environmental relevance (including metabolites, bioactive peptides, amino acids and small peptides, sugars and glycans, lipids and phospholipids, drugs, pesticides, surfactants, and various contaminants).
Table I.
Reproducibility (Relative Standard Deviation, RSD) of the relative ion abundances (RA) of the 50 SRM 1950 metabolites shown in the extracted chromatograms of figure 1 (conventional HPLC/Q-TOF platform).
| Metabolite | HMDB ID | Precursor Type |
Precursor Formula |
Precursor m/z |
RA (%) |
RSD (%) |
|---|---|---|---|---|---|---|
| Creatinine | HMDB00562 | [M+H]+ | C4H8N3O | 114.0662 | 0.33 | 3.31 |
| L-Valine | HMDB00883 | [M+H]+ | C5H12NO2 | 118.0863 | 0.18 | 1.23 |
| Isoleucine | HMDB00172 | [M+H]+ | C6H14NO2 | 132.1019 | 3.12 | 0.87 |
| Octopamine | HMDB04825 | [M+H-H2O]+ | C8H10NO | 136.0757 | 2.35 | 1.78 |
| Hypoxanthine | HMDB00157 | [M+H]+ | C5H5N4O | 137.0458 | 0.16 | 2.77 |
| 2-Amino-1-phenylethanol | HMDB01065 | [M+H]+ | C8H12NO | 138.0913 | 0.03 | 2.35 |
| 3-Phenyllactic acid | HMDB00748 | [M+H]+ | C9H9O2 | 149.0597 | 0.28 | 0.32 |
| Methionine | HMDB00696 | [M+H]+ | C5H12NO2S | 150.0583 | 0.22 | 0.56 |
| Gabapentin | HMDB05015 | [M+H-H2O]+ | C9H16NO | 154.1226 | 1.17 | 0.71 |
| Carnitine | HMDB14721 | [M+H]+ | C7H16NO3 | 162.1125 | 0.57 | 0.91 |
| Phenylalanine | HMDB00159 | [M+H]+ | C9H12NO2 | 166.0863 | 12.36 | 0.07 |
| Diphenylamine | HMDB32562 | [M+H]+ | C12H12N | 170.0964 | 0.52 | 1.64 |
| Arginine | HMDB00517 | [M+H]+ | C6H15N4O2 | 175.1190 | 0.82 | 0.35 |
| Paraxanthine | HMDB01860 | [M+H]+ | C7H9N4O2 | 181.0720 | 0.87 | 0.35 |
| Tyrosine | HMDB00158 | [M+H]+ | C9H12NO3 | 182.0817 | 1.45 | 1.83 |
| L-Kynurenine | HMDB00684 | [M+H-NH3]+ | C10H10NO3 | 192.0655 | 0.09 | 0.87 |
| Caffeine | HMDB01847 | [M+H]+ | C8H11N4O2 | 195.0877 | 3.00 | 1.29 |
| NG,NG-Dimethyl-L- arginine |
HMDB01539 | [M+H]+ | C8H19N4O2 | 203.1503 | 1.01 | 0.21 |
| Acetyl-carnitine | HMDB00201 | [M+H]+ | C9H18NO4 | 204.1400 | 2.07 | 0.60 |
| L-Tryptophan | HMDB30396 | [M+H]+ | C11H13N2O2 | 205.0791 | 8.44 | 0.29 |
| Indole-3-lactic acid | HMDB00671 | [M+H]+ | C11H12NO3 | 206.0812 | 1.19 | 0.25 |
| Dodecanedioic acid | HMDB00623 | [M+H-H2O]+ | C12H21O3 | 213.1491 | 1.16 | 1.13 |
| Propionylcarnitine | HMDB00824 | [M+H]+ | C10H20NO4 | 218.1387 | 0.69 | 4.24 |
| Gly-Phe | [M+H]+ | C11H15N2O3 | 223.1077 | 0.36 | 1.10 | |
| Ile-Pro | [M+H]+ | C11H21N2O3 | 229.1547 | 2.71 | 0.85 | |
| Pantothenic acid | HMDB00210 | [M+Na]+ | C9H17NO5Na | 242.0999 | 0.13 | 0.97 |
| Valerylcarnitine | HMDB13128 | [M+H]+ | C12H24NO4 | 246.1627 | 1.96 | 0.63 |
| Hydroxybupropion | HMDB12235 | [M+H]+ | C13H19ClNO2 | 256.1099 | 0.65 | 0.24 |
| Hexanoylcarnitine | HMDB00705 | [M+H]+ | C13H26NO4 | 260.1818 | 1.43 | 2.78 |
| Atenolol | HMDB01924 | [M+H]+ | C14H23N2O3 | 267.1767 | 1.65 | 0.15 |
| Heptanoylcarnitine | HMDB13238 | [M+H]+ | C14H28NO4 | 274.1978 | 0.18 | 0.23 |
| Glu-Met | [M+H]+ | C10H19N2O5S | 279.1009 | 0.36 | 1.32 | |
| Benzoylecgonine | HMDB41836 | [M+H]+ | C16H20NO4 | 290.1387 | 9.28 | 1.69 |
| D-Sphingosine | HMDB00252 | [M+H]+ | C18H38NO2 | 300.2897 | 1.64 | 0.18 |
| cis-5,8,11-Eicosatrienoic acid |
HMDB10378 | [M+H]+ | C20H35O2 | 307.3600 | 0.21 | 0.68 |
| α-L-Glu-L-Tyr | [M+H]+ | C14H19N2O6 | 311.1238 | 2.39 | 0.20 | |
| Ranitidine | HMDB01930 | [M+H]+ | C13H23N4O3S | 315.1485 | 5.19 | 0.26 |
| Trp-Glu | [M+H-H2O]+ | C16H18N3O4 | 316.1292 | 0.08 | 0.08 | |
| His-Trp | [M+H]+ | C17H20N5O3 | 342.1561 | 0.04 | 0.20 | |
| Hydrocortisone | HMDB14879 | [M+H]+ | C21H31O5 | 363.2166 | 4.18 | 0.21 |
| Cholesterol | HMDB06740 | [M+H-H2O]+ | C27H45 | 369.3566 | 0.78 | 1.56 |
| Trazodone | HMDB14794 | [M+H]+ | C19H23ClN5O | 372.1591 | 2.62 | 1.32 |
| Deoxycholic acid | HMDB00626 | [M+H]+ | C24H41O4 | 393.2999 | 1.59 | 0.42 |
| Cholic acid | HMDB00619 | [M+H]+ | C24H41O5 | 409.2949 | 1.76 | 0.99 |
| 1-Myristoyl-sn-glycero-3- phosphocholine |
HMDB10379 | [M+H]+ | C22H47NO7P | 468.3085 | 3.01 | 0.27 |
| Lyso-PC(16:0) | HMDB10382 | [M+H]+ | C24H51NO7P | 496.3398 | 6.51 | 1.80 |
| Lyso-PE(0:0/20:1) | HMDB11482 | [M+H]+ | C25H51NO7P | 508.3398 | 0.46 | 0.86 |
| 1-(9Z-octadecenoyl)-sn- glycero-3-phosphocholine |
HMDB02815 | [M+H]+ | C26H53NO7P | 522.3560 | 4.46 | 1.58 |
| 1-Octadecanoyl-sn- glycero-3-phosphocholine |
HMDB11128 | [M+H]+ | C26H55NO7P | 524.3711 | 1.53 | 1.46 |
| Lyso-PAF C-18 | [M+Na]+ | C26H56NO6PNa | 532.3737 | 1.53 | 1.94 |
The results indicated that repeatability and intermediate precision on the same instrument are excellent. The sample preparation and analysis procedures were highly repeatable as 90% of the peak intensities in Table I have a RSD < 2 % for the five replicates. (A few outliers have been removed from the calculation of the RSD using a modified Z-score[40]). Intermediate precision for three replicate experiments (sample prepared in three different batches on different days) was similar to results for repeatability with RSD < 2%.
Table I also shows that there was not always a strong correlation between higher abundance and lower RSD. Some abundant peaks such as those of phenylalanine and tryptophan have very small RSDs, but some lower abundance peaks such as methionine and kynurenine also have very small RSDs. This suggests that major differences in RSD are due to the ionization efficiencies of the small molecules and not to their relative abundances (see Table III and later discussion).
Table III.
Identifications belonging to different chemical classes on different LC-MS platforms.
| Chemical Classes | Number of Identifications (%) | ||||
|---|---|---|---|---|---|
| Q-TOF nano |
Q-TOF Conv |
Q-TOF UHPLC |
Orbitrap nano |
Orbitrap Conv |
|
| Amino Acids and Related Molecules | 20 (21%) |
27 (26) | 49 (32) | 29 (40) | 31 (36) |
| Carbohydrates | 4 (4) | 0 (0) | 1 (1) | 6 (8) | 2 (2) |
| Hormones and Steroids | 6 (6) | 7 (7) | 15 (10) | 2 (3) | 14 (16) |
| Lipids and Phospholipids | 6 (6) | 24 (23) | 18 (12) | 7 (10) | 16 (19) |
| Others | 58 (62) | 45(44) | 72 (46) | 28 (39) | 23 (27) |
| Total | 94 | 103 | 155 | 72 | 86 |
As mentioned above, while results from the same platform have excellent repeatability and intermediate precision, the analysis of the same samples with different chromatographic separations (conventional HPLC, UHPLC and nanoLC) also produced different results, which mirrors our findings above when processing the data including all molecular features (identified and identified). A Venn diagram of the metabolite identifications for the Q-TOF mass spectrometer with different chromatographic separations is shown in Figure 2 (details of the experiments and data processing are given in the Experimental Methods section and Supporting Information).
For the conventional HPLC, UHPLC, and nanoLC separations, 103, 155, and 94 metabolites were identified, respectively from a single experiment (the first replicate). Different LC-MS platforms produced large differences in the metabolite identifications. Only 27 metabolites were identified in all three separations for this single replicate. In part, these differences depend on the quality of the experimental spectra, the library searching algorithm performance, and the limited coverage of the library to identify all components in a complex mixture. Different compounds can be identified in replicate runs because of the numerous low abundance compounds that are present in the material or also because of missing data, which has been previously reported[41]. The comparison of replicates shows an overlap of about 85% of the identifications. The average number of IDs and their deviations for all replicates are not very representative of the overall variability because in different replicates some missing values are replaced by different identifications.
The degree of overlap between nanoLC and the two other chromatographic separations was very poor. NanoLC is not very popular in metabolomics applications, so regarding these separations we shall limit the discussion to those elements relevant to the cross-platform comparison. The differences between the nanoLC separation and the other two can be explained in part by the fact that a different packing material (see Experimental Section) was used for the separation. The packing material used for the HPLC and UHPLC columns was not available for the nano scale. In addition, the theoretical base of nanoLC is currently underdeveloped which makes the scaling up of the chromatographic processes difficult. In general, the chromatography at the nano scale showed poor separation at the beginning of the run (hydrophilic region) having very broad peaks. The high organic elution region also shows depleted signals compared to the conventional HPLC and UHPLC analyses. Phospholipids and probably some other large molecules are partially retained by the trap column of the nano chip. This is supported by the observation that initial data collected on a new column will show depletion and then eventually, a large mass of hydrophobic components will be eluted in a single injection. Although the chromatograms are also reproducible, the phospholipid retention can be a major problem affecting the retention of the small molecules in the long term. For this reason, in order to eliminate the retained material, a 90% B wash was run after each injection. It is not clear how these variations actually lead to different identifications, but most hydrophilic compounds elute earlier in nanoLC using the same gradient as the conventional HPLC (e.g., phenylalanine and tryptophan have roughly half of the retention times). This, in conjunction with the broad peaks and weak signal strengths, explain in part why fewer identifications are found using the nanoLC. More experiments are necessary to improve and validate the use of nanospray techniques for this kind of complex metabolite mixture analysis. Due to the small size of the columns, nanoLC requires cleaner samples previously subjected to solid phase extraction (SPE) or a different sample preparation protocol (not protein precipitation), things not performed in this work. However, in general, the observed peak capacities of the nano-columns in this work are smaller than the conventional HPLC and UHPLC columns. It is likely due to these reasons that, nanoLC separations are rarely used in metabolomics and would be recommended only in cases of limited sample amount and for relatively clean samples.
For conventional HPLC and UHPLC analyses the same packing material was used in a different particle size (3.5 µm and 1.8 µm, respectively) where the gradient and flow rate were scaled to maintain the separation performance, and the scan speed of the mass spectrometer was also scaled, so the differences are only due to particle size. The UHPLC column shows a significantly higher estimated peak capacity than the conventional column (187 vs 165). The peak capacity[42] was estimated based on the average baseline peak width of the 20 most intense peaks. This higher peak capacity resulted in a better separation and more metabolite identifications for the UHPLC runs. These runs consistently showed a better MS2 sampling and better peak purity.
Q-TOF vs Orbitrap Analyses
At first sight the chromatographic patterns derived from Orbitrap Elite data were clearly different from the ones from the Q-TOF. As stated before, repeatability and intermediate and precision of analyses[18] of SRM 1950 within a LC-MS platform were deemed acceptable to excellent depending on the relative abundance of the components (see Table I and Table II below). Also, we determined that, when identified and unidentified compounds were included in the analysis the major source of variability resulted from low intensity peaks, a finding previously shown in the analysis of a material with a simpler matrix, urine[43]. However, significant differences were found between different LC-MS platforms suggesting that the results obtained should be cautiously analyzed. In fact, data shows great disparity between LC-MS platforms when comparing the total number of molecular features, the nature of the molecular features and the intensity patterns of the molecular features that are common to both analyses.
Table II.
Repeatibility (RSD) of relative ion abundances of SRM 1950 metabolites from Orbitrap data (these metabolites were consistently found in both conventional analyses: Orbitrap and Q-TOF mass spectrometers).
| Metabolite | HMDB ID | Precursor Type |
Precursor Formula |
Precursor m/z |
RA (%) |
RSD (%) |
|---|---|---|---|---|---|---|
| Phthalic anhydride | [M+H]+ | C8H5O3 | 149.0233 | - | ||
| Methionine | HMDB00696 | [M+H]+ | C5H12NO2S | 150.0583 | 0.28 | 20.47 |
| Carnitine | HMDB14721 | [M+H]+ | C7H16NO3 | 162.1125 | 0.44 | 0.97 |
| Phenylalanine | HMDB00159 | [M+H]+ | C9H12NO2 | 166.0863 | 3.75 | 5.80 |
| Diphenylamine | HMDB32562 | [M+H]+ | C12H12N | 170.0964 | 0.52 | 0.39 |
| Arginine | HMDB00517 | [M+H]+ | C6H15N4O2 | 175.1190 | 0.01 | - |
| Tyrosine | HMDB00158 | [M+H]+ | C9H12NO3 | 182.0817 | 2.31 | 1.50 |
| Ecgonine | HMDB06548 | [M+H]+ | C9H16NO3 | 186.1125 | 0.01 | 7.25 |
| L-Tryptophan | HMDB30396 | [M+H]+ | C11H13N2O2 | 205.0791 | 1.27 | 1.39 |
| Indole-3-lactic acid | HMDB00671 | [M+H]+ | C11H12NO3 | 206.0812 | 0.02 | 2.99 |
| L-Kynurenine | HMDB00684 | [M+H]+ | C10H13N2O3 | 209.0921 | 0.04 | 0.15 |
| Pantothenic acid | HMDB00210 | [M+H]+ | C9H18NO5 | 220.1179 | 0.03 | 31.41 |
| Leu-Pro | [M+H-H2O]+ | C11H19N2O2 | 211.1441 | 0.07 | 41.53 | |
| Atenolol | HMDB01924 | [M+H]+ | C14H23N2O3 | 267.1767 | 0.20 | 0.07 |
| Glu-Met | [M+H]+ | C10H19N2O5S | 279.1009 | 0.02 | 1.25 | |
| 9-Octadecenamide, (Z)- | HMDB02117 | [M+H]+ | C18H36NO | 282.2791 | 0.16 | 7.61 |
| Retinol | HMDB00305 | [M+H-H2O]+ | C20H29 | 269.2264 | 0.90 | 1.56 |
| p-hydroxybenzoylecgonine | [M+H]+ | C16H20NO5 | 306.1336 | 0.04 | 15.57 | |
| Ranitidine | HMDB01930 | [M+H]+ | C13H23N4O3S | 315.1485 | 0.01 | 13.05 |
| Trp-Asn | [M+H]+ | C15H19N4O4 | 319.1401 | 0.02 | 32.93 | |
| Citalopram | HMDB05038 | [M+H]+ | C20H22FN2O | 325.1711 | 0.08 | - |
| Trp-Glu | [M+H-H2O]+ | C16H18N3O4 | 316.1292 | 0.02 | 6.05 | |
| Phe-Trp | [M+H]+ | C20H22N3O3 | 352.1656 | 0.31 | 3.73 | |
| Hydrocortisone | HMDB14879 | [M+H]+ | C21H31O5 | 363.2166 | 0.51 | - |
| Cholic acid | HMDB00619 | [M+H]+ | C24H41O5 | 409.2949 | 0.29 | 33.01 |
| Glycodeoxycholic acid | HMDB00631 | [M+H]+ | C26H44NO5 | 450.3214 | 0.27 | 4.91 |
| 1-Myristoyl-sn-glycero-3- phosphocholine |
HMDB10379 | [M+H]+ | C22H47NO7P | 468.3085 | 3.23 | 4.74 |
| 1-(1Z-hexadecenyl)-sn- glycero-3-phosphocholine |
[M+H]+ | C24H51NO6P | 480.3449 | 3.38 | - | |
| 1-Octadecanoyl-sn-glycero- 3-phosphoethanolamine |
[M+Na]+ | C23H48NO7PNa | 504.3061 | 0.10 | 35.71 | |
| Lyso-PC(16:0) | HMDB10382 | [M+H]+ | C24H51NO7P | 496.3398 | 33.18 | 0.08 |
| 1-Eicosatrienoyl-sn-glycero- 3-phosphoethanolamine |
[M+H]+ | C25H47NO7P | 504.3085 | 0.10 | 3.56 | |
| Lyso-PE(0:0/20:1) | HMDB11482 | [M+H]+ | C25H51NO7P | 508.3398 | 3.11 | |
| 1-(9Z-octadecenoyl)-sn- glycero-3-phosphocholine |
HMDB02815 | [M+H]+ | C26H53NO7P | 522.356 | 16.65 | 3.88 |
| 1-Octadecanoyl-sn-glycero- 3-phosphocholine |
HMDB11128 | [M+H]+ | C26H55NO7P | 524.3711 | 14.96 | 3.12 |
| 1-nonadecanoyl-sn-glycero- 3-phosphocholine |
[M+H]+ | C27H57NO7P | 538.3867 | 1.73 | 2.96 |
Molecular features from the conventional analysis of SRM 1950 in two different platforms (HPLC Agilent 1290 / Agilent QTOF 6530 and HPLC Agilent 1290 / Thermo Orbitrap Elite), with absolute peak heights greater than 5000 counts, were selected for feature alignment, data processing and multivariate statistical analysis in XCMS. Agilent raw data files were also processed using Agilent MassHunter Qualitative Analysis Software (version B.05.00). The untargeted molecular feature extraction algorithm[44], was used to generate features based on the elution profile of identical mass and retention times (mass accuracy <5 ppm). Only molecular features found in at least 75% of the samples were taken in account.
As mentioned before, the average numbers of extracted molecular features from the conventional QTOF data using XCMS was 6455. On the other hand, 3229 molecular features were found in the conventional Orbitrap data and 1593 molecular features were common to both analyses. This number was reduced to 821 when a constraint to count only features found in all replicates was applied. Again, these numbers do not show much variation across batches.
Figure 3 shows a principal component analysis (PCA) that was applied to find molecular features showing statistical differences across chromatographic separations on both platforms. The Benjamini-Horchberg procedure was applied to adjust for false-positive discovery, arising from multiple testing of p-values (adjusted for predicted p<0.01). The PCA analysis showed that the LC separations are clearly statistically different without any explicit reference to the identified features. The first principal component captures over 75% of the variance. As mentioned above, the major source of variability across chromatographic separations are the signal intensities.
Figure 3.
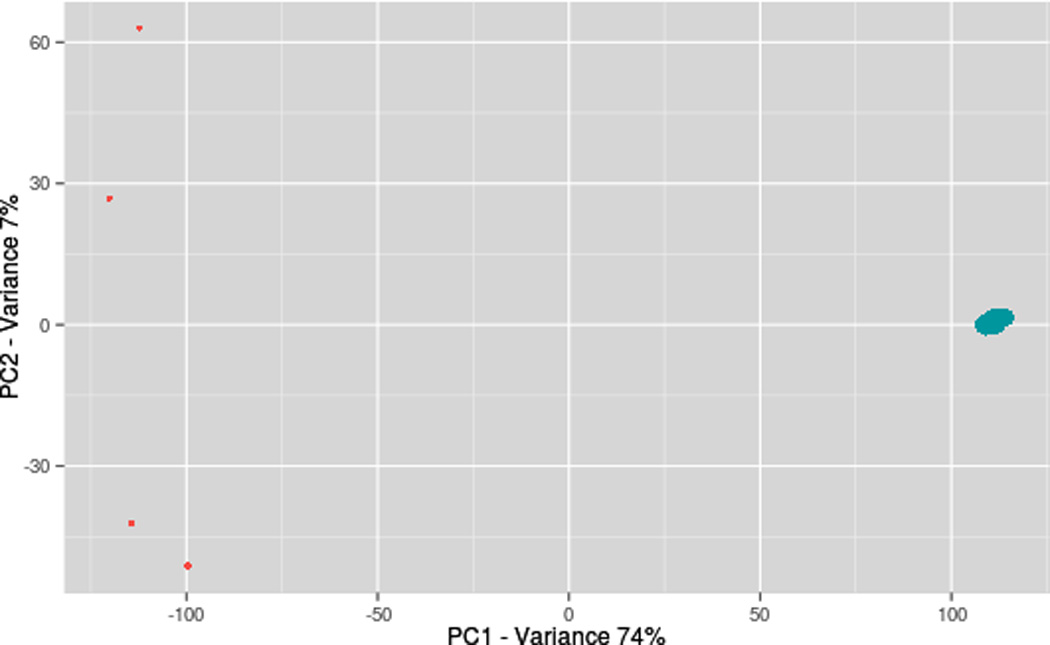
Although PCA is a useful technique for linear dimension reduction, it is difficult to use with spectral data derived from complex samples due to the information loss caused by frequent missing values. However, it is still useful for capturing the main modes of variability of the data when dealing with large datasets. For example, Figure 3 shows more variability between Q-TOF experiments than between Orbitrap experiments.
In order to find a unique chemical signature transferable across platforms a library searching against the SRM 1950 tandem mass spectral library of recurrent spectra was run. 335 of the common molecular features found a library match (match factor > 600 out of 999), which represents a drastic reduction of the number of features. Nevertheless, there was a significant overlap considering the limitations of the libraries at this point to contain all spectra of the material. More importantly, the most prominent features of both chromatograms were found among these features.
Regarding only MS/MS identifications, for the conventional HPLC and nanoLC separations on the Orbitrap Elite a total of 86 and 72 metabolites were identified, respectively. Also, similar differences among the different chromatographic separations were observed in the Orbitrap results. The nanoLC/Orbitrap chromatograms did not show apparent depletion at the end of the run (in the hydrophobic region) as the nanoLC/Q-TOF, but there were still fewer identifications in this region compared to the conventional HPLC. This difference is likely due to the fact that the sample was directly injected onto the analytical column for the Orbitrap separation while it was first trapped on a trap column in the chip for the Q-TOF separation.
As we reported before[18], nanoLC systems allowed us to generate many ion features, but lead to a greater uncertainty in MS/MS identification. In that work, we were able to identify many components with the nanoLC/Orbitrap Elite platform, but more than 50% of the identifications represent borderline identifications (match factor between 500 and 600). Frequently, the experimental spectra were of poor quality to be fully interpretable because of low signal strength.
Experiments of conventional HPLC chromatography using the same LC coupled to the Orbitrap mass spectrometer show also significant differences with the Q-TOF experiments. Only 45% of the identifications obtained for the conventional HPLC separation on the Orbitrap were also found in the conventional HPLC separation on the Q-TOF (see also NIST pipeline outputs in the supplemental information, Tables SI–3 and SI–4). Therefore, since the samples were analyzed with the same LC, same column and same amount of sample injected, we assume that the major differences between the platforms are determined by the ion source and the MS settings.
Table II shows the repeatability of relative ion abundances of 35 metabolites that are consistently found in both platforms (Q-TOF and Orbitrap) with the 2.1 mm column. The RSDs of the ion abundances of these components in some cases reaches 30% or more. This is likely due to the fact that despite the chromatographic patterns of the main molecular features being very reproducible, the extracted features from medium and low abundance components with poor ionizing power add high variability to the analysis. This occurs frequently with significant fluctuations in the peak composition and peak shape. A similar behavior has been observed across batches. In general, repeatability and intermediate precision depend on the analytical platform and it is recommended to determine these before drawing conclusions about any biological significance of the data. Standard Reference Materials are especially useful in this regard.
Since the MS settings were optimized using experimental design to maximize the number of identifications we intuit that most differences are due to the differences in the ion sources for the two instruments.
Table III shows the identifications belonging to different chemical classes on different LC-MS platforms. Amino acids and related metabolites represent around 30% of all identifications in most platforms. The milder conditions set in nanoLC instruments allow us to identify more carbohydrates than in other instruments such as the Q-TOF with the JetStream source or even a more conventional ESI source where these ions are more subject to in-source fragmentation. In contrast, these sources allow identifying higher molecular mass molecules, such as steroids, lipids and phospholipids.
It is worth mentioning at this point that the unsupervised analysis of non-targeted experiments requires the use of optimized software settings for each platform for the data processing; otherwise many spurious differences might be reported. An account of a study performed along this line has been recently published[45]. The authors used three AB Sciex proprietary software packages and one open source software package to compare the peak picking performance on the same LC-MS data. Despite the fact that the data was from an AB Sciex instrument they found only about 10% overlapping between the peak lists generated for different packages. One could object that the analysis was almost exclusively limited to data and software from one vendor or that the package parameters were not properly optimized, but it still raises serious questions regarding the processing of metabolomics data. This setting-dependence is reduced using the above-mentioned material-oriented spectral libraries.
Semi-quantitative Aspects of the Plasma Metabolite Profiling across Platforms
In a separate series of experiments, we tested the reliability of the relative semi-quantitation across platforms for those metabolites identified in both platforms by matching the library of recurrent spectra. (We refer to this as a semi-quantitative approach because no internal standards were used; the only purpose of this experiment was to verify if both platforms at least show proportional metabolite variations). Different dilutions of SRM 1950 were injected and separated in a similar way to the UHPLC metabolite profiling experiments described in the previous sections. Five injections were made yielding the following amounts on column (2.1×150mm): 254, 507, 761, 1015, and 1269 µg. The ions identified as recurrent features in both platforms were extracted from each of the five separations and the maximum intensities were collected. Linear regression analysis using the two set of intensities (Orbitrap vs. Q-TOF) shows that both platforms render similar metabolite variations for most components. Table SI_5 in the supplemental information shows a list of the molecular features that were extracted, the maximum intensities of each feature, and the Pearson correlation coefficients between the variables, Orbitrap and Q-TOF intensities. Out of the 1299 common features extracted in this experiment, 612 had correlation coefficients between 0.9 and 1.0 and 847 had correlation coefficients that were higher than 0.7. It means, 50% of the shared ions had correlation coefficients greater than 0.9. It was also observed that the correlation of the relative semi-quantitation for multiple components across platforms depends on the metabolite abundances, and the size of the injection (see Table SI_5 of the supplemental information). It is worth mentioning that 96 relevant metabolites have been quantified in SRM 1950 and reported in its Certificate of Analysis, so it could be used as an external standard for the quantitation of these and other similar metabolites.
Complicating Factors in Metabolite Identification
The above metabolite identifications are MS/MS library identifications and not authentic identifications (where the pure compound is injected onto the platform and identification is confirmed by retention time also). In addition, the use of retention times as a second dimension for the identification is limited by the poor transferability and reproducibility across different chromatographic conditions, chromatographic instruments, and mass spectrometers. Therefore, all hits were manually curated and the poor matches removed. In the process of manually curating the data, we have found many complicating factors in the identification process based on LC-MS data that are relevant to metabolite profiling. These are worth discussing individually. Some of these important factors include metabolite co-elution which is a problem for near isobaric ions, ion suppression, multiple ions of the same compound, contaminants, in-source fragmentation and artifacts originating from chemical reactions in the ion source or the collision cell.
As an illustrative example of co-elution, we have chosen six metabolites that have been reported as possible prostate cancer biomarkers[12], sarcosine, uracil, kynurenine, glycerol-3-phosphate, leucine, and proline. This finding still remains controversial[46, 47]. In our separations, we found in all cases co-eluted analytes contributing to the spectrum, but we were not able to properly identify all the co-elutants. Moreover, two of these metabolites, sarcosine and kynurenine, co-elute with compounds generating precursor ions with the same m/z. Sarcosine co-elutes with alanine and kynurenine with an unknown compound of m/z 209. A strategy to deal with unknowns is discussed below.
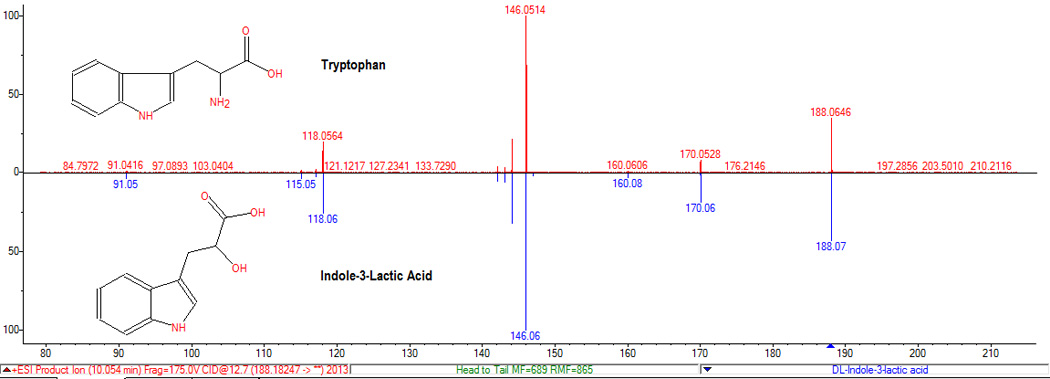
In-source fragmentation, adduct formation and large differences in ionization efficiencies in the ion source are a major challenge for the proper identification of components in a complex mixture. Figure 4 shows a zoomed portion of a 3D plot (retention time, m/z, relative abundance) of the SRM 1950 sample chromatogram (known: blue, unknown: red) that shows phenylalanine. Phenylalanine is not only detected as the protonated form, but also as the sodiated form, and the protonated form with formic acid, ammonia, and ammonia plus water losses. In all, we see five different ions for a single metabolite.
Figure 4.
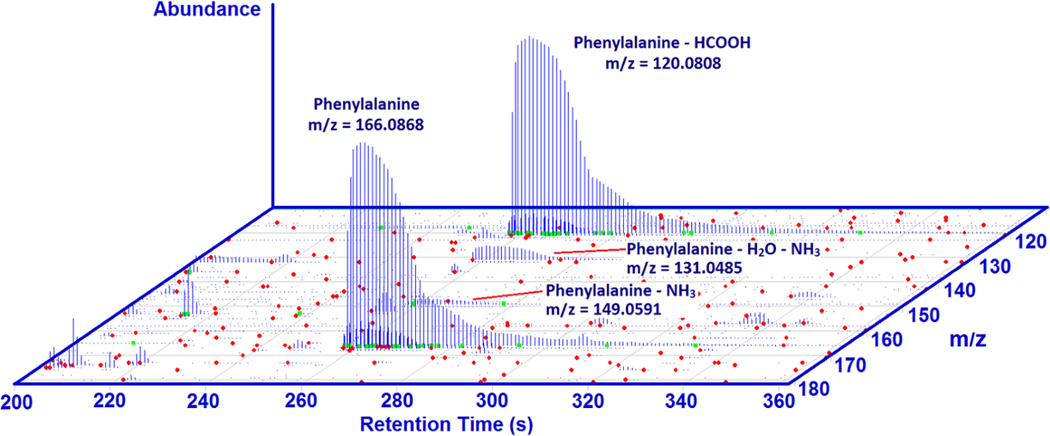
As mentioned above, it is typical to observe several peaks with similar retention times (co-elution), but frequently the MS1 information and the spectral libraries can help to distinguish between related peaks and unrelated compounds. Another complication that is observed is that a tandem mass spectrum for one ion can match the spectrum of other ions. For example, the loss of formic acid from phenylalanine produces an ion that has the same tandem mass spectrum as the one derived from the loss of water of phenylethanolamine (m/z 120.08). A similar situation is illustrated for tryptophan as the loss of ammonia matches the spectrum of the ion generated by loss of water from indole-3-lactic acid (see figure 5). Both compounds appear to be present in the samples and the retention times are similar. The library search software yields a slightly higher score for the identification of the product ion derived from tryptophan as phenylethanolamine. In this case the fragment ion information is redundant because both compounds were identified from their parent ion spectra, but in many cases fragment ion spectra are the only spectra available. In these cases, manual inspection and prior probability information are necessary.
Figure 5.
The NIST pipeline is able to retrieve information on multiple ions, typical adducts or contaminants, etc., automatically from the MS1 spectra. There are many other complicating factors in the identification process that are more difficult to detect automatically without previous knowledge of unexpected spectral modifications. For example, reactions that can take place in the ion source or collision cell and modify mass and tandem mass spectra. In our laboratory we have documented reactions of many protonated molecules and fragment ions with residual water or collision gas[48, 49] in the collision cell.
Cross-platform Transferable Chemical Signatures
Perhaps the main problem in the LC-MS analysis of any biological fluid is that it produces a very large number of species that are not confidently identified. These molecular features are used in many studies ‘as is’ assuming at best that exact mass and biological relevance are good enough for the purposes of metabolomics. Figure 6a shows the molecular features that are found in a sample of SRM 1950 using the conventional HPLC 1290 / Orbitrap platform. It reveals that the universe of unknowns is vast and we were not able to identify more than 10% of the components of this complex sample even after reducing the number of components by counting multiple ions, adducts, artifacts and others. It is worth mentioning that many adducts, artifacts, in-source transformations, etc., are also specific to the LC-MS platform used.
Figure 6.
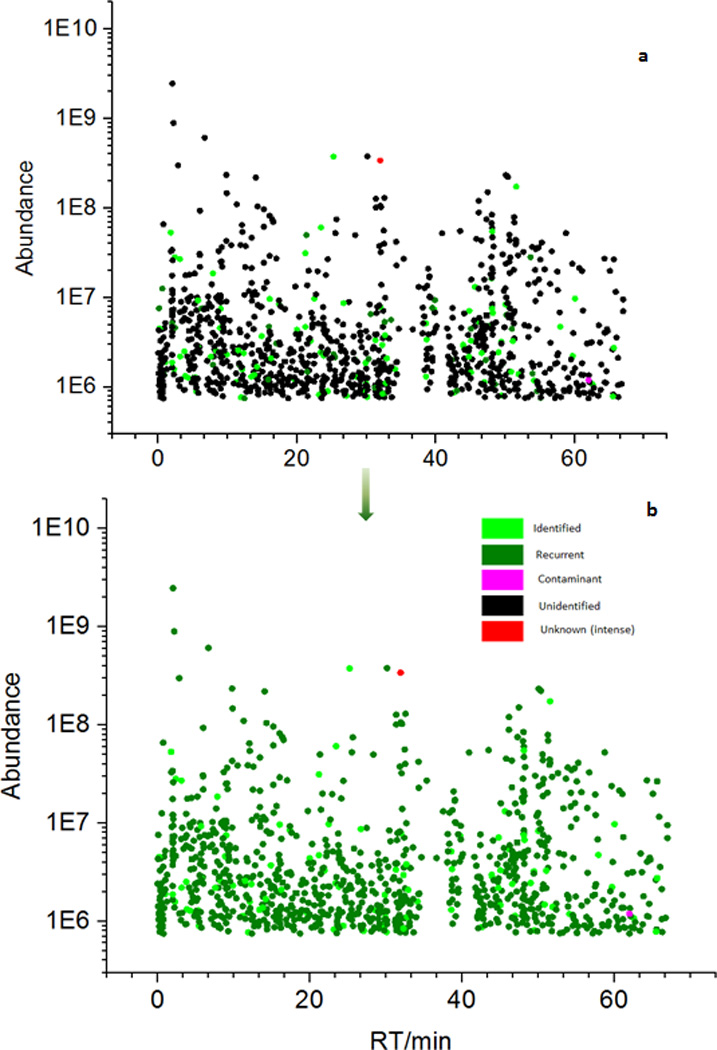
Figure 6b also shows the usefulness of the tandem mass spectral libraries of recurrent spectra in managing the uncertainty related to unknown compounds. Once the components in a material are properly labeled with a fragmentation spectrum, even if an authentic identification is not possible, consistent chemical signatures can be generated across platforms. Also, the library assigns prior probabilities of finding particular components in a material. Thus, chemical signatures based on the library match and prior probabilities are most likely cross-platform transferable. In general, the uses of recurrent unidentified spectra are multiple and include applications such as helping to connect samples, e.g., case vs. control, analyzing variation across laboratories, across methods, across platforms, identifying sample preparation artifacts, identifying new targets and finding chemical classes of unknowns.
After this work was completed, a paper dealing with the reproducibility of metabolomics studies was published[50]. The authors observed large differences in the number of spectral features across methods and analytical platforms, but also significant overlap among them.
CONCLUSIONS
The metabolite profiling of SRM 1950, Metabolites in Human Plasma, was performed using different LC-MS platforms. High repeatability, expressed as a RSD < 2% for relative abundances of the main components, was found while working within a particular LC-MS platform. However, large variability is observed in the metabolite profiling of the same material (including using the same sample) on different LC-MS platforms. Most of the variability is associated with the ionization process in the interface (ion source) and not with the chromatographic behavior (LC) of the sample or the nature of the mass analyzer (MS). The differences in the metabolite profiling on different platforms may have serious implications for the interpretation of metabolomics results. Several factors affecting the identification process have been discussed including and co-elution, multiple ions for each component, in-source fragmentation, the formation of adducts and reactions in the collision cell. Tandem mass spectral libraries of human plasma (SRM 1950) including all recurrent spectra (identified and unidentified) have been built. These libraries have been used for generating unique chemical signature that are transferable across different LC-MS platforms. Also, the relative semi-quantitation of signature metabolites renders similar results on different LC-MS platforms. All programs and data are freely available upon request.
Supplementary Material
Footnotes
Certain commercial instruments are identified in this document. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the products identified are necessarily the best available for the purpose.
References
- 1.Patti GJ, Yanes O, Siuzdak G. Nature Reviews Molecular Cell Biology. 2012;13:263. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yanes O, Tautenhahn R, Patti GJ, Siuzdak G. Anal Chem. 2011;83:2152. doi: 10.1021/ac102981k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B. PLoS One. 2011;6:e16957. doi: 10.1371/journal.pone.0016957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuehnbaum NL, Britz-McKibbin P. Chem Rev. 2013;113:2437. doi: 10.1021/cr300484s. [DOI] [PubMed] [Google Scholar]
- 5.Buscher JM, Czernik D, Ewald JC, Sauer U, Zamboni N. Anal Chem. 2009;81:2135. doi: 10.1021/ac8022857. [DOI] [PubMed] [Google Scholar]
- 6.Vaughan AA, Dunn WB, Allwood JW, Wedge DC, Blackhall FH, Whetton AD, Dive C, Goodacre R. Anal Chem. 2012;84:9848. doi: 10.1021/ac302227c. [DOI] [PubMed] [Google Scholar]
- 7.Beckonert O, Keun HC, Ebbels TM, Bundy J, Holmes E, Lindon JC, Nicholson JK. Nat Protoc. 2007;2:2692. doi: 10.1038/nprot.2007.376. [DOI] [PubMed] [Google Scholar]
- 8.Lawton KA, Berger A, Mitchell M, Milgram KE, Evans AM, Guo L, Hanson RW, Kalhan SC, Ryals JA, Milburn MV. Pharmacogenomics. 2008;9:383. doi: 10.2217/14622416.9.4.383. [DOI] [PubMed] [Google Scholar]
- 9.Patterson AD, Maurhofer O, Beyoğlu D, Lanz C, Krausz KW, Pabst T, Gonzalez FJ, Dufour J-F, Idle JR. Cancer research. 2011;71:6590. doi: 10.1158/0008-5472.CAN-11-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urayama S, Zou W, Brooks K, Tolstikov V. Rapid Communications in Mass Spectrometry. 2010;24:613. doi: 10.1002/rcm.4420. [DOI] [PubMed] [Google Scholar]
- 11.Fan L, Zhang W, Yin M, Zhang T, Wu X, Zhang H, Sun M, Li Z, Hou Y, Zhou X, Lou G, Li K. Acta Oncol. 2012;51:473. doi: 10.3109/0284186X.2011.648338. [DOI] [PubMed] [Google Scholar]
- 12.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y. Nature. 2009;457:910. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 13.Cai XM, Dong J, Zou LJ, Xue XY, Zhang XL, Liang XM. Chromatographia. 2011;74:391. [Google Scholar]
- 14.Cai XM, Zou L, Dong J, Zhao L, Wang Y, Xu Q, Xue X, Zhang X, Liang X. Anal Chim Acta. 2009;650:10. doi: 10.1016/j.aca.2009.01.054. [DOI] [PubMed] [Google Scholar]
- 15.Want EJ, O'Maille G, Smith CA, Brandon TR, Uritboonthai W, Qin C, Trauger SA, Siuzdak G. Anal Chem. 2006;78:743. doi: 10.1021/ac051312t. [DOI] [PubMed] [Google Scholar]
- 16.Tulipani S, Llorach R, Urpi-Sarda M, Andres-Lacueva C. Analytical Chemistry. 2013;85:341. doi: 10.1021/ac302919t. [DOI] [PubMed] [Google Scholar]
- 17.Bruce SJ, Tavazzi I, Parisod V, Rezzi S, Kochhar S, Guy PA. Analytical Chemistry. 2009;81:3285. doi: 10.1021/ac8024569. [DOI] [PubMed] [Google Scholar]
- 18.Simón-Manso Y, Lowenthal MS, Kilpatrick LE, Sampson ML, Telu KH, Rudnick PA, Mallard WG, Bearden DW, Schock TB, Tchekhovskoi DV, Blonder N, Yan X, Liang Y, Zheng Y, Wallace WE, Neta P, Phinney KW, Remaley AT, Stein SE. Anal Chem. 2013;85:11725. doi: 10.1021/ac402503m. [DOI] [PubMed] [Google Scholar]
- 19.Ballihaut G, Kilpatrick LE, Davis WC. Anal Chem. 2011;83:8667. doi: 10.1021/ac2021147. [DOI] [PubMed] [Google Scholar]
- 20.McGaw EA, Phinney KW, Lowenthal MS. J Chromatogr A. 2010;1217:5822. doi: 10.1016/j.chroma.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 21.Reiner JL, Phinney KW, Keller JM. Anal Bioanal Chem. 2011;401:2899. doi: 10.1007/s00216-011-5380-x. [DOI] [PubMed] [Google Scholar]
- 22.Quehenberger O, Armando AM, Brown AH, Milne SB, Myers DS, Merrill AH, Bandyopadhyay S, Jones KN, Kelly S, Shaner RL, Sullards CM, Wang E, Murphy RC, Barkley RM, Leiker TJ, Raetz CR, Guan Z, Laird GM, Six DA, Russell DW, McDonald JG, Subramaniam S, Fahy E, Dennis EA. J Lipid Res. 2010;51:3299. doi: 10.1194/jlr.M009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gowda GA, Tayyari F, Ye T, Suryani Y, Wei S, Shanaiah N, Raftery D. Anal Chem. 2010;82:8983. doi: 10.1021/ac101938w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prendergast JL, Sniegoski LT, Welch MJ, Phinney KW. Analytical and Bioanalytical Chemistry. 2010;397:1779. doi: 10.1007/s00216-010-3710-z. [DOI] [PubMed] [Google Scholar]
- 25.Phinney KW, Ballihaut G, Bedner M, Benford BS, Camara JE, Christopher SJ, Davis WC, Dodder NG, Eppe G, Lang BE, Long SE, Lowenthal MS, McGaw EA, Murphy KE, Nelson BC, Prendergast JL, Reiner JL, Rimmer CA, Sander LC, Schantz MM, Sharpless KE, Sniegoski LT, Tai SSC, Thomas JB, Vetter TW, Welch MJ, Wise SA, Wood LJ, Guthrie WF, Hagwood CR, Leigh SD, Yen JH, Zhang NF, Chaudhary-Webb M, Chen HP, Fazili Z, LaVoie DJ, McCoy LF, Momin SS, Paladugula N, Pendergrast EC, Pfeiffer CM, Powers CD, Rabinowitz D, Rybak ME, Schleicher RL, Toombs BMH, Xu M, Zhang M, Castle AL. Analytical Chemistry. 2013;85:11732. doi: 10.1021/ac402689t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elias JE, Haas W, Faherty BK, Gygi SP. Nat Methods. 2005;2:667. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
- 27.Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. Analytical Chemistry. 2006;78:779. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 28.Tautenhahn R, Bottcher C, Neumann S. Bmc Bioinformatics. 2008;9:504. doi: 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benton P, Want EJ, Ebbels TMD. Bioinformatics. 2010;26:2488. doi: 10.1093/bioinformatics/btq441. [DOI] [PubMed] [Google Scholar]
- 30.Rudnick PA, Clauser KR, Kilpatrick LE, Tchekhovskoi DV, Neta P, Blonder N, Billheimer DD, Blackman RK, Bunk DM, Cardasis HL, Ham AJ, Jaffe JD, Kinsinger CR, Mesri M, Neubert TA, Schilling B, Tabb DL, Tegeler TJ, Vega-Montoto L, Variyath AM, Wang M, Wang P, Whiteaker JR, Zimmerman LJ, Carr SA, Fisher SJ, Gibson BW, Paulovich AG, Regnier FE, Rodriguez H, Spiegelman C, Tempst P, Liebler DC, Stein SE. Mol Cell Proteomics. 2010;9:225. doi: 10.1074/mcp.M900223-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rutala WA, Weber DJ. Centers for Disease Control (US) 2008 [Google Scholar]
- 32.Roberts PL, Lloyd D. Biologicals. 2007;35:343. doi: 10.1016/j.biologicals.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 33.Guillarme D, Nguyen DT, Rudaz S, Veuthey JL. Eur J Pharm Biopharm. 2007;66:475. doi: 10.1016/j.ejpb.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 34.Guillarme D, Nguyen DT, Rudaz S, Veuthey JL. Eur J Pharm Biopharm. 2008;68:430. doi: 10.1016/j.ejpb.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 35.NIST tandem mass spectral library. 2014 [Google Scholar]
- 36.Stein SE. J. Am. Soc. Mass Spectrom. 1994;5:316. doi: 10.1016/1044-0305(94)85022-4. [DOI] [PubMed] [Google Scholar]
- 37.Stein SE, Scott DR. J. Am. Soc. Mass Spectrom. 1994;5:859. doi: 10.1016/1044-0305(94)87009-8. [DOI] [PubMed] [Google Scholar]
- 38.Mallard WG, Andriamaharavo NR, Mirokhin YA, Halket JM, Stein SE. Analytical Chemistry. 2014;86:10231. doi: 10.1021/ac502379x. [DOI] [PubMed] [Google Scholar]
- 39.Wallace WE, Srivastava A, Telu KH, Simón-Manso Y. Analytica Chimica Acta. 2014;841:10. doi: 10.1016/j.aca.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 40.Iglewicz B, Hoaglin D. In: The ASQC Basic References in Quality Control: Statistical Techniques, Vol. 16. Mykytka EF, editor. 1993. [Google Scholar]
- 41.Hrydziuszko O, Viant MR. Metabolomics. 2012;8:S161. [Google Scholar]
- 42.Dolan JW, Snyder LR, Djordjevic NM, Hill DW, Waeghe TG. Journal of chromatography A. 1999;857:1. doi: 10.1016/s0021-9673(99)00765-7. [DOI] [PubMed] [Google Scholar]
- 43.Gika HG, Macpherson E, Theodoridis GA, Wilson ID. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;871:299. doi: 10.1016/j.jchromb.2008.05.048. [DOI] [PubMed] [Google Scholar]
- 44.Sana TR, Waddell K, Fischer SM. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;871:314. doi: 10.1016/j.jchromb.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 45.Rafiei A, Sleno L. Rapid Commun Mass Spectrom. 2015;29:119. doi: 10.1002/rcm.7094. [DOI] [PubMed] [Google Scholar]
- 46.Jentzmik F, Stephan C, Lein M, Miller K, Kamlage B, Bethan B, Kristiansen G, Jung K. Journal of Urology. 2010;185:706. doi: 10.1016/j.juro.2010.09.077. [DOI] [PubMed] [Google Scholar]
- 47.Wagner C, Luka Z. Journal of Urology. 2010;185:385. doi: 10.1016/j.juro.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 48.Liang Y, Neta P, Simón-Manso Y, Stein SE. Rapid Commun Mass Spectrom. 2015;29:629. doi: 10.1002/rcm.7147. [DOI] [PubMed] [Google Scholar]
- 49.Neta P, Farahani M, Simón-Manso Y, Liang Y, Yang X, Stein SE. Rapid Commun Mass Spectrom. 2014;28:2645. doi: 10.1002/rcm.7055. [DOI] [PubMed] [Google Scholar]
- 50.Martin JC, Maillot M, Mazerolles G, Verdu A, Lyan B, Migne C, Defoort C, Canlet C, Junot C, Guillou C, Manach C, Jabob D, Bouveresse DJ, Paris E, Pujos-Guillot E, Jourdan F, Giacomoni F, Courant F, Fave G, Le Gall G, Chassaigne H, Tabet JC, Martin JF, Antignac JP, Shintu L, Defernez M, Philo M, Alexandre-Gouaubau MC, Amiot-Carlin MJ, Bossis M, Triba MN, Stojilkovic N, Banzet N, Molinie R, Bott R, Goulitquer S, Caldarelli S, Rutledge DN. Metabolomics. 2015;11:807. doi: 10.1007/s11306-014-0740-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.