

Abstract
The microbiome has an important role in human health. Changes in the microbiota can confer resistance to or promote infection by pathogenic bacteria. Antibiotics have a profound impact on the microbiota that alters the nutritional landscape of the gut and can lead to the expansion of pathogenic populations. Pathogenic bacteria exploit microbiota-derived sources of carbon and nitrogen as nutrients and regulatory signals to promote their own growth and virulence. By eliciting inflammation, these bacteria alter the intestinal environment and use unique systems for respiration and metal acquisition to drive their expansion. Unravelling the interactions between the microbiota, the host and pathogenic bacteria will produce strategies for manipulating the microbiota against infectious diseases.
Appreciation of the important role of the microbiota in human health and nutrition has grown steadily in the past decade. Initial studies focused on cataloguing the microbial species that comprise the microbiota and correlating the composition of the microbiota with the health or disease state of the host. The present period of renaissance has resulted in technologies and interdisciplinary research that are conducive to mechanistic studies and, in particular, those that focus on associations between the microbiota, the host and pathogenic bacteria. Exciting research is now starting to unravel how the composition of the microbiota can offer either resistance or assistance to invading pathogenic species. The majority of these studies were conducted in the gastrointestinal tract, in which associations between the host and microbes are of paramount importance. The gut microbiota of each individual is unique at the genus and species levels; however, it is more generally conserved at the phylum level, which is populated most prominently by Bacteroidetes and Firmicutes, followed by Proteobacteria and Actinobacteria. Host genetics, diet and environmental insults such as treatment with antibiotics alter the microbiota1–4, which can lead to varying susceptibility to infectious diseases between individuals5.
The microbiota can promote resistance to colonization by pathogenic species6–9. For instance, mice that are treated with antibiotics or that are bred in sterile environments (known as germ-free mice) are more susceptible to enteric pathogenic bacteria such as Shigella flexneri, Citrobacter rodentium, Listeria monocytogenes and Salmonella enterica serovar Typhimurium10–13. And some microbiotas can lead to the expansion or enhanced virulence of pathogenic populations7. A notable example concerns how differences in the composition of microbiotas determine the susceptibility of the mice to infection with C. rodentium: the transplantation of microbiotas from strains of mice that are susceptible to infection induced similar susceptibility in animals that were previously insusceptible, and the transplantation of microbiotas from resistant animals led to resistance to infection in previously susceptible animals14,15. Epidemiological surveys reinforce this idea. For example, differential susceptibility to infection with Campylobacter jejuni was shown to depend on the species composition of the microbiotas in a study of Swedish adults16. Individuals with a higher diversity within their microbiotas, and with an abundance of bacteria from the genera Dorea and Coprococcus, were significantly recalcitrant to C. jejuni infection compared with people who had low-diversity microbiotas and non-abundance of Dorea and Coprococcus.
The host’s diet profoundly affects the composition of the microbiota, with repercussions for the physiology, immunity and susceptibility to infectious diseases of the host17. Dietary choices have been shown to affect colonization by enterohaemorrhagic Escherichia coli (EHEC) serotype O157:H7 and the severity and length of its resulting disease18, and supplementation of the diet with phytonutrients promotes the expansion of beneficial Clostridia species that protect mice from colonization by C. rodentium19.
The use of innovative technologies, in combination with more conventional approaches, is driving our understanding of the interactions between the microbiota, the host and pathogenic bacteria. The genetic tractability of several species of bacteria, as well as of their mammalian hosts (such as mice), allows for the mechanistic investigation of these relationships. The investigation of changes in the composition of microbiotas has been driven by next-generation sequencing, which also facilitated the analysis of transcriptomes. The growing power and finesse of metabolomics studies are quickly expanding our knowledge of the impact of both the microbiota and of pathogenic bacteria on the metabolic landscape of the gut. Here, we review advances in our understanding of the complex relationships that determine the severity and outcome of gastrointestinal infections. The majority of the mechanistic studies that investigate these interactions have been conducted in S. Typhimurium, EHEC and Clostridium difficile: therefore, these pathogenic organisms are covered more extensively than others in this Review.
Antibiotics
Antibiotics revolutionized medicine and were justifiably dubbed ‘magic bullets’ against bacterial infections. However, conventional antibiotics are generally bacteriostatic or bactericidal, which means that they indiscriminately kill or prevent the growth of both pathogenic and beneficial microbes. Antibiotics can alter the taxonomic, genomic and functional features of the microbiota, and their effects can be rapid and sometimes everlasting20. They can decrease the diversity of the microbiota, which compromises resistance to colonization by incoming pathogenic bacteria20 — most notably leading to an expansion of C. difficile that can cause diarrhoea that leads to potentially fatal colitis21.
C. difficile is a spore-forming bacterium that, on germination, colonizes the large intestine and causes colitis through the action of two toxins: TcdA and TcdB. The majority of C. difficile infections are nosocomial, but there has also been an increase in community-acquired infections, mainly due to the ubiquitous presence of C. difficile spores. C. difficile can colonize the mammalian intestine without causing disease, but one of the most important risk factors for colitis that is mediated by C. difficile is the use of antibiotics21. The antibiotics-mediated loss of resistance to colonization also allows colonization by S. Typhimurium and the development of disease22. Both C. difficile and S. Typhimurium catabolize sialic acid as a source of carbon in the lumen to promote their expansion23. They rely on saccharolytic members of the microbiota, such as Bacteroides thetaiotaomicron, to make this sugar freely available in the intestinal lumen. Treatment with antibiotics increases the abundance of host-derived free sialic acid as well as enhancing its release into the lumen by B. thetaiotaomicron, which promotes the expansion of the two pathogenic bacteria23. Antibiotic use also triggers production of the organic acid succinate, another microbiota-derived nutrient that confers a growth advantage to C. difficile. It is often present at a low concentration in the microbiotas of conventional mice, but its presence increases on treatment with antibiotics, which promotes a bloom of C. difficile24 (Fig. 1).
Figure 1. The impact of antibiotics on the microbiota and the expansion of enteric pathogens.

a, A diverse and non-disturbed microbiota confers resistance to colonization by enteric pathogens in the intestinal epithelium. b, Treatment with antibiotics decreases the diversity of the microbiota and leads to expansion of the C. difficile population. Toxins that are released from C. difficile (TcdA and TcdB) enter and damage the cells of the epithelium, which leads to inflammation (colitis) and cell death. c, Treatment with antibiotics also leads to an increase in the levels of free sialic acid (from the host) and succinate (from the microbiota) in the lumen of the intestine. Elevated sialic acid promotes the expansion of the S. Typhimurium population, which can lead to inflammation (gastroenteritis) if the bacterium invades the cells of the intestinal epithelium. Elevated levels of sialic acid and succinate further promote the expansion of the C. difficile population and the development of colitis and cell death.
Knowledge of how microbiota disruption affects the ability of bona fide or opportunistic pathogenic organisms to infect hosts is still in its infancy. However, two underlying themes converge: microbiota-induced changes in the metabolite landscape of the gut and inflammation.
Utilization of nutrients
Simple dietary sugars are absorbed in the small intestine, which means that they are unavailable as sources of carbon for the microbiota and pathogenic bacteria in the colon. The most abundant members of the microbiota are those that are able to utilize the undigested plant polysaccharides and host glycans that are present in the colon25.
The gut epithelium is protected by a layer of mucus that is composed of proteins known as mucins that are rich in fucose, galactose, sialic acid, N-acetylgalactosamine, N-acetylglucosamine and mannose. These sugars are harvested by saccharolytic members of the microbiota, such as Bacteroidales in the gut, which makes them available to species within the microbiota that lack this capability26. However, pathogenic bacteria in the gut can also exploit the availability of these sugars to promote their own expansion. Several studies have used B. thetaiotaomicron as a model Bacteroides in which to investigate these syntrophic links. Sialic acid is a terminal sugar of some mucosal glycans, and B. thetaiotaomicron has sialidase activity but lacks the catabolic pathway for sialic-acid utilization. The bacterium therefore releases sialic acid to gain access to underlying glycans that it can use as a source of carbon. The sialic acid that B. thetaiotaomicron releases from the mucus can be catabolized by both C. difficile and S. Typhimurium, which provides them with a growth advantage23. The ability of the microbiota to use sialic acid therefore depends on the action of B. thetaiotaomicron, and mutants that lack sialidase fail to enhance the growth of these two pathogenic bacteria23.
B. thetaiotaomicron also releases fucose from the mucus. It harbours multiple enzymes that can cleave fucose from host glycans, so its presence results in the high availability of fucose in the lumen of the gut27–30. This free fucose can also be used as a source of carbon by S. Typhimurium23. Importantly, B. thetaiotaomicron can promote the fucosylation of mucosal glycans when introduced into monoassociated germ-free mice31,32.
The microbiota resides in the lumen and the outer mucus layer of the intestine. EHEC, however, aims to achieve a unique niche by closely adhering to the enterocytes of the intestinal epithelium. To achieve its goal, EHEC must successfully compete with the microbiota for nutrients. B. thetaiotaomicron does not need to compete with EHEC, however, because it can utilize polysaccharides; EHEC can only utilize monosaccharides and disaccharides13,33. EHEC’s main competitors are commensal E. coli, which preferentially utilizes fucose as a source of carbon when growing in the mammalian intestine13,33. To circumvent this competition, EHEC utilizes other sources of sugar, such as galactose, the hexuranates, mannose and ribose, which commensal E. coli cannot catabolize optimally33,34 (Fig. 2).
Figure 2. Modulation of enterohaemorrhagic E. coli virulence through nutrients provided by the microbiota.

a, The microbiota resides in the lumen and outer mucus layer of the intestine. The saccharolytic bacterium Bacteroides thetaiotaomicron is a prominent member of the microbiota. It can release fucose from the mucus and makes the sugar available to other bacteria. When EHEC senses fucose through the FusKR signalling system, it represses both its use of the sugar and the expression of genes that encode the T3SS, a protein-translocation apparatus that enables the bacterium to secrete effector proteins into host cells. This repression prevents EHEC from competing for fucose with commensal E. coli and from expending energy unnecessarily on T3SS expression. b, Metabolites that are provided by B. thetaiotaomicron, such as succinate, lead to an increase in the expression by EHEC of the enzyme mucinase, which obliterates the mucus layers of the intestine. EHEC is then able to reach the intestinal epithelium. B. thetaiotaomicron then begins to secrete succinate and other metabolites that are required for gluconeogenesis into the now nutrient-poor environment. The compounds are sensed by EHEC, which upregulates its expression of the T3SS to enable the bacterium to attach to the epithelial cells of the host intestine and form lesions that cause diarrhoea.
EHEC uses fucose as a signalling molecule with which to adjust its metabolism and to regulate the expression of its virulence repertoire in the lumen and the outer mucus layer of the colon35. It horizontally acquired a pathogenicity island of genes that encode a fucose-sensing signalling-transduction system35. This system is unique to EHEC and to C. rodentium35 (which is used extensively in mouse models as a surrogate for the human pathogen EHEC36). It is composed of the membrane-bound histidine sensor kinase FusK, which specifically autophosphorylates in response to fucose. FusK then transfers its phosphate to a response regulator called FusR, which is a transcription factor. Phosphorylation activates FusR, which represses the expression of the fucose utilization genes in EHEC, and helps EHEC to avoid the need to compete for this nutrient with commensal E. coli35. To prevent the unnecessary expenditure of energy by EHEC, FusR represses the genes that encode the EHEC virulence machinery, a syringe-like apparatus known as a type III secretion system (T3SS), which the bacterium uses to adhere itself to enterocytes and highjack the function of these host cells35. EHEC therefore uses fucose, a host-derived signal that is made available by the microbiota, to sense the environment of the intestinal lumen and to modulate its own metabolism and virulence.
To reach the lining of the epithelium, EHEC and C. rodentium produce mucinases37, which cleave the protein backbone of mucin-type glycoproteins. Expression of these enzymes is increased by metabolites that are produced by B. thetaiotaomicron38. Because mucus is one of the main sources of sugar in the colon, where EHEC and C. rodentium colonize, obliteration of the mucus layer creates a nutrient-poor environment near the epithelium that is referred to as gluconeogenic. The colonization of mice by B. thetaiotaomicron therefore profoundly changes the metabolic landscape of the mouse gut because it raises the levels of organic acids such as succinate24,38,39. Moreover, several metabolites that indicate a gluconeogenic environment, such as lactate and glycerate, are also elevated38. EHEC and C. rodentium sense this gluconeogenic and succinate-rich environment through the transcriptional regulator Cra. On receiving the cue that they have reached the lining of the gut epithelium, these bacteria activate the expression of their T3SSs38. EHEC therefore exploits metabolic cues from B. thetaiotaomicron, and probably other members of the microbiota, to precisely programme its metabolism and virulence (Fig. 2).
Other pathogenic bacteria can also adjust their gene expression in the presence of microbiota-produced succinate. C. difficile induces a pathway that converts succinate to butyrate, which confers a growth advantage in vivo24. Populations of C. difficile mutants that are unable to convert succinate fail to expand in the gut in the presence of B. thetaiotaomicron24.
Several short-chain fatty acids that are produced by the microbiota, are important determinants of interactions between the microbiota and pathogenic bacteria in the gut. The abundance and composition of short-chain fatty acids is distinct in each compartment of the intestine, and the ability to sense these differences might help pathogenic bacteria in niche recognition. The most abundant short-chain fatty acids in the gut are acetate, propionate and butyrate. S. Typhimurium preferably colonizes the ileum40, which generally contains acetate at a concentration of 30 mM. This acetate concentration enhances the expression of the S. Typhimurium Salmonella pathogenicity island 1 (SPI-1)-encoded T3SS (T3SS-1), which is involved in the bacterium’s invasion of the host. Conversely, 70 mM propionate and 20 mM butyrate, concentrations typical of the colon, suppress the expression of the T3SS-1 (ref. 41). Propionate and butyrate seem to affect the T3SS-1 regulatory cascade at various levels. However, the detailed mechanism of this regulation is yet to be unravelled. In EHEC, exposure to the levels of butyrate found in the colon increases the expression of the EHEC T3SS through post-transcriptional activation of the transcriptional regulator Lrp42. Exposure to the concentrations of acetate and propionate that are found in the small intestine does not significantly affect the virulence of EHEC.
Diet has a profound effect on the composition of the microbiota and the concentration of short-chain fatty acids in the gut17. A diet that is high in fibre results in the enhanced production of butyrate by the gut microbiota. That increases the host’s expression of globotriaosylceramide, which is a receptor for the Shiga toxin that is produced by EHEC18. Shiga toxin can lead to the development of haemolytic uraemic syndrome (HUS) and is the cause of the morbidity and mortality associated with outbreaks of EHEC43. Consequently, animals that are fed a high-fibre diet are more susceptible to Shiga toxin than are those on a low-fibre diet and develop more severe disease18. Conversely, increased levels of microbiota-derived acetate protect animals from disease that is caused by the toxin. Certain species of Bifidobacteria contribute to higher levels of acetate in the gut, which helps to improve the barrier function of the intestinal epithelium and to prevent Shiga toxin from reaching the bloodstream44.
Enteric pathogenic bacteria also use other nutrients to successfully overcome the microbiota’s resistance to their colonization. Ethanolamine is abundant in the mammalian intestine45. It can be used as a source of carbon and of nitrogen by a number of pathogenic species46, and food-borne bacteria are particularly adept at using it. However, it cannot be metabolized by the majority of commensal species47. S. Typhimurium, EHEC and L. monocytogenes gain a growth advantage in the intestine through their ability to use this compound45,48,49. Ethanolamine is also used as a signal by EHEC and S. Typhimurium to activate the expression of virulence genes50,51. And S. Typhimurium uses hydrogen produced by the microbiota as an energy source to enhance its growth during the initial stage of infection52.
The exploitation of microbiota-derived molecules as both nutrients and signals is crucial for the successful infection of the host by pathogenic bacteria. Although such organisms have clearly developed many strategies through which to circumvent the microbiota’s resistance to colonization, and in many cases even employ its help, the microbiota pushes back, which creates an intense competition for resources. The ability of EHEC to colonize the intestine stems from differences in the sources of sugar that are used by EHEC and by commensal E. coli. For example, the presence of multiple strains of commensal E. coli with overlapping nutritional requirements interferes with the colonization of the mouse intestine by EHEC53. This study uses a streptomycin-treated mouse model of EHEC and three distinct commensal strains of E. coli to assess differential sugar requirements for the successful colonization of the intestines53. EHEC could colonize mice that were pre-colonized with any one of the commensal strains, but it could not colonize mice that were pre-colonized with all three strains53. EHEC has evolved to exploit distinct sources of sugar during colonization of the gut. It utilizes catabolic pathways for the hexuronates glucuronate and galacturonate and for sucrose that are not employed by commensal E. coli within the gut33,53. It can also metabolize several sugars simultaneously. The loss of multiple catabolic pathways has an additive effect on colonization. This phenomenon is not observed in commensal E. coli, however, which suggests that E. coli uses available sugars in a stepwise fashion54. EHEC therefore differs from commensal E. coli in metabolic strategy and the use of nutrients for the colonization of the mammalian intestine.
C. rodentium is outcompeted and then cleared from the mouse gut through a bloom in the population of commensal E. coli, which competes with C. rodentium for monosaccharides for nutrition13. By contrast, C. rodentium is not cleared by B. thetaiotaomicron in germ-free mice that are fed a diet that contains both monosaccharides, which can be used by Enterobacteriacae such as C. rodentium, and polysaccharides, which can be used by Bacteroides. However, when the mice are switched to a diet that consists only of monosaccharides, B. thetaiotaomicron and C. rodentium are forced to compete for sugars, and B. thetaiotaomicron outcompetes C. rodentium13. The ability of pathogenic bacteria to successfully compete with commensal species for nutrients is therefore important for their establishment in the gut.
Interception of signals from the microbiota and the host
The microbiota affects the risks and courses of enteric diseases. Vibrio cholerae is a major cause of explosive diarrhoea in which there is extensive disruption of the intestinal population of microbes. Metagenomic studies of the faecal microbiota of people with cholera in Bangladesh show that recovery is characterized by a certain microbiota signature. Reconstitution of this microbiota in germ-free mice restricts the infectivity of V. cholerae. Specifically, the presence of Ruminococcus obeum can hamper the colonization of the intestines by V. cholerae through the production of the furanone signal autoinducer-2, which causes the repression of several V. cholerae colonization factors55.
Another example of the effect of microbiota-derived signals on host colonization is their use by EHEC in the colonization of its ruminal reservoir. EHEC exclusively colonizes the recto–anal junction of adult cattle. Through the sensor protein SdiA, EHEC detects acyl-homoserine lactone signals from the rumen microbiota, which it uses to reprogram itself to survive the acidic pH of the animal’s stomachs and to successfully colonize the rectoanal junction56.
As well as being able to directly detect signals that are derived from the microbiota, pathogenic bacteria can detect host-derived signals that have been modified by the microbiota to modulate their virulence. V. cholerae has a type VI secretion system (T6SS), which it uses to kill other bacteria. During its colonization of the intestine, V. cholerae comes in contact with the mucosal microbiota, which can affect the composition of bile acids in the intestine. For example, Bifidobacterium bifidum negatively regulates the T6SS activity of V. cholerae through the metabolic conversion of three bile acids (glycodeoxycholic acid, taurodeoxycholic acid and cholic acid) into the bile acid deoxycholic acid. Deoxycholic acid, but not its unmodified salts, decreases the expression of T6SS genes. This leads to a decrease in the killing of E. coli by V. cholerae owing to bile-acid conversion by other commensals, which decreases the activity of the T6SS57.
Another microbiota-modified host signal that is detected by pathogenic bacteria is the neurotransmitter noradrenaline. The gut is highly innervated, and neurotransmitters are important signals in the gastrointestinal tract, where they modulate peristalsis, the flow of blood and the secretion of ions58. The microbiota affects the availability of neurotransmitters in the intestinal lumen, as well as their biosynthesis. For example, the microbiota induces biosynthesis of serotonin59, and microbiota-derived enzymatic activities increase the levels of active noradrenaline in the gut lumen60. Noradrenaline is synthesized by the adrenergic neurons of the enteric nervous system61 and it is inactivated by the host through conjugation with glucuronic acid (to produce a glucuronide). Microbiota-produced enzymes known as glucuronidases then deconjugate glucuronic acid from noradrenaline, which increases the amount of active noradrenaline in the lumen of the intestine60. Several pathogenic bacteria of the gut, including EHEC, S. Typhimurium and V. parahaemolyticus, sense noradrenaline to activate the expression of virulence genes62–65. Two adrenergic sensors have been identified in bacteria: the membrane-bound histidine kinases QseC and QseE66,67. QseC also detects the microbiota-produced signal autoinducer-3 (refs 64 and 66), so the sensing of signals from both the host and the microbiota converge at the level of a single receptor, a process known as inter-kingdom signalling.
Inflammation
Although diet and the composition of the microbiota heavily influence the availability of nutrients in the gut, the host also has an important part to play. A crucial driver of changes in the gut environment is the inflammatory response of the host. Intestinal inflammation in people is associated with an imbalance in the microbiota, known as dysbiosis, and is characterized by a reduced diversity of microbes, a reduced abundance of obligate anaerobic bacteria and an expansion of facultative anaerobic bacteria in the phylum Proteobacteria, mostly members of the family Enterobacteriaceae68–73. Similar changes in the composition of the gut microbiota are observed in mice with chemically induced colitis74 and genetically induced colitis75. These changes in the structure of the microbiota probably reflect an altered nutritional environment that is created by the inflammatory response of the host.
The availability of nutrients in the large intestine is altered during inflammation through changes in the composition of mucous carbohydrates. Interleukin (IL)-22, a cytokine that is prominently induced in the intestinal mucosa when mice and rhesus macaques are infected with S. Typhimurium76,77, stimulates the epithelial expression of galactoside 2-α-L-fucosyltransferase 2 and enhances the α(1,2)-fucosylation of mucus carbohydrates78,79. The gut microbiota can liberate fucose from mucus carbohydrates23,80, which leads to the induction of genes for fucose utilization in E. coli78. Similarly, increased fucosylation of glycans is observed during S. Typhimurium-induced colitis in mice, which correlates with elevated synthesis of the proteins involved in fucose utilization81. Mucus fucosylation that is induced during infection with C. rodentium causes changes in the composition of the gut microbiota that help to protect the host from the expansion and epithelial translocation of the pathobiont Enterococcus faecalis79.
Another driver of changes in the nutritional environment of the gut is the generation of reactive oxygen species and reactive nitrogen species during inflammation. Pro-inflammatory cytokines such as interferon-γ (IFN-γ) activate dual oxidase 2 in the intestinal epithelium, which produces hydrogen peroxide82. Increased expression of DUOX2, the gene that encodes dual oxidase 2, in the intestinal mucosa of patients with Crohn’s disease and ulcerative colitis correlates with an expansion of Proteobacteria in the gut microbiota83. IFN-γ also induces epithelial expression of the gene Nos2 (ref. 84), which encodes inducible nitric oxide synthase, the enzyme that catalyses the production of nitric oxide from L-arginine85. As a result, the concentration of nitric oxide is elevated in gases from the colons of people with inflammatory bowel disease86–88. Although reactive oxygen and nitrogen species have antimicrobial activity, these radicals quickly form non-toxic compounds in the lumen of the gut as they diffuse away from the epithelium. For example, when they are generated during inflammation by host enzymes in the intestinal epithelium, these species react to form nitrate89. This by-product of inflammation is present at elevated concentrations in the intestines of mice with chemically induced colitis90 (Fig. 3). Nitrate reductases, enzymes that are broadly conserved among the Enterobacteriaceae, couple the reduction of nitrate to energy-conserving electron transport systems for respiration, a process termed nitrate respiration. However, the genes that encode them are absent from the genomes of obligate anaerobic Clostridia or Bacteroidia91. Nitrate respiration drives the Nos2-dependent expansion of commensal E. coli in mice with chemically or genetically induced colitis, but not in animals without signs of intestinal inflammation91. Respiratory electron acceptors that are generated as a by-product of the host inflammatory response therefore create a niche in the lumen of the intestines that supports the uncontrolled expansion of commensal Enterobacteriaceae rather than of obligate anaerobic bacteria91. The resulting bloom in the inflamed intestine is one of the most consistent and robust ecological patterns that has been observed in the gut microbiota92.
Figure 3. The effect of intestinal inflammation on nutrient availability.

S. Typhimurium uses its virulence factors (T3SS-1 and T3SS-2) to trigger intestinal inflammation. Cytokines that are released during inflammation, such as IL-22 and IFN-γ, trigger the release of antimicrobial molecules lipocalin-2, reactive oxygen species (ROS) and reactive nitrogen species (RNS) from the intestinal epithelium. Lipocalin-2 can block the growth of commensal Enterobacteriaceae that rely on the siderophore enterobactin for the acquisition of iron (Fe3+). It does not bind to the S. Typhimurium siderophone salmochelin, however, which confers the bacterium with resistance to its effects on growth. RNS and ROS react to form nitrate, which drives the growth of Enterobacteriaceae through nitrate respiration. Microbiota-derived hydrogen sulfide is converted to thiosulfate by colonic epithelial cells. Neutrophils that migrate into the lumen of the intestine during inflammation generate ROS that convert endogenous sulfur compounds (thiosulfate) into an electron acceptor (tetrathionate) that further boosts the growth of S. Typhimurium through tetrathionate respiration.
The creation of a niche for respiratory nutrients during inflammation is also an important driver of the strategies that pathogenic bacteria from the family Enterobacteriaceae use to invade the gut ecosystem. In the absence of inflammation or treatment with antibiotics, members of the gut microbiota occupy all available nutrient niches, which makes it very challenging for pathogenic Enterobacteriaceae to enter the community. One solution is for these bacteria to trigger intestinal inflammation, which would coerce the host into creating a fresh niche of respiratory nutrients that is suitable for its expansion — an approach that is used by S. Typhimurium93. On ingestion, S. Typhimurium uses T3SS-1 to invade the intestinal epithelium94 and T3SS-2 to survive in the tissue of the host95. Both of these processes trigger acute intestinal inflammation in cattle and in mouse models of gastroenteritis96–98 (Fig. 3). The inflammatory response of the host drives the expansion of S. Typhimurium in the lumen of the gut99, which is required for the transmission of this pathogenic species to a new host through the faecal–oral route100.
Although such expansion allows S. Typhimurium to side-step competition with obligate anaerobic Clostridia and Bacteroidia, this strategy forces the bacterium into battle with commensal Enterobacteriaceae over limited resources. For example, S. Typhimurium expands in the inflamed gut through nitrate respiration101,102, which results in rivalry with commensal Enterobacteriaceae that pursue a similar strategy91. S. Typhimurium can gain an edge in this competition through its ability to utilize a broader range of inflammation-derived electron acceptors than its rivals. A source of one such electron acceptor is sulfate-reducing species of Desulfovibrio from the microbiota, which release hydrogen sulfide, a compound that is converted to thiosulfate by the epithelium of the colon to avoid toxicity103. Deployment of the virulence factors of pathogenic bacteria leads to the recruitment of neutrophils to the intestinal mucosa, which is the histopathological hallmark of S. Typhimurium-induced gastroenteritis96. A fraction of these recruited neutrophils migrate into the lumen of the intestine — a diagnostic marker of inflammatory diarrhoea104. In the lumen, neutrophils help to protect the mucosa by engulfing bacteria in the vicinity of the epithelium105, but reactive oxygen species that are generated by the phagocyte-produced NADPH oxidase 2 (also known as cytochrome b-245 heavy chain) convert thiosulfate into tetrathionate, a respiratory electron acceptor that supports the expansion of S. Typhimurium in the lumen of the inflamed gut106 (Fig. 3). Although tetrathionate respiration is a characteristic of Salmonella serovars and has been used empirically in their isolation in clinical microbiology laboratories since 1923 (ref. 107), insights into the respiratory nutrient niche that Salmonella occupies suggest that this property is part of a strategy to edge out competing commensal Enterobacteriaceae in the inflamed gut106.
The inflammatory response of the host also ignites competition between commensal and pathogenic Enterobacteriaceae over trace elements such as iron, which is less available during inflammation. IL-22 induces the release of the antimicrobial protein lipocalin-2 (also known as neutrophil gelatinase-associated lipocalin) from the epithelium in mice and rhesus macaques108,109. Lipocalin-2 reduces iron availability by binding to enterobactin, a low-molecular-weight iron chelator (or siderophore) that is produced by Enterobacteriaceae110,111. To overcome this, S. Typhimurium and some commensal E. coli secrete a glycosylated derivative of enterobactin, termed salmochelin, which is not bound by lipocalin-2 (ref. 108). By producing salmochelin as well as two further siderophores that are not bound by lipocalin-2, yersiniobactin and aerobactin, the probiotic E. coli strain Nissle 1917 can limit the expansion of S. Typhimurium in the lumen of the inflamed gut112. Conversely, lipocalin-2 secretion by the epithelium generates an environment that enables S. Typhimurium to edge out commensal Enterobacteriaceae that depend solely on enterobactin for the acquisition of iron109 (Fig. 3).
Through its limitation of iron availability, intestinal inflammation also sets the stage for battles between Enterobacteriaceae that use protein-based toxins known as colicins113 that affect a narrow range of hosts. Iron limitation induces the synthesis of siderophore receptor proteins for the bacterial outer membrane113, which also commonly serve as receptors for colicins114–116. Expression of a siderophore receptor protein termed the colicin I receptor (CirA) confers commensal E. coli with sensitivity to colicin Ib produced by S. Typhimurium113. The respiratory nutrient niche that is generated by the inflammatory response of the host is therefore a battleground on which commensal and pathogenic Enterobacteriaceae struggle for dominance using a diverse arsenal of nutritional and antimicrobial strategies.
Perspective and the future
The study of the microbiome began more than a century ago. equencing of 16S rRNA genes provided the first insights into the taxonomic composition of microbial communities. Later, sequencing of the complete metagenome of microbial communities provided a more detailed insight into the full genetic capacity of such a community. The use of germ-free animals, either alone or in combination with emerging technologies such as laser-capture microdissection and transcriptomics, enabled mechanistic studies of the associations between the microbiota, the host and pathogenic bacteria117. Multi-taxon insertion sequencing now allows researchers to investigate both the assembly and the shared and strain-specific dietary requirements of communities of microbes, and it has also facilitated the informed manipulation of such communities through diet118. The development of quantitative imaging technologies has provided insight into the localization of microbes within the gastrointestinal tract, and it has also enabled studies on the proximity of and interactions between microbes119. The increasing refinement and power of metabolomics, imaging mass spectrometry and three-dimensional mapping of mass-spectrometry data provide a high-resolution image of the complex chemistry landscape of the interactions between microbes and the host, which sets the stage for manipulating this chemistry to prevent or treat infectious diseases24,38,120–127. A marriage of metagenomics and mathematical modelling promises to enhance the precision of microbiome reconstitution, which has proven successful for tackling C. difficile infections in mice128. In these exciting times, the expansion of multidisciplinary research is rapidly generating new technologies and mechanistic insights into interactions between the microbiota, the host and pathogenic bacteria (Box 1).
BOX 1. Microbiota interventions as therapeutic strategies to limit pathogen expansion.
An imbalance in the gut microbiota might underlie many human diseases but, in most cases, the development of treatment options is still in its infancy. This could be in part because the mechanisms that lead to adverse effects in the host differ for each disease, which means that intervention strategies must be developed for each. The treatment options for antibiotic-induced dysbiosis are perhaps the most advanced, mainly because faecal microbiota transplantation can reverse this imbalance in the gut microbiota129. Nonetheless, the mechanisms through which treatment with antibiotics encourages an uncontrolled expansion of the obligate anaerobe C. difficile differ markedly from those that stimulate the growth of the facultative anaerobes Enterobacteriaceae, which has implications for the development of precision microbiome interventions.
Mice that are treated with streptomycin have a reduced abundance of members of the class Clostridia130, which are credited with producing the lion’s share of the short-chain fatty acid butyrate in the large intestine131. The resulting depletion of short-chain fatty acids drives an expansion of Enterobacteriaceae through mechanisms that are not fully resolved44,132. Depletion of Clostridia-derived butyrate affects the metabolism of enterocytes in the colon, which derive most of their energy by butyrate respiration133. The depletion of short-chain fatty acids also leads to a contraction in the pool of regulatory T cells in the colonic mucosa134–136. These changes in the host physiology increase the inflammatory tone of the mucosa, as indicated by the elevated expression of Nos2, the gene that encodes inducible nitric oxide synthase, and contributes to the expansion of commensal E. coli through nitrate generation137. Although other mechanisms probably contribute to the post-antibiotic expansion of certain populations of bacteria in the gut126, the transfer of Clostridia, with their capacity for producing short-chain fatty acids, represents the most effective treatment for limiting the growth of E. coli in streptomycin-treated mice138.
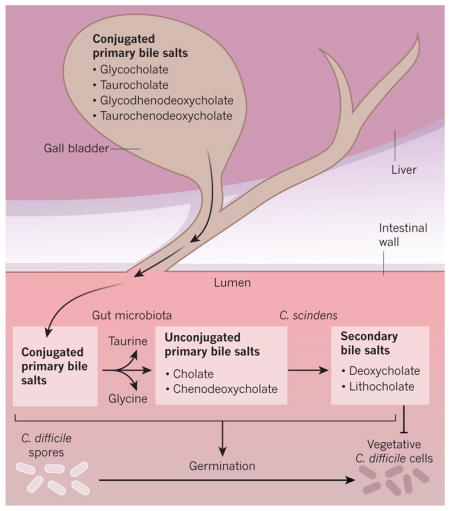
By contrast, the post-antibiotic expansion of the C. difficile population is driven by a depletion of secondary bile salts. The liver produces the primary bile salts cholate and chenodeoxycholate, which are conjugated to the amino acids taurine (to produce taurocholate and taurochenodeoxycholate) or glycine (to produce glycocholate and glycochenodeoxycholate) and then secreted into the gut. Bile salt hydrolases, enzymes that are produced by many members of the gut microbiota, remove the conjugated amino acid from the primary bile salt. C. scindens is one of a limited number of species of bacteria that can actively transport cholate and chenodeoxycholate into its cytosol, where these unconjugated primary bile salts are converted into the secondary bile salts deoxycholate and lithocholate, which are subsequently secreted into the extracellular environment139 (Box Fig.). Although both primary and secondary bile salts induce the germination of C. difficile spores, only secondary bile salts efficiently prevent the growth of vegetative C. difficile cells140. By significantly reducing the abundance of species that are capable of producing deoxycholate and lithocholate, treatment with antibiotics causes a depletion of these secondary bile salts and promotes the expansion of vegetative C. difficile cells in the large intestine141,142. Faecal microbiota transplantation restores the production of secondary bile salts and therefore prevents the expansion of C. difficile143. Direct supplementation of the diet with secondary bile salts warrants caution because increased concentrations of bile salts have been linked to gastrointestinal cancers144. However, inoculation with only the secondary-bile-salt-producing C. scindens confers mice with resistance to C. difficile expansion following treatment with antibiotics128. This remarkable observation opens the door to novel precision microbiome interventions that aim to prevent or treat the colitis that is associated with C. difficile infection after antibiotic therapy.
Acknowledgments
Work in the V.S. laboratory is supported by US National Institutes of Health (NIH) grants AI053067, AI077613, AI05135 and AI114511. Work in the A.J.B. laboratory is supported by US Department of Agriculture grant 2015-67015-22930 and NIH grants AI044170, AI096528, AI112445, AI114922 and AI117940. The contents of this Review are solely the responsibility of the authors and do not necessarily represent the official views of the NIH National Institute of Allergy and Infectious Diseases.
Footnotes
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of this paper at go.nature.com/28ix4vg.
References
- 1.Eckburg PB, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank DN, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The Human Microbiome Project Consortium. Structure function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 5.Yurist-Doutsch S, Arrieta MC, Vogt SL, Finlay BB. Gastrointestinal microbiota-mediated control of enteric pathogens. Annu Rev Genet. 2014;48:361–382. doi: 10.1146/annurev-genet-120213-092421. [DOI] [PubMed] [Google Scholar]
- 6.Sassone-Corsi M, Raffatellu M. No vacancy: how beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. J Immunol. 2015;194:4081–4087. doi: 10.4049/jimmunol.1403169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cameron EA, Sperandio V. Frenemies: signaling and nutritional integration in pathogen–microbiota–host interactions. Cell Host Microbe. 2015;18:275–284. doi: 10.1016/j.chom.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pacheco AR, Sperandio V. Enteric pathogens exploit the microbiota-generated nutritional environment of the gut. Microbiol Spectr. 2015;3 doi: 10.1128/microbiolspec.MBP-0001-2014. MBP-0001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohnhoff M, Drake BL, Miller CP. Effect of streptomycin on susceptibility of intestinal tract to experimental Salmonella infection. Proc Soc Exp Biol Med. 1954;86:132–137. doi: 10.3181/00379727-86-21030. [DOI] [PubMed] [Google Scholar]
- 10.Ferreira RB, et al. The intestinal microbiota plays a role in Salmonella-induced colitis independent of pathogen colonization. PLoS ONE. 2011;6:e20338. doi: 10.1371/journal.pone.0020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sprinz H, et al. The response of the germfree guinea pig to oral bacterial challenge with Escherichia coli and Shigella flexneri. Am J Pathol. 1961;39:681–695. [PMC free article] [PubMed] [Google Scholar]
- 12.Zachar Z, Savage DC. Microbial interference and colonization of the murine gastrointestinal tract by Listeria monocytogenes. Infect Immun. 1979;23:168–174. doi: 10.1128/iai.23.1.168-174.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamada N, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. This study showed that enteric pathogens use virulence genes to compete with the microbiota for nutrients in the gut. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh S, et al. Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am J Physiol Gastrointest Liver Physiol. 2011;301:G39–G49. doi: 10.1152/ajpgi.00509.2010. [DOI] [PubMed] [Google Scholar]
- 15.Willing BP, Vacharaksa A, Croxen M, Thanachayanont T, Finlay BB. Altering host resistance to infections through microbial transplantation. PLoS ONE. 2011;6:e26988. doi: 10.1371/journal.pone.0026988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kampmann C, Dicksved J, Engstrand L, Rautelin H. Composition of human faecal microbiota in resistance to Campylobacter infection. Clin Microbiol Infect. 2016;22:61.e1–61.e8. doi: 10.1016/j.cmi.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zumbrun SD, et al. Dietary choice affects Shiga toxin-producing Escherichia coli (STEC) O157:H7 colonization and disease. Proc Natl Acad Sci USA. 2013;110:E2126–E2133. doi: 10.1073/pnas.1222014110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wlodarska M, Willing BP, Bravo DM, Finlay BB. Phytonutrient diet supplementation promotes beneficial Clostridia species and intestinal mucus secretion resulting in protection against enteric infection. Sci Rep. 2015;5:9253. doi: 10.1038/srep09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Modi SR, Collins JJ, Relman DA. Antibiotics and the gut microbiota. J Clin Invest. 2014;124:4212–4218. doi: 10.1172/JCI72333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leffler DA, Lamont JT. Clostridium difficile infection. N Engl J Med. 2015;373:287–288. doi: 10.1056/NEJMc1506004. [DOI] [PubMed] [Google Scholar]
- 22.Pavia AT, et al. Epidemiologic evidence that prior antimicrobial exposure decreases resistance to infection by antimicrobial-sensitive Salmonella. J Infect Dis. 1990;161:255–260. doi: 10.1093/infdis/161.2.255. [DOI] [PubMed] [Google Scholar]
- 23.Ng KM, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502:96–99. doi: 10.1038/nature12503. This study showed that treatment with antibiotics enhances the abundance of host sialic acid that can be harvested by the microbiota, which promotes the expansion of enteric pathogens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreyra JA, et al. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe. 2014;16:770–777. doi: 10.1016/j.chom.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferreyra JA, Ng KM, Sonnenburg JL. The enteric two-step: nutritional strategies of bacterial pathogens within the gut. Cell Microbiol. 2014;16:993–1003. doi: 10.1111/cmi.12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rakoff-Nahoum S, Coyne MJ, Comstock LE. An ecological network of polysaccharide utilization among human intestinal symbionts. Curr Biol. 2014;24:40–49. doi: 10.1016/j.cub.2013.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alverdy J, Chi HS, Sheldon GF. The effect of parenteral nutrition on gastrointestinal immunity. The importance of enteral stimulation. Ann Surg. 1985;202:681–684. doi: 10.1097/00000658-198512000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischbach MA, Sonnenburg JL. Eating for two: how metabolism establishes interspecies interactions in the gut. Cell Host Microbe. 2011;10:336–347. doi: 10.1016/j.chom.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chow WL, Lee YK. Free fucose is a danger signal to human intestinal epithelial cells. Br J Nutr. 2008;99:449–454. doi: 10.1017/S0007114507812062. [DOI] [PubMed] [Google Scholar]
- 30.Bourlioux P, Koletzko B, Guarner F, Braesco V. The intestine and its microflora are partners for the protection of the host: report on the Danone Symposium “The Intelligent Intestine,” held in Paris, June 14, 2002. Am J Clin Nutr. 2003;78:675–683. doi: 10.1093/ajcn/78.4.675. [DOI] [PubMed] [Google Scholar]
- 31.Bry L, Falk PG, Midtvedt T, Gordon JI. A model of host–microbial interactions in an open mammalian ecosystem. Science. 1996;273:1380–1383. doi: 10.1126/science.273.5280.1380. [DOI] [PubMed] [Google Scholar]
- 32.Hooper LV, Xu J, Falk PG, Midtvedt T, Gordon JI. A molecular sensor that allows a gut commensal to control its nutrient foundation in a competitive ecosystem. Proc Natl Acad Sci USA. 1999;96:9833–9838. doi: 10.1073/pnas.96.17.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fabich AJ, et al. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun. 2008;76:1143–1152. doi: 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Autieri SM, et al. L-fucose stimulates utilization of D-ribose by Escherichia coli MG1655 ΔfucAO and E. coli Nissle 1917 ΔfucAO mutants in the mouse intestine and in M9 minimal medium. Infect Immun. 2007;75:5465–5475. doi: 10.1128/IAI.00822-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pacheco AR, et al. Fucose sensing regulates bacterial intestinal colonization. Nature. 2012;492:113–117. doi: 10.1038/nature11623. This paper showed that sugars from the mucus that are released by the microbiota can be perceived as signals by enteric pathogens to regulate the expression of virulence genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schauer DB, Falkow S. Attaching and effacing locus of a Citrobacter freundii biotype that causes transmissible murine colonic hyperplasia. Infect Immun. 1993;61:2486–2492. doi: 10.1128/iai.61.6.2486-2492.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szabady RL, Lokuta MA, Walters KB, Huttenlocher A, Welch RA. Modulation of neutrophil function by a secreted mucinase of Escherichia coli O157:H7. PLoS Pathog. 2009;5:e1000320. doi: 10.1371/journal.ppat.1000320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curtis MM, et al. The gut commensal Bacteroides thetaiotaomicron exacerbates enteric infection through modification of the metabolic landscape. Cell Host Microbe. 2014;16:759–769. doi: 10.1016/j.chom.2014.11.005. This study revealed that metabolites that are produced by the microbiota can be exploited as cues to increase the virulence of enteric pathogens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macy JM, Ljungdahl LG, Gottschalk G. Pathway of succinate and propionate formation in Bacteroides fragilis. J Bacteriol. 1978;134:84–91. doi: 10.1128/jb.134.1.84-91.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carter PB, Collins FM. The route of enteric infection in normal mice. J Exp Med. 1974;139:1189–1203. doi: 10.1084/jem.139.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawhon SD, Maurer R, Suyemoto M, Altier C. Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol Microbiol. 2002;46:1451–1464. doi: 10.1046/j.1365-2958.2002.03268.x. [DOI] [PubMed] [Google Scholar]
- 42.Takao M, Yen H, Tobe T. LeuO enhances butyrate-induced virulence expression through a positive regulatory loop in enterohaemorrhagic Escherichia coli. Mol Microbiol. 2014;93:1302–1313. doi: 10.1111/mmi.12737. [DOI] [PubMed] [Google Scholar]
- 43.Karmali MA, Petric M, Lim C, Fleming PC, Steele BT. Escherichia coli cytotoxin, haemolytic-uraemic syndrome, and haemorrhagic colitis. Lancet. 1983;2:1299–1300. doi: 10.1016/s0140-6736(83)91167-4. [DOI] [PubMed] [Google Scholar]
- 44.Fukuda S, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469:543–547. doi: 10.1038/nature09646. This study showed that the acetate that is produced by certain probiotic bacteria can enhance the barrier function of the gut epithelium to protect the host from enteric infections. [DOI] [PubMed] [Google Scholar]
- 45.Bertin Y, et al. Enterohaemorrhagic Escherichia coli gains a competitive advantage by using ethanolamine as a nitrogen source in the bovine intestinal content. Environ Microbiol. 2011;13:365–377. doi: 10.1111/j.1462-2920.2010.02334.x. [DOI] [PubMed] [Google Scholar]
- 46.Garsin DA. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nature Rev Microbiol. 2010;8:290–295. doi: 10.1038/nrmicro2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korbel JO, et al. Systematic association of genes to phenotypes by genome and literature mining. PLoS Biol. 2005;3:e134. doi: 10.1371/journal.pbio.0030134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thiennimitr P, et al. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci USA. 2011;108:17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joseph B, et al. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol. 2006;188:556–568. doi: 10.1128/JB.188.2.556-568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kendall MM, Gruber CC, Parker CT, Sperandio V. Ethanolamine controls expression of genes encoding components involved in interkingdom signaling and virulence in enterohemorrhagic Escherichia coli O157:H7. mBio. 2012;3:00050–12. doi: 10.1128/mBio.00050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson CJ, Clark DE, Adli M, Kendall MM. Ethanolamine signaling promotes Salmonella niche recognition and adaptation during infection. PLoS Pathog. 2015;11:e1005278. doi: 10.1371/journal.ppat.1005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maier L, et al. Microbiota-derived hydrogen fuels Salmonella Typhimurium invasion of the gut ecosystem. Cell Host Microbe. 2013;14:641–651. doi: 10.1016/j.chom.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 53.Maltby R, Leatham-Jensen MP, Gibson T, Cohen PS, Conway T. Nutritional basis for colonization resistance by human commensal Escherichia coli strains HS and Nissle 1917 against E. coli O157:H7 in the mouse intestine. PLoS ONE. 2013;8:e53957. doi: 10.1371/journal.pone.0053957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fabich AJ, et al. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun. 2008;76:1143–1152. doi: 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsiao A, et al. Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature. 2014;515:423–426. doi: 10.1038/nature13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hughes DT, et al. Chemical sensing in mammalian host–bacterial commensal associations. Proc Natl Acad Sci USA. 2010;107:9831–9836. doi: 10.1073/pnas.1002551107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bachmann V, et al. Bile salts modulate the mucin-activated type VI secretion system of pandemic Vibrio cholerae. PLoS Negl Trop Dis. 2015;9:e0004031. doi: 10.1371/journal.pntd.0004031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hörger S, Schultheiss G, Diener M. Segment-specific effects of epinephrine on ion transport in the colon of the rat. Am J Physiol. 1998;275:G1367–G1376. doi: 10.1152/ajpgi.1998.275.6.G1367. [DOI] [PubMed] [Google Scholar]
- 59.Yano JM, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015;161:264–276. doi: 10.1016/j.cell.2015.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Asano Y, et al. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1288–G1295. doi: 10.1152/ajpgi.00341.2012. [DOI] [PubMed] [Google Scholar]
- 61.Furness JB. Types of neurons in the enteric nervous system. J Auton Nerv Syst. 2000;81:87–96. doi: 10.1016/s0165-1838(00)00127-2. [DOI] [PubMed] [Google Scholar]
- 62.Curtis MM, Sperandio V. A complex relationship: the interaction among symbiotic microbes, invading pathogens, and their mammalian host. Mucosal Immunol. 2011;4:133–138. doi: 10.1038/mi.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moreira CG, Weinshenker D, Sperandio V. QseC mediates Salmonella enterica serovar Typhimurium virulence in vitro and in vivo. Infect Immun. 2010;78:914–926. doi: 10.1128/IAI.01038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sperandio V, Torres AG, Jarvis B, Nataro JP, Kaper JB. Bacteria–host communication: the language of hormones. Proc Natl Acad Sci USA. 2003;100:8951–8956. doi: 10.1073/pnas.1537100100. This paper describes how signals from the microbiota and host converge to enhance virulence in enteric pathogens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakano M, Takahashi A, Sakai Y, Nakaya Y. Modulation of pathogenicity with norepinephrine related to the type III secretion system of Vibrio parahaemolyticus. J Infect Dis. 2007;195:1353–1360. doi: 10.1086/513275. [DOI] [PubMed] [Google Scholar]
- 66.Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V. The QseC sensor kinase: a bacterial adrenergic receptor. Proc Natl Acad Sci USA. 2006;103:10420–10425. doi: 10.1073/pnas.0604343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reading NC, Rasko DA, Torres AG, Sperandio V. The two-component system QseEF and the membrane protein QseG link adrenergic and stress sensing to bacterial pathogenesis. Proc Natl Acad Sci USA. 2009;106:5889–5894. doi: 10.1073/pnas.0811409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seksik P, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut. 2003;52:237–242. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJ. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. 2006;44:4136–4141. doi: 10.1128/JCM.01004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baumgart M, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007;1:403–418. doi: 10.1038/ismej.2007.52. [DOI] [PubMed] [Google Scholar]
- 71.Walker AW, et al. High-throughput clone library analysis of the mucosaassociated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11:7. doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gevers D, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chiodini RJ, et al. Microbial population differentials between mucosal and submucosal intestinal tissues in advanced Crohn’s disease of the ileum. PLoS ONE. 2015;10:e0134382. doi: 10.1371/journal.pone.0134382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lupp C, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2:119–129. doi: 10.1016/j.chom.2007.06.010. corrigendum 2, 204 (2007). This paper reported that the induction of inflammation in the host disrupts the microbiota and promotes a bloom of Enterobacteriaceae. [DOI] [PubMed] [Google Scholar]
- 75.Garrett WS, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raffatellu M, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nature Med. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Godinez I, et al. T cells help to amplify inflammatory responses induced by Salmonella enterica serotype Typhimurium in the intestinal mucosa. Infect Immun. 2008;76:2008–2017. doi: 10.1128/IAI.01691-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pickard JM, et al. Rapid fucosylation of intestinal epithelium sustains host–commensal symbiosis in sickness. Nature. 2014;514:638–641. doi: 10.1038/nature13823. This paper showed that cytokines that are induced by enteric infection promote fucosylation in the host. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pham TA, et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe. 2014;16:504–516. doi: 10.1016/j.chom.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sonnenburg JL, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–1959. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 81.Ansong C, et al. Studying Salmonellae and Yersiniae host–pathogen interactions using integrated ’omics and modeling. Curr Top Microbiol Immunol. 2013;363:21–41. doi: 10.1007/82_2012_247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harper RW, et al. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 2005;579:4911–4917. doi: 10.1016/j.febslet.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 83.Haberman Y, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124:3617–3633. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Salzman A, et al. Induction and activity of nitric oxide synthase in cultured human intestinal epithelial monolayers. Am J Physiol. 1996;270:G565–G573. doi: 10.1152/ajpgi.1996.270.4.G565. [DOI] [PubMed] [Google Scholar]
- 85.Palmer RM, Rees DD, Ashton DS, Moncada S. L-arginine is the physiological precursor for the formation of nitric oxide in endothelium-dependent relaxation. Biochem Biophys Res Commun. 1988;153:1251–1256. doi: 10.1016/s0006-291x(88)81362-7. [DOI] [PubMed] [Google Scholar]
- 86.Lundberg JO, Weitzberg E, Lundberg JM, Alving K. Intragastric nitric oxide production in humans: measurements in expelled air. Gut. 1994;35:1543–1546. doi: 10.1136/gut.35.11.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Singer II, et al. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology. 1996;111:871–885. doi: 10.1016/s0016-5085(96)70055-0. [DOI] [PubMed] [Google Scholar]
- 88.Enocksson A, Lundberg J, Weitzberg E, Norrby-Teglund A, Svenungsson B. Rectal nitric oxide gas and stool cytokine levels during the course of infectious gastroenteritis. Clin Diagn Lab Immunol. 2004;11:250–254. doi: 10.1128/CDLI.11.2.250-254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bai P, et al. Protein tyrosine nitration and poly(ADP-ribose) polymerase activation in N-methyl-N-nitro-N-nitrosoguanidine-treated thymocytes: implication for cytotoxicity. Toxicol Lett. 2007;170:203–213. doi: 10.1016/j.toxlet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 90.Dudhgaonkar SP, Tandan SK, Kumar D, Raviprakash V, Kataria M. Influence of simultaneous inhibition of cyclooxygenase-2 and inducible nitric oxide synthase in experimental colitis in rats. Inflammopharmacology. 2007;15:188–195. doi: 10.1007/s10787-007-1603-3. [DOI] [PubMed] [Google Scholar]
- 91.Winter SE, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339:708–711. doi: 10.1126/science.1232467. This study revealed that nitrate respiration drives the expansion of commensal E. coli during colitis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Winter SE, Baumler AJ. Why related bacterial species bloom simultaneously in the gut: principles underlying the ‘like will to like’ concept. Cell Microbiol. 2014;16:179–184. doi: 10.1111/cmi.12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rivera-Chávez F, Baumler AJ. The pyromaniac inside you: Salmonella metabolism in the host gut. Annu Rev Microbiol. 2015;69:31–48. doi: 10.1146/annurev-micro-091014-104108. [DOI] [PubMed] [Google Scholar]
- 94.Galán JE, Curtiss R., III Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc Natl Acad Sci USA. 1989;86:6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hensel M, et al. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269:400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- 96.Tsolis RM, Adams LG, Ficht TA, Baumler AJ. Contribution of Salmonella typhimurium virulence factors to diarrheal disease in calves. Infect Immun. 1999;67:4879–4885. doi: 10.1128/iai.67.9.4879-4885.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barthel M, et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. 2003;71:2839–2858. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Coburn B, Li Y, Owen D, Vallance BA, Finlay BB. Salmonella enterica serovar Typhimurium pathogenicity island 2 is necessary for complete virulence in a mouse model of infectious enterocolitis. Infect Immun. 2005;73:3219–3227. doi: 10.1128/IAI.73.6.3219-3227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stecher B, et al. Salmonella enterica serovar Typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lawley TD, et al. Host transmission of Salmonella enterica serovar Typhimurium is controlled by virulence factors and indigenous intestinal microbiota. Infect Immun. 2008;76:403–416. doi: 10.1128/IAI.01189-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lopez CA, et al. Phage-mediated acquisition of a type III secreted effector protein boosts growth of Salmonella by nitrate respiration. mBio. 2012;3:e00143–12. doi: 10.1128/mBio.00143-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lopez CA, Rivera-Chavez F, Byndloss MX, Baumler AJ. The periplasmic nitrate reductase NapABC supports luminal growth of Salmonella enterica serovar Typhimurium during colitis. Infect Immun. 2015;83:3470–3478. doi: 10.1128/IAI.00351-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Levitt MD, Furne J, Springfield J, Suarez F, DeMaster E. Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J Clin Invest. 1999;104:1107–1114. doi: 10.1172/JCI7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Harris JC, Dupont HL, Hornick RB. Fecal leukocytes in diarrheal illness. Ann Intern Med. 1972;76:697–703. doi: 10.7326/0003-4819-76-5-697. [DOI] [PubMed] [Google Scholar]
- 105.Loetscher Y, et al. Salmonella transiently reside in luminal neutrophils in the inflamed gut. PLoS ONE. 2012;7:e34812. doi: 10.1371/journal.pone.0034812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Winter SE, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010;467:426–429. doi: 10.1038/nature09415. This paper showed that tetrathionate is a unique electron acceptor that is induced through inflammation that promotes expansion of S. Typhimurium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Muller HJ. Partial list of biological institutes and biologists doing experimental work in Russia at the present time. Science. 1923;57:472–473. doi: 10.1126/science.57.1477.472. [DOI] [PubMed] [Google Scholar]
- 108.Raffatellu M, et al. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe. 2009;5:476–486. doi: 10.1016/j.chom.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Behnsen J, et al. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity. 2014;40:262–273. doi: 10.1016/j.immuni.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goetz DH, et al. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 111.Flo TH, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 112.Deriu E, et al. Probiotic bacteria reduce Salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe. 2013;14:26–37. doi: 10.1016/j.chom.2013.06.007. This paper revealed that probiotic strains of E. coli restrict infection of the gut with S. Typhimurium by competing for sources of iron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cascales E, et al. Colicin biology. Microbiol Mol Biol Rev. 2007;71:158–229. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guterman SK. Colicin B: mode of action and inhibition by enterochelin. J Bacteriol. 1973;114:1217–1224. doi: 10.1128/jb.114.3.1217-1224.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cardelli J, Konisky J. Isolation and characterization of an Escherichia coli mutant tolerant to colicins Ia and Ib. J Bacteriol. 1974;119:379–385. doi: 10.1128/jb.119.2.379-385.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Patzer SI, Baquero MR, Bravo D, Moreno F, Hantke K. The colicin G, H and X determinants encode microcins M and H47, which might utilize the catecholate siderophore receptors FepA, Cir, Fiu and IroN. Microbiology. 2003;149:2557–2570. doi: 10.1099/mic.0.26396-0. [DOI] [PubMed] [Google Scholar]
- 117.Hooper LV, et al. Molecular analysis of commensal host–microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. This was the first report to show that the microbiota can change the expression of mammalian genes to modulate the immune system of the host. [DOI] [PubMed] [Google Scholar]
- 118.Wu M, et al. Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science. 2015;350:aac5992. doi: 10.1126/science.aac5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Earle KA, et al. Quantitative imaging of gut microbiota spatial organization. Cell Host Microbe. 2015;18:478–488. doi: 10.1016/j.chom.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bouslimani A, et al. Molecular cartography of the human skin surface in 3D. Proc Natl Acad Sci USA. 2015;112:E2120–E2129. doi: 10.1073/pnas.1424409112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Marcobal A, et al. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 2013;7:1933–1943. doi: 10.1038/ismej.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lau W, Fischbach MA, Osbourn A, Sattely ES. Key applications of plant metabolic engineering. PLoS Biol. 2014;12:e1001879. doi: 10.1371/journal.pbio.1001879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marcobal A, et al. Metabolome progression during early gut microbial colonization of gnotobiotic mice. Sci Rep. 2015;5:11589. doi: 10.1038/srep11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rath CM, et al. Molecular analysis of model gut microbiotas by imaging mass spectrometry and nanodesorption electrospray ionization reveals dietary metabolite transformations. Anal Chem. 2012;84:9259–9267. doi: 10.1021/ac302039u. This study used imaging mass spectrometry to map differences in the gut metabolic landscape that are promoted by the microbiota. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dorrestein PC, Mazmanian SK, Knight R. Finding the missing links among metabolites, microbes, and the host. Immunity. 2014;40:824–832. doi: 10.1016/j.immuni.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Antunes LC, et al. Antivirulence activity of the human gut metabolome. mBio. 2014;5:e01183–14. doi: 10.1128/mBio.01183-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Antunes LC, et al. Impact of Salmonella infection on host hormone metabolism revealed by metabolomics. Infect Immun. 2011;79:1759–1769. doi: 10.1128/IAI.01373-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Buffie CG, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. doi: 10.1038/nature13828. This paper showed that the introduction of a single species of bacteria that can produce secondary bile salts confers the host with resistance to the expansion of C. difficile after treatment with antibiotics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Koenigsknecht MJ, Young VB. Faecal microbiota transplantation for the treatment of recurrent Clostridium difficile infection: current promise and future needs. Curr Opin Gastroenterol. 2013;29:628–632. doi: 10.1097/MOG.0b013e328365d326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sekirov I, et al. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect Immun. 2008;76:4726–4736. doi: 10.1128/IAI.00319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Louis E, Libioulle C, Reenaers C, Belaiche J, Georges M. Genomics of inflammatory bowel diseases: basis for a new molecular classification and new therapeutic strategies of these diseases [in French] Rev Med Liege. 2009;64:S1, 24–28. [PubMed] [Google Scholar]
- 132.Meynell GG. Antibacterial mechanisms of the mouse gut. II. The role of Eh and volatile fatty acids in the normal gut. Br J Exp Pathol. 1963;44:209–219. [PMC free article] [PubMed] [Google Scholar]
- 133.Donohoe DR, Wali A, Brylawski BP, Bultman SJ. Microbial regulation of glucose metabolism and cell-cycle progression in mammalian colonocytes. PLoS ONE. 2012;7:e46589. doi: 10.1371/journal.pone.0046589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Atarashi K, Umesaki Y, Honda K. Microbiotal influence on T cell subset development. Semin Immunol. 2011;23:146–153. doi: 10.1016/j.smim.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 135.Atarashi K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 136.Furusawa Y, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 137.Spees AM, et al. Streptomycin-induced inflammation enhances Escherichia coli gut colonization through nitrate respiration. mBio. 2013;4:e00430–13. doi: 10.1128/mBio.00430-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Itoh K, Freter R. Control of Escherichia coli populations by a combination of indigenous clostridia and lactobacilli in gnotobiotic mice and continuous-flow cultures. Infect Immun. 1989;57:559–565. doi: 10.1128/iai.57.2.559-565.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wells JE, Hylemon PB. Identification and characterization of a bile acid 7α-dehydroxylation operon in Clostridium sp. strain TO-931, a highly active 7α-dehydroxylating strain isolated from human feces. Appl Environ Microbiol. 2000;66:1107–1113. doi: 10.1128/aem.66.3.1107-1113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wilson KH. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J Clin Microbiol. 1983;18:1017–1019. doi: 10.1128/jcm.18.4.1017-1019.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol. 2008;190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Theriot CM, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nature Commun. 2014;5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Weingarden AR, et al. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am J Physiol Gastrointest Liver Physiol. 2014;306:G310–G319. doi: 10.1152/ajpgi.00282.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. 2005;589:47–65. doi: 10.1016/j.mrrev.2004.08.001. [DOI] [PubMed] [Google Scholar]