

Abstract
Edges are special sites in nanomaterials. The atoms residing on the edges have different environments compared to those in other parts of a nanomaterial and, therefore, they may have different properties. Here, recent progress in nanomaterial fields is summarized from the viewpoint of the edges. Typically, edge sites in MoS2 or metals, other than surface atoms, can perform as active centers for catalytic reactions, so the method to enhance performance lies in the optimization of the edge structures. The edges of multicomponent interfaces present even more possibilities to enhance the activities of nanomaterials. Nanoframes and ultrathin nanowires have similarities to conventional edges of nanoparticles, the application of which as catalysts can help to reduce the use of costly materials. Looking beyond this, the edge structures of graphene are also essential for their properties. In short, the edge structure can influence many properties of materials.
Keywords: nanomaterial morphology, catalytic active centers, interfaces, nanoframes, ultrathin nanowires
1. Introduction
This is an open access article under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Tailoring the morphologies of nanomaterials has achieved great success in catalyst optimization. What are the key factors to tune the catalytic properties? The answers may be complicated. From the view of band theory, the morphology varies the energy bands of particles.1 From the point of chemistry, size effect in performance is related with higher content of surface atoms.2 However, these kinds of explanations are not always suitable in every case. Band theory is usually applied in reactions with light as the energy input.3 Particles with smaller size possess larger amount of surface atoms, however, it is not always true that smaller particles show better performance.4 Unveiling the mystery underneath the high catalytic activity is a tough but meaningful and valuable work. From the view of geometry, any particles are enclosed by several facets, the intersection of facets is an edge, and the intersection of two edges is a corner. Tuning the morphology is actually tuning the facets, edges and corners. There are many reviews concerning the facets of nanoparticles as key points for properties,5, 6, 7 however, the edges are less discussed.
Atoms on edges usually show different properties comparing with atoms on inner faces (here “inner” refers to “on the surface but not on the edge”). Increasing the content of edge sites may help to further optimize the activities for catalysts. It is well accepted that edges of MoS2, other than basal plane, are active for many catalytic reactions.8, 9 This is not an isolate case, and the edges of noble metals are also linked with high activities. Looking beyond this, the edge structure of graphene plays a vital role for determining the energy band and other related properties.10 Here we consider that the edges are keys for catalytic reactions to organize the review. At first, we begin with an overall picture of nanoparticles (NPs) that serve as catalysts, then several examples of conventional edges in nanomaterials (which refer to natural boundaries of surfaces) as active sites are presented in section 3. Here we mainly discuss reactions such as hydrogen evolution reaction, which is important for energy conversion, and CO2 reduction, which is essential for environment protection. We do not limit other scope in homogeneous NPs, the edge sites in heterostructures are more intricate and an introduction to these is presented. Many catalytic reactions require cooperation between various components, thus the activities may be ascribed to the novel interface. However, the lattices on both sides of the interface would greatly hinder the approach of substrates, thus almost no activity could be found at the interior interface. This is to say that the edges of interface are pivotal for activities, so we also review this area. There are also other kinds of edge structures which cannot be simply explained as natural boundary of surfaces. If the edges of NPs are active for applications, an intuitional method to reduce the use of expensive catalyst is to construct nanoframes (NFs) which only contain edges. NFs, together with ultrathin nanowires (UNWs), are considered as atypical edges of novel structures in this review. Untangling the intersecting lines (edges) in NFs can lead to nanowires (NWs). UNWs are an emerging hot topic of material fabrication and applications. However, the detailed structures of UNWs are far from well‐known, so it is hard to determine whether UNWs could resemble conventional edges in nanoparticles or not now. But with the fast developing speed of high resolution and non‐destructive analytic methods, we should be confident that the answer would be soon solved. In the sections afterward, we present some areas concerning edges not only for the application as catalyst, edges of graphene could determine various properties. The researches on their atomic structures may shed light on research of other materials' edge structures. In conclusion, there are many types of edge structures, a comprehensive review of them could help with their applications.
2. Catalysts and Nanoparticles
Chemical reactions happen almost everywhere from life to industry. Among them, catalysts play vital roles in most situations. Active enzymes which could accelerate reactions in cells ensure that organs could function. Industrial catalysts from petroleum refining to car exhaust catalysts are key steps to convenient modern life. Catalysts are also essential in academic research. Proper catalysts could help to activate C‐H bonds, couple new C‐C bonds in desired position, and alter function groups.11 These kinds of reactions are usually achieved via the introduction of soluble metal complex in the same phase with the reactants, which are called homogeneous catalysts. Homogeneous catalysts have a number of advantages such as high selectivity, good yield and easy to optimize. The active center of homogeneous catalysts is always related with the metal center, and its coordinated environments, which make them easy to optimize. However, catalysts for industry also require easy recovery or recycle. Many homogeneous catalytic systems cannot be commercialized because of the difficulties encountered in separating the catalyst from the final products.12 Otherwise the metal contamination could affect the safety of products, especially for the pharmaceutical industry. This disobeys the requirements of green chemistry.13
Heterogeneous catalysts that react with reactants in different phases might be a promising method to overcome the obstacle of separation while maintaining the yield of products. In fact, there are many important industrial homogeneous catalysts in biphasic systems,14 which are fixed on supports (usually are “metal on support” catalysts), in order to suit the demand of easy separation. Another hot topic of heterogeneous catalysts is using NPs to serve as highly active catalysts.15 Academic researches of heterogeneous catalysis aim to realize undeveloped reactions16, 17 or optimize the activities of developing reactions.18, 19, 20 The boundary between homogeneous or heterogeneous is the size of catalyst. While the functional dimension of a chemical bond in reaction typically has a length of about 0.2 nm, a larger catalyst would not do much help to combine atoms which are too far from each other if the migration of atoms are hampered, which is usually true in real reactions.21
The catalysis effect rellies on the lowering of activation energy, by altering the intermediated steps of reaction. This process usually involves the binding of substrates or intermediates with catalysts, the release of the following intermediates or products and the recycle of catalysts. When the proper metal core is selected, optimization of homogeneous catalyst relies mainly on altering the ligands around metal to tune the electronic structure to achieve suitable binding or releasing. For “metal on support” catalyst, to tune the performance requires the optimization of metals and selection of supports, while the supports are not innocent during catalytic reactions in many cases.22, 23, 24 For NPs as catalysts, the optimizations of their performances are mainly achieved by tailoring their morphologies. Energy band of crystal stems from the Bragg diffraction of electron, thus tuning the sizes and morphologies could tune the electronic structures of nanocrystals and influence the activities. On the other hand, the large amount of surface atoms could enhance the regional catalyst concentration. As researches go deeper, cooperation between various metals are found to be important in reactions.25 Many natural enzymes possess multi‐metal core, such as nitrogenase.26 The cooperation of various metals is also true in organic synthesis.27 NPs feature vast atoms on surface, cooperation between them are inevitable, and the distance between surface atoms could be tailored by exposing different facets. So using NPs as catalyst are easy to optimize, thus they are promising for application.
However, there are still some obstacles hindering the realization of NPs as high efficient catalysts. First of all, the capping agents on NPs' surfaces, which are used in most NP synthesis, may prevent the contact between reactant and particles, thus lowering the efficiency. Secondly, the deterioration of NPs may hamper the recycle of catalysts. When noble metals are used as catalysts, the internal atoms seem useless in reaction, however, increase the total cost of materials. Last but not the least, the real mechanisms of reactions are unknown in many cases, thus add extra difficulties for optimizing. In short, the active center of NPs need to be debunked. With the knowledge of active center, rational optimization or design of high efficient catalysts is not impossible.
When NPs are used as catalysts, the surface atoms are usually considered as active centers. They are able to contact and combine with substrates, and reactions may happen at surface under suitable conditions. Thus the manner that substrates bind with NPs may influence the yield and selectivity of reactions.28 The surface structure of NPs could also affect the contact with substrate.29 The composition of composite NPs also complicates the detailing of active centers. These factors make the study of active centers more complicated.
To combine with substrates, the surface atoms must have unsaturated coordination, the coordination number (CN) may dramatically influence performance. Edges and corners of NPs feature lower CN than atoms on the surface, thus increasing the amount of them can help to tune the activity. Considering the very low content of corner sites of NPs, edges are better solutions to enhance the activities of catalysts. On the other hand, the chemical environment of edges in nanosheets or multicomponent interfaces is different with other parts, thus could be utilized to tune the electronic structure, as well as catalytic performance.
3. Edges as Active Sites
Here we present some prominent works that utilize the edges of nanomaterials as active centers, the optimization of edge structure could greatly enhance the performance of materials. In this section, we introduce three types of conventional edges in nanostructures, and a brief summary of them is presented in Table 1 . Taking MoS2 nanosheets as an example of 2D structure, the edges of them mainly refer to the boundary of sheets, and their chemical environments are different with atoms on basal plane because of the termination of atoms. The unique structure makes edges of 2D materials special and could be utilized as catalyst. For NPs, edges are the intersections of surfaces, they feature low CN and are active to bind some of the substrates or intermediates in catalytic reactions. As for the edges of multicomponent interface, they are specialized as the external boundary of conjunction of different components. The contact of two particles makes a two dimensional interface, so the edges of them may feature similar characteristics with 2D structures, such as a special chemical environment. The “basal plane” of interface is not approachable due to the existence of particles, thus cannot simply be used as active sites. The more details and methods to enhance the relative catalytic performance of these three types of edges are present in the following discussions.
Table 1.
A brief comparison of conventional edges
| 2D structure | Nanoparticles | Multicomponent interface | |
|---|---|---|---|
| Structural feature | The boundary of sheet | The intersection of surfaces | The external boundary of conjunction of different components |
| Chemical feature | Special chemical environment | Low CN | Special chemical environment |
| Method to enhance catalytic properties | Tune the structure; expose more edges | Enlarge the edge content | Select proper components; tune the conjunction manner; optimize the edge content |
3.1. MoS2 and Hydrogen Evolution Reaction
Searching for cheap and high efficient catalysts of hydrogen evolution reaction (HER) is required for water splitting and energy storage,30 lots of experimental31 and computational32 works have been done in this area. The noble metal, Pt, shows perfect activity for HER, however it is expensive and scarcity on earth. Meanwhile some natural enzymes which contains much less noble metals are also effective HER catalysts.33, 34 By comparing the active center of nitrogenase with different inorganic compounds, the Nørskov group found that the MoS2 edge possess similar structure and was chosen as a candidate for HER.35 From then on, vast researches of MoS2 for HER have been carried out to identify the active sites and further enhance the activities. In order to figure out the active sites, a combination of surface sensitive methods and reactivity studies must be sophisticatedly designed.36 Fresh and clean MoS2 polygonal single layer sheets with different sizes and area coverages could be obtained by a physical vapor deposition and followed by annealing.37 After characterizing by scanning tunneling microscopy (STM) under ultrahigh vacuum chamber (Figure 1 a) to get structure information, the products was tested for HER activity respectively. Figure 1c is the activities indicated by the exchange current density, larger value indicates a better performance. For sheets with different sizes, the exchange current densities are quite different, however it shows almost no relationship between basal sizes and activities, the reason is that the basal plane of MoS2 has no catalytic activity.8 By using edge length as the abscissa, a clear linear relationship appears, higher edge length shows higher activity. This direct and well‐designed experiment clearly identified that the edge of MoS2 is activity sites for HER.
Figure 1.
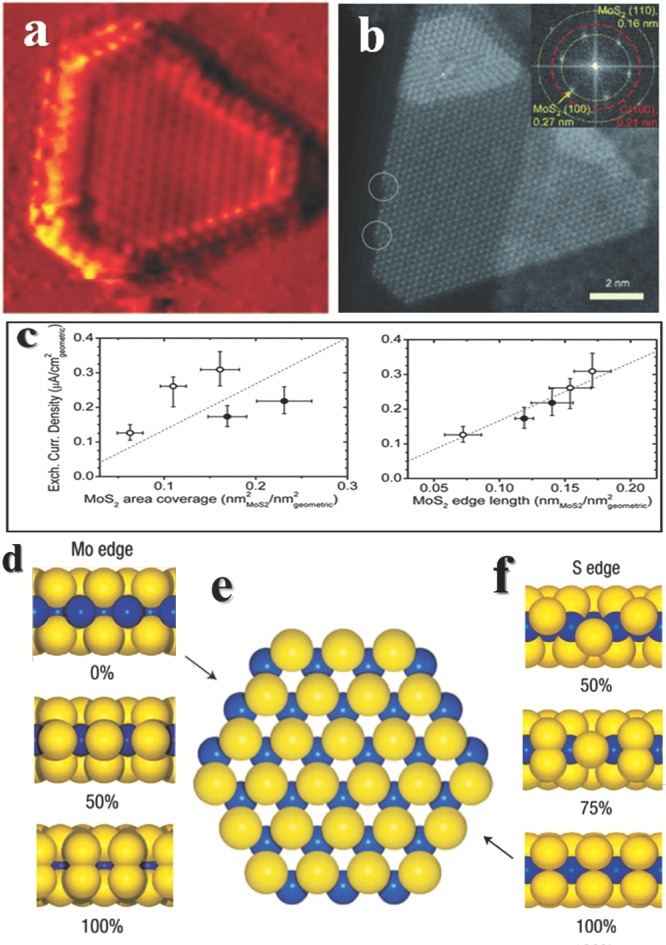
The structures of MoS2 and their activities for HER. a) Atomically resolved MoS2 particle by STM and c) their exchange current density versus MoS2 area coverage (left) or MoS2 edge length (right). In both figures, open circles and filled circles are samples prepared under different conditions. The exchange current density does not correlate with the area coverage of MoS2, whereas it shows a linear dependence on the MoS2 edge length. Reproduced with permission.36 Copyright 2007, American Association for the Advancement of Science. b) High‐resolution STEM image of an industrial MoS2 supported on a graphite support. The insert is a fast Fourier transform (FFT) of the image and shows hexagonally arranged spots at the 0.27 nm and 0.16 nm lattice distances, corresponding to the MoS2 (100) and (110) lattice planes, respectively. Moreover, a hexagonal set of lattice distances at 0.21 nm, corresponding to graphite (100), is also revealed. At the low‐indexed edges, kink sites may be present as indicated by white circles. The observation matches the 50% sulfur covered Mo edge structure. Reproduced with permission.39 d) Side view of the Mo edge, showing the fully stripped 0% edge and two almost equally stable edge configurations with 50% or 100% sulphur coverage, respectively. e) Ball model of the hexagonal MoS2 nanocluster. f) Side view of the S edge, showing three configurations representing 50%, 75% or 100% sulphur coverage, respectively. Reproduced with permission.42 Copyright 2007, Macmillan Publishers Ltd.
It is well known that enzymes must adopt a specific configuration to achieve efficient catalytic performances, unravelling details of MoS2 edge structures would gain our knowledge to better understand the reactions. 2D MoS2 nanosheets can exist in various polymorphs, natural MoS2 favors an edge‐sharing MoS6 trigonal prisms,38 named 2H phase. For such configuration, two types of low‐index edge terminations are possible, one is (100) Mo and the other is (010) S edges (Figure 1d‐f), but a triangular product implies only one edge type is energetically favored in fact. As for industrial‐style MoS2, high resolution scanning transmission electron microscopy (STEM) reveals the nanocatalyst suits the 50% sulfur cover Mo edge structures (Figure 1b).39 The edges of single‐layer MoS2 have two localized metallic states which indicate subtle electronic structure changes close to the edges,40 and result in the light lines in STM images. More STM studies reveal the coordination structures of Mo‐S at edge are a little different with bulk MoS2, a so‐called “S2 dimer” is formed by two outermost S atoms which contract in the direction perpendicular to the basal plane.41 Not to our surprise, the structures of MoS2 with different sizes are not exact the same, they possess different Mo or S edge coverage. A rearrangement of the S atoms at edges would influence bonding strength of Mo‐S, thus changes the stability and catalytic activity.42 Another phase of MoS2 is called 1T phase, which is constructed by edge‐sharing MoS6 octahedra. It is a metastable structure, and can be prepared by ultrasound‐promoted hydration of lithium‐intercalated compounds.43
This 1T‐MoS2 can dramatically decrease the Tafel slope comparing with 2H‐MoS2 in the same acidic environment (Figure 2 f).18 However, the best way to optimizing activity is not keeping on exploring 1T‐MoS2 or some other unstable structures,44 but on other practical methods to maintain high stability while keeping accessible activity. There are three basic and comprehensive rules to design more active HER catalysts45: increasing the activity of single active site, or increasing the amount of active sites, and the third rule is to increase the conductivity of catalyst. 1T‐MoS2 may be activity for HER, but it is not stable, thus greatly hinder the real use. Another way is to design new catalyst with similar active center, since we already know that the active sites are edges and we already know their structures. The research began from the mimic of nitrogenase, and now can go back to small molecule to optimize the active center. A large ligand called PY5Me2 [PY5Me2 = 2,6‐bis(1,1‐bis(2‐pyridyl)ethyl)pyridine] could generate a new HER catalyst with Mo and S2 dimmer (Figure 2d).46 This alternative strategy may open new gate for catalyst design. The second rule seems easier to achieve: Jaramillo group synthesized a highly ordered double‐gyroid MoS2 bicontinuous network with nanoscale pores, the high surface curvature exposes abundant edges47; Xie group prepared defect‐rich MoS2 nanosheets which force the formation of edges (Figure 2b).48 As for enhancing the conductivity, Dai group fabricated a strong coupling MoS2–graphene sheets, and the conducting network facilitate the catalytic reaction (Figure 2a).49
Figure 2.
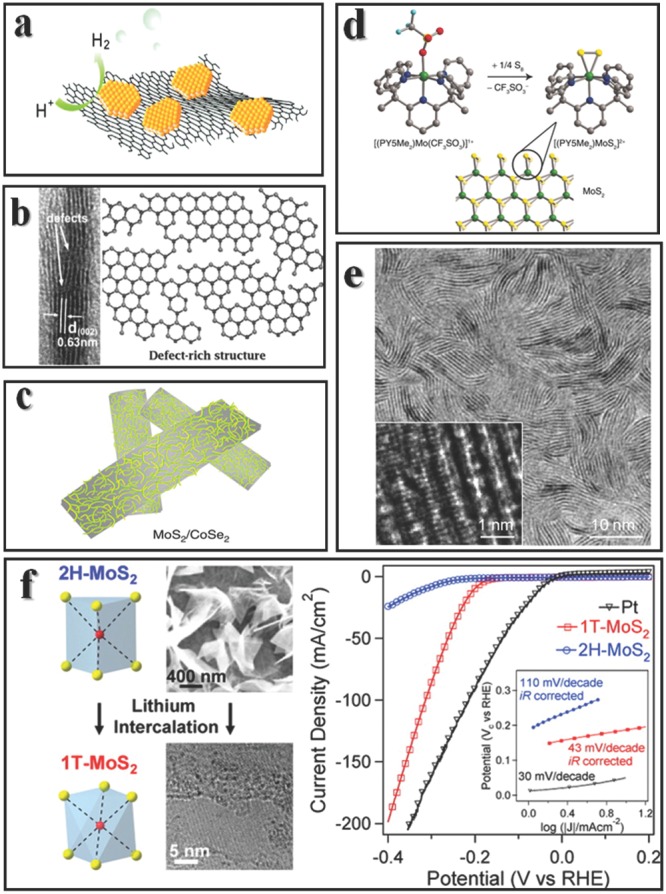
Methods to optimize HER properties. a) MoS2 grown on graphene can help to increase the conductivity. Reproduced with permission.49 Copyright 2011, American Chemistry Society. b) Defect rich structure can create more edges. Reproduced with permission.48 c) Schematic representation of MoS2 edges on CoSe2 belts, this is the best HER catalyst among non‐noble metal to date. Reproduced with permission.54 Copyright 2015, Macmillan Publishers Ltd. d) A molecular mimic of MoS2 edges. Reproduced with permission.46 Copyright 2012, American Associaton for the Advancement of Science. e) Verticle aligned MoS2 structures. Reproduced with permission.51 Copyritht 2013, American Chemical Society. f) 1T phase of MoS2 can be prepared via lithium intercalation in 2H phase (left), the performances are shown at right. Reproduced with permission.18 Copyright 2013, American Chemical Society.
Synthesis of large area thin MoS2 is not easy,50 fortunately highly active HER catalysts demand vast edges but not large area. Vertical aligned MoS2 layers grown on electrode would maximally expose edges as well as increasing the conductivity, although they are not easy to fabricate. The edges are thermodynamically unfavorable with basal plane by 2 orders of magnitude of surface energy, the vertical aligned structure could still be formed through a kinetically controlled rapid growth method (Figure 2e).51 In the chemical vapor deposition MoS2 synthesis, chemical conversion is a fast step comparing with gas diffusion, if diffusion along the layers through basal plane is much faster than diffusion across layers, then vertical growth is favored. The vertical aligned layers indeed enhanced the activity.52 Meanwhile such vertical structures are best platforms to investigate the edge properties, transition metal could dope at edge sites but not on basal planes,53 and Li2S nanoparticles mainly deposit on the edges versus basal planes in Li‐S batteries test. To date, the best HER catalyst among non‐noble metal is constructed by in situ growth of MoS2 edges on CoSe2 belt (Figure 2c), which possesses onset potential of ‐11 mV and Tafel slope of 36 mV dec‐1, comparable to commercial Pt/C catalyst.54
Another practical method to decorate edges in layered structures is achieved via edge overgrowth through screw dislocation driven growth55 (Figure 3 ). Crystals can be enlarged through screw dislocations under proper supersaturations in two competing directions, directions of in‐plane and vertical to plane.56 For layered structures, the growth rates of these two directions are inherently different, thus the growth manner can be proper controlled. By introducing a growth inhibitor in the growth of layered structure, the growth rate in direction of vertical to plane might be more affected than direction of in‐plane. Then after the depletion of growth inhibitor in reaction media, the growth rate of vertical to plane is released and the edge overgrowth might happen. This is a promising method to sophisticatedly design the edge structures of layered materials. As a proof of concept, ultrathin spiral nanosheets with overgrown edges of NiFe, CoFe and CoNi bimetallic hydroxides are achieved now, and they show promising electrochemical activities for oxygen evolution reaction. The growth mechanism and influencing parameters are easy to accessible, and taking the achievement of several other spiral layered structures57, 58 into account, the fabrication of more spiral nanosheets with overgrown edges can be foreseen in the future. Since structure determines properties, the properties of such novel structures might be quite interesting.
Figure 3.
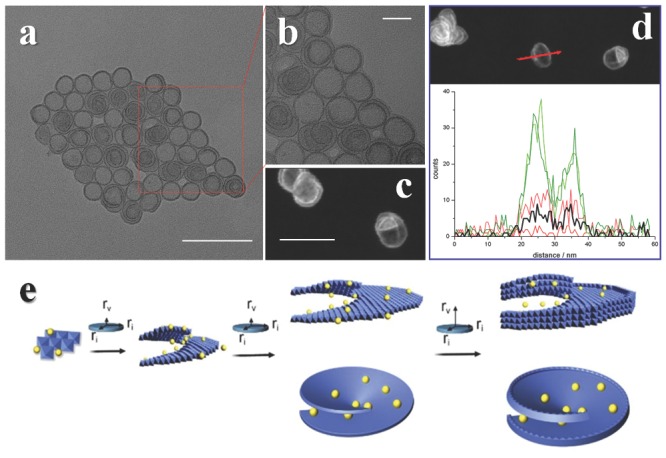
Spiral ultrathin nanosheets with overgrown edges (SUNOE). a,b) TEM and c,d) STEM images of NiFe SUNOE, the element line scan (d, down) indicates the ratios of metal to growth inhibitor vary with position (black line stands for growth inhibitor and other lines stands for metals). e) Growth mechanism of SUNOE, yellow balls represent growth inhibitor, after the concentration of inhibitors in solution decrease a lot, the growth rate of vertical to plane is released and cause an overgrowth at edges. Scale bars: a) 100 nm; b) 20 nm; and c) 50 nm. Reproduced with permission.55 Copyright 2015, Royal Society of Chemistry.
The edges of MoS2 can do more than HER. They can be used in catalyzing hydrodesulfurization,8 and methane conversion.59 More researches need to be done in designing new structures utilizing the convenience of edge properties and searching new catalytic reactions. Other transition metal dichalogenides (TMD) also experimentally showed promising activities for HER.60, 61 Meanwhile theoretical study suggests the edge sites of MoSe2 and WSe2 should be comparable or even better performance than MoS2 for HER.62 However, the exact edge structures of such compounds under reaction conditions still need to be more explored in order to link with properties.
3.2. Metals and CO2 Conversion
Au may be the most fascinating metal in history as it can be used as coin metal, mainly because it is inert and scarce on earth. Early experiments using gold as catalyst did not show superior to other catalysts.63 But now gold has become a never‐ending frontier in homogeneous or heterogeneous catalyst since the finding of low‐temperature oxidation of CO.64 One of the most concerning worldwide problems in the rapidly developing world is the overproduction of the greenhouse gas carbon dioxide led by ever‐increasing worldwide consumption of fossil fuels.65 The perfect solution may be conversion of CO2 to other useful compounds by using sunlight,66, 67, 68 like what has been done by plants. However efficient catalysts are still in fancy. Finding a feasible method to converse CO2 to other carbon forms open a new area for catalyst research. So far, electrochemical reduction of CO2 is considered as a promising method and Au is a good platform for mechanism study.
By thermodynamic calculation, the half reactions potential electrochemical reduction of CO2 to CO is –0.1 V, but experimentally very negative potential (usually –0.6 V to –1 V) must be applied to initiate CO2 reduction. Meanwhile such large negative potential would also trigger the evolution of H2 since the half reaction potential to form H2 is 0 V. The sluggish in overpotential is chiefly caused by the multi‐step processes of reduction, density function theory (DFT) studies suggests the mechanism include the following steps69, 70, 71, 72:
| (1) |
| (2) |
| (3) |
where * denotes a free active site. From these elementary reactions, we can intuitively conclude that materials which could both stabilize COOH* and destabilize CO* would be a prominent catalyst for reduction of CO2 to CO.
Many efforts have been made to search high active catalysts and determine active sites in reactions. Small Au25 clusters experimentally show much better performance than bulk Au particles, and computational data illustrate the banding energy of CO2 with Au cluster vary from sites to sites.73 For Au particles with different sizes, the ratios of atoms at edges or surface are different. An optimized ratio is required to reconcile the requirement of maximizing active sites for CO2 reduction and minimizing the active sites for HER. Sun group4 produced monodisperse Au NPs with different sizes, and found the 8 nm Au particles showed maximum Faradic efficiency (FE) at –0.67 V to convert to CO (Figure 4 a,b). The 8 nm Au particles with 4 nm crystallite diameter appeared to meet the demand for optimizing the edge fraction. While Cuenya group found that Au particles below 5 nm all showed significantly better performance than bulk Au, and particles larger than 5 nm were similar with bulk gold (Figure 4c,d).74 The disparate activities in these two results may stem from different methods for measurement of size, Sun used transmission electron microscopy (TEM) while Cuenya used atomic force microscope (AFM). However, a similar conclusion made in this two cases is that the edge sites or low coordinate sites are active for reactions, the method to further enhance the activities may be enlarge the fraction of edges in particles or create more edges. This could be achieved by ultrathin nanowires (UNWs), ultrathin structure would maximally expose edges and long nanowire would further elongate the edges (Figure 4e). The ultrathin Au nanowire with diameter of 2.1 nm and length of 500 nm reaches 94% of FE at relative low potential (–0.35 V) (Figure 4f,g),75 putting forward the realization of CO2 reduction. In computation results, Au(211) is the best for COOH* stabilization, but the binding energy of CO* offset the advantage (Figure 4h). Taking the whole energy effects into account, ultrathin nanowire is a promising candidate for CO formation.
Figure 4.
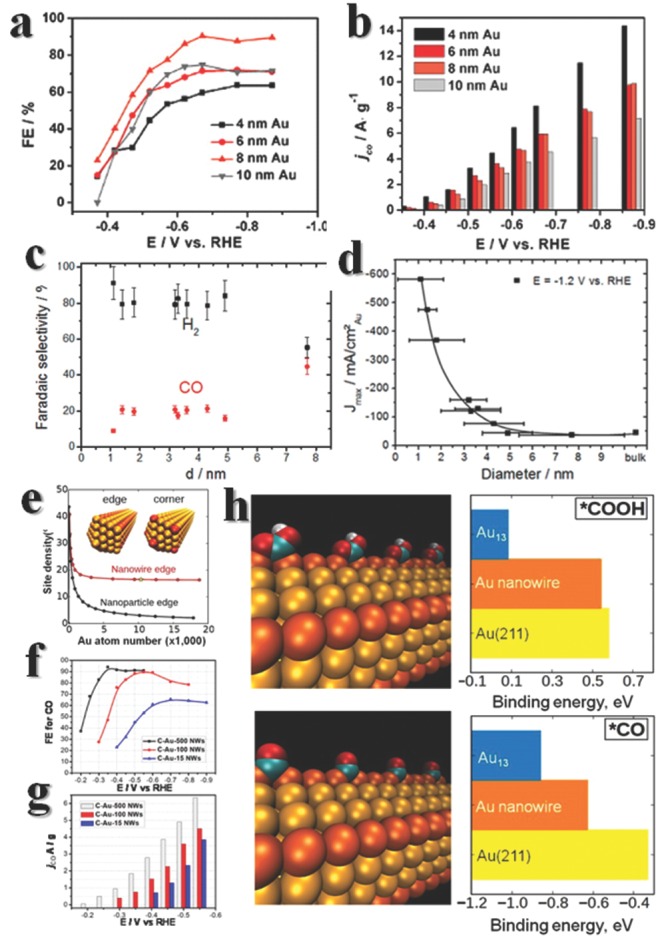
Electrochemical conversion of CO2 results of Au nanomaterials. Here show the performances of gold nanoshperes characterized by a,b) TEM and c,d) STM. Data in (c) were acquired at E = −1.2 V vs RHE. Reproduced with permission.4, 74 Copyright 2013, American Chemical Society. e) While the UNWs get longer, the edge density is much larger than that in nanoparticles. f,g) The property varies with the length of nanowires, the longer the better. C‐Au‐500 (100 or 15) NWs means Au UNWs of 500 (100 or 15) nm length mixed with carbon, respectively. h) Computational results on CO2 reduction and CO formation. Configurations of the adsorbed COOH (up) and CO (down) on a Au NW are portrayed here. The edge‐type sites are highlighted in orange. In the Au‐COOH bonding mode, both C and O (carbonyl) bind to Au directly with OH tilting away from the O=C bond. In the Au‐CO bonding mode, one CO binds to two Au atoms with CO serving as the bridge. Binding energies of the key COOH and CO intermediates calculated on the Au NW, Au13 cluster, and Au(211) are shown in the right. Reproduced with permission.75 Copyright 2013, American Chemical Society.
Edges, on one hand, are the active centers, but on the other hand, may be an obstacle for experimental measuring the molecules' binding energy on surface. For commercial metal films, the surfaces are not perfect flat or exposing pure lattice, and there are always terraces and edges in one optical flat surface. However, the adsorption energies are not the same at terraces and edges, and this would lead to disparities for experimentally determining the adsorption energy on facets. Taking CO on Pt(111) as an example, the adsorption energy was found to be 32.3 ± 1 kcal mol‐1 by Ertl et al.76, but 45 ± 3 kcal mol‐1 by King et al.,77 and 31 ± 0.5 kcal mol‐1 by Campbell.78 Bartels group79 developed the velocity selected residence time method to remove the effects of different surface structures or defects. The well‐designed technics corroborated that the edges of Pt are special, and bind stronger with CO than Pt(111) facets.
What if the reactive sites bind CO too tightly? The electrochemical reduction would keep on going and CO2 would be further reduced to hydrocarbons. Tests on Au, Ag, Zn, Cu, Ni, Pt, and Fe films all showed the capability to produce methane or methanol.80 A volcano plot with CO binding strength as abscissa and CO2 reduction current density as vertical coordinates was made by experimental data (Figure 5 ), suggesting the CO binding is the vital step for CO2 conversion. The disparate trends beside the peak may be driven by competition of abilities to stabilize CO* and reduction of CO*.
Figure 5.
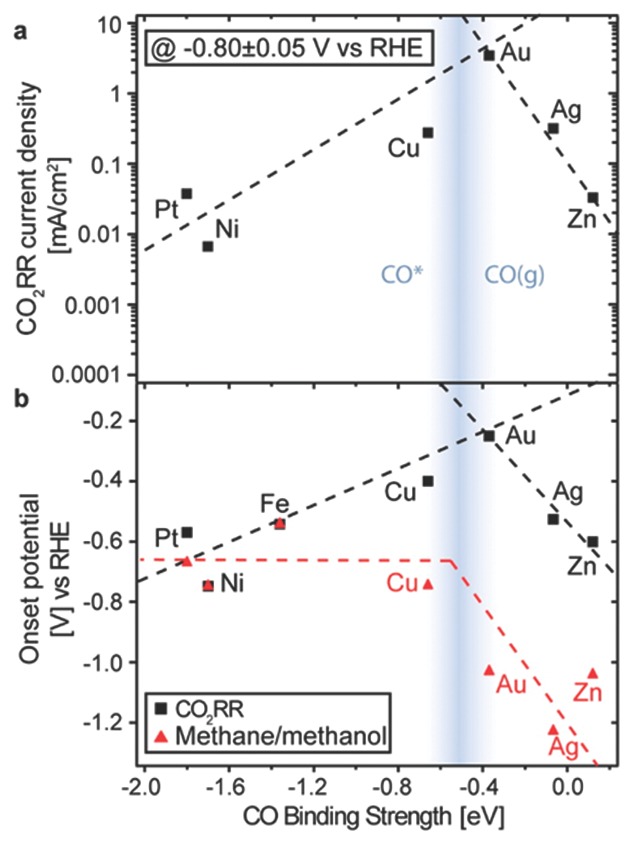
a) Volcano plot of partial current density for CO2 reduction reaction (RR) at −0.8 V vs CO binding strength. b) Two distinct onset potentials plotted vs CO binding energy: the overall CO2RR, and methane or methanol. Dashed lines are to guide the eye. Reproduced with permission.80 Copyright 2014, American Chemical Society.
Copper is a widely studied metal for CO2 conversion into hydrocarbon. The fully reduction of CO2 requires an eight electron transfer process, thus lots of intermediates and final products may exists in the reaction. A systematic computation study involving 41 different intermediate steps on copper surface illustrated numerous routes may be possible to produce CO, HCOOH, CH4 and C2H4.70 Additionally, it is well accepted that Cu(211) facet is more active than (111), (100) facets to generate CH4.71 One more thing to note is that (211) facet of face center cubic (fcc) packing is not a close packing layer, and expose well‐packed edges of (111) facets. As for the reduction to ethylene, there may be two distinct mechanisms: one is through the formation of a surface‐adsorbed CO dimer at (100) facet at relatively low overpotentials,81 and the other has a methane intermediate at both (111) and (100) facets.82 To further corroborate the mechanism in real condition, in situ study is required.83 Since CO2 conversion is a multistep process, we can design multicomponent catalysts to work cooperatively to stabilize or destabilize some intermediates, and lead the reaction to reach a single products.84 One advantage of NPs to serve as catalysts is that the amount of atoms on surface is enlarged, meanwhile the edge fraction is also increased. The 7.0 nm Cu NPs reach up to 4 times methanation current densities comparing with high purity copper foil electrodes.85 However, they are not stable under electric treatment. Alloying Cu nanoparticles with Au would enhance the stability, however, it would decrease the selectivity.86
3.3. Reactions at Edges of a Multicomponent Interface
As we can see from previous discussion, a catalytic reaction usually requires the stabilization of some intermediates and destabilization of some other intermediates. Such observation enlightens us to pay attention to multicomponent interface, especially the edge of the interface. Elaborately selecting various active compounds and carefully tailoring the combination of multi components would realize lots of unexpected performance. This trial has started long time ago, Tauster et al. found strong metal‐support interactions (SMSI) in TiO2‐support noble metals, and the CO and H2 absorption capacities was lost at high temperature.87 The SMSI effects are detrimental in many cases,88, 89 thus researchers would like to avoid them and choose inert supports in early studies. Recently, more and more examples16, 22, 23, 90, 91 proved that the supports might promote some reactions and enhance the total catalytic activities.
Taking HER as an example, metal like Pt can easily adsorb atomic hydrogen and subsequent form H2, so the last thing for HER catalyst design is choosing a proper compound to dissociate water and coupling these two materials in a proper manner. Ni(OH)2 is a good candidate for such demand. A controlled arrangement of Ni(OH)2 clusters on Pt(111) (Figure 6 a) can enhance the activity by a factor of 8 compared to Pt.92 The active sites sit on the well‐arranged edges of Ni(OH)2, which remind us to think about multi active centers in homogeneous reactions. In both areas, one compound plays only a part in the catalytic reaction by its own advantages, and multi‐compounds are precisely placed in order to achieve the well communication of intermediate steps. The difference of them are their complete structures, and homogeneous catalysts with multiple active centers usually have elegant molecule constructions with certain constitutions, while in heterogeneous catalysts, they only have well‐controlled active centers.
Figure 6.
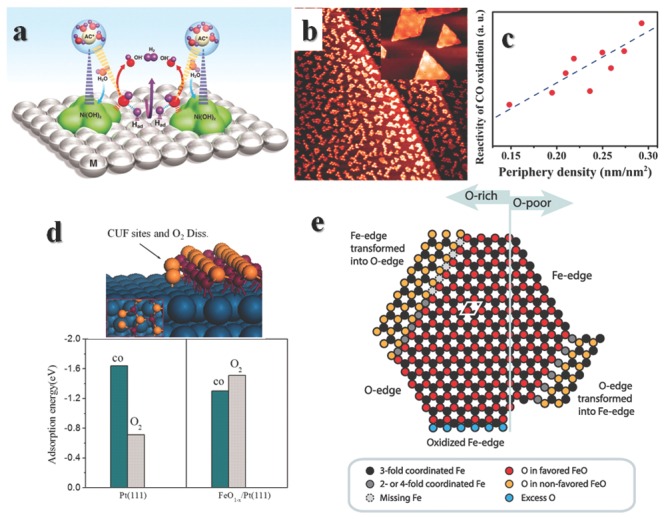
a) Schematic illustration of the cooparation of Pt and Ni(OH)2 to enhance HER properties. Reproduced with permission.92 Copyright 2011, American Association for the Advancement of Science. b) STM image (200 nm × 200 nm) from the FeO1‐x/Pt(111) surface. Inset is an atomic‐resolution STM image of FeO monolayer nanoislands (25 nm × 20.8 nm) recorded at liquid N2 temperature. c) Dependence of reactivity of CO oxidation on the periphery density (edge density). d) Schematic structure of the Coordinatively unsaturated ferrous (CUF) sites and calculated transition states of O2 dissociation (the inset shows the top view) at the boundary between FeO and Pt(111). The calculated adsorption energies (in eV) for CO and O2 molecules on Pt(111) and FeO1‐x/Pt(111) surfaces are different. Reproduced with permission.94 Copyright 2010, American Association for the Advancement of Science. e) There can be found five types of FeO1‐x/Pt edge structures. Here is a brief summary of them. Reproduced with permission.24 Copyright 2015, American Chemical Society.
Composite structures with transition metal oxides on noble metals are called inverse catalyst,93 in order to distinguish them from conventional metal on support catalyst. A well‐studied case of inverse catalyst is ferrous oxides on Pt substrates (Figure 6b).94 Pt catalyst is easily poisoned by CO because of the strong adsorption by an adsorption energy of –1.64 eV on (111). Such a strong interaction would block all the surface Pt atoms and prevent other substrate adsorption, thus no other reactions would happen on Pt(111) in the present of CO. However, on the edges of FeO1‐x monolayer islands, the binding energies change a lot (Figure 6d), and preferentially adsorb and activate O2. Then spontaneous oxidation of CO would validate even at room temperature, depending on the periphery density or edge density of FeO1‐x islands (Figure 6c). Fully covering the Pt(111) with FeO1‐x exhibits minimal reactivity at room temperature due to the lack of edges of interface.93 NiO‐on‐Pt95, 96 and CoO‐on‐Pt97 can also be obtained in similar methods. Among this three catalysts, FeO1‐x/Pt(111) shows the best performance of preferential oxidation reaction of CO in the presence of H2 (PROX), while NiO1‐x/Pt(111) is best for oxidation of CO in the absent of H2 (COOX).
The reaction demands elaborate collaboration between two functional compounds on the edges of one. However, ionic or metallic bonds are not similar with covalent bonds. They cannot be fixed into a certain direction, and defects or distortions cannot usually be avoided in real materials. There have been found 5 different edges structures of FeO monolayer islands on Pt(111) (Figure 6e),24 depending on the synthesis environments.98 Computational99, 100 or experimental101, 102, 103 works have shown activities with different O‐edges or Fe‐edges are distinct. Meanwhile, oxygen vacancies near edge would also affect the oxidation mechanisms, “spectator” mechanism or “carboxyl” mechanism of reduction to C2 compounds are possible depending on edge structures.104 Besides FeO monolayer islands on Pt(111), CeOx and TiOx on Au(111),105 FeO on Ru(0001),106 CoO on Ir(100)107 and NiO on Rh(111)108 have also been synthesized and studied in various situations, more comprehensive studies could be find in reviews by Freund et al.109 and Surnev et al.110
In such inverse catalysts, the metal oxides are usually composed of monolayer or several layers, the active sites reside on the edges of metal oxides. The multicomponent interface may owe their origin of activities to the “coordinate unsaturated”.93 In other cases, there are many beneficial SMSI effects on metal oxides support metals,23, 111 and sometimes the enhanced performance cannot simply derive from “coordinate unsaturated”. When using reductive metal oxides as supports, the reducing nature of supports would whether donate or withdraw charges from metals near the metal/oxide interface, depending on the work functions of two materials.22 SMSI effects cannot be simply concluded. The Pt NPs on iron oxide may undergo single layer encapsulation with Fe migration driven by SMSI.112 This effect is similar with under potential deposition in electrochemistry.113 To some extends, we may consider such interface as a new chemical compound, since it possesses new characters comparing with each single materials.
In a hybrid structure of CuO covered Ag interface, the CO oxidation activities is linked with the interfacial lengths.114 Ag is inert for CO oxidation, while CuO can oxidize CO to CO2 with an activation energy of about 62.8 kJ mol–1. However, the partial covered structures possess apparent activation energies range from 36.7 to 42.7 kJ mol‐1. DFT calculations reveal that charge redistributes in the interface (Figure 7 b), the charge is enriched at CuO surface near interface (or edge of CuO), while for space far away from interface, the charge density almost keeps same. In many cases, the interactions are more complicate, the adhesion of gold on TiO2(110) differs with different treatments of TiO2,115 dual catalytic sites in the composite are possible for CO oxidation,116 Au16 on TiO2 (110) for CO oxidation involves the generation of oxygen vacancies (Figure 7a).90 In another case, a simple synergistic effect of different active compounds without charge redistribution may be essential for selective catalytic oxidative dehydrogenation of carboxylic acids.117 The study of SMSI or interface need to be more studied, then more rational rules can be drawn.
Figure 7.
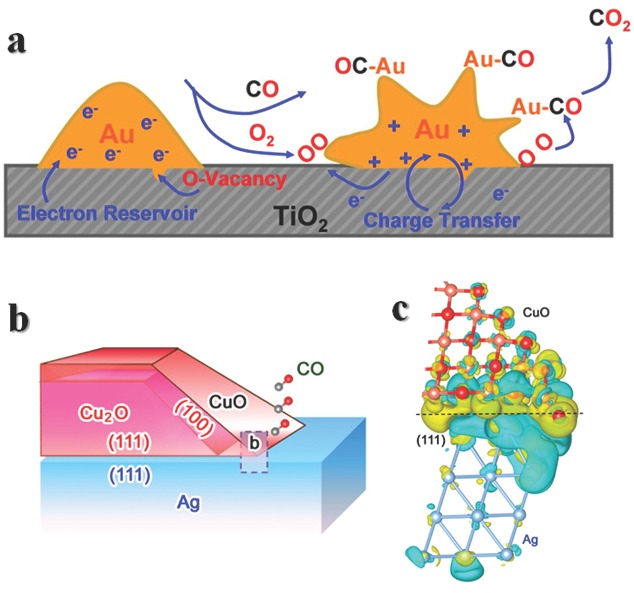
a) Schematic illustration of Au16 on TiO2 (110) for CO oxidation involved the generation of oxygen vacancies. Reproduced with permission.22 Copyright 2013, American Chemical Society. b) Schematic illustration for the Ag‐CuO/Cu2O hybrid structures. c) Differential charge density by first‐principles simulations illustrates the increase (olive color) and decrease (cyan color) of electron distributions. Reproduced with permission.114 Copyright 2014, American Chemical Society.
4. Novel Structures and Atypical Edges
Vast morphologies of nanomaterials from 0D to 3D and their composites have been achieved in the past several decades.30, 91, 118, 119, 120 Among them, novel structures like nanoframe (NF) and ultrathin nanowire (UNW) have attracted lots of attention both in nanostructure fabrication and applications. NFs are NPs with interior atoms etched out and atoms on edges preserved. From the sight of pristine NPs, the NFs are absolutely the edges of them, and the research on NFs represents the study of edges of pristine NPs. However, if we consider NFs as newly formed NPs, the edges of them are difficult to define, the structures of NFs are not easy to detailing. For convenience, we consider the whole NFs as atypical edges. UNW is a kind of unique one dimensional material. The atomic structures of UNW are, in many cases, not quite clear yet, and thus it would be difficult to define the edge of UNW, or determine whether the UNW as an entirety could resemble edge. From the sight of geometry, both UNW and conventional edges are one dimensional structures, and they may both feature low CN, too. So it might be proper to consider the UNW as edges. A brief comparison between these two novel structures, which could be seen as atypical edges, with conventional edges is made in Table 2 , the specified discussions of them are made in the following parts.
Table 2.
A brief comparison between conventional edges and two kinds of atypical edges
| Conventional edges of nanostructures | Nanoframe | Ultrathin nanowire | |
|---|---|---|---|
| Structural feature | Natural ends of surfaces in nanostructures | Structures with internal atoms on surfaces and in body of particles etched out and atoms on edges reserved | Ultrathin one dimensional materials |
| Chemical feature | Low CN and large edge content | Low CN (maybe) | |
| Advantage | Easy accessible and abundant types | High activity and low use of costly materials | Enhanced activity |
| Limits | Difficulty in fabrication; Not all NFs are stable | Dimness in detailing structures |
4.1. Nanoframes and their Unique Properties
Reaction on metal surface depends on the nanoscale structure of metal.29 For atoms on metal surface, the CN changes from corners, edges to surfaces. Lower CN is always related to high active center. Thus a stable structure with numerous low CN sites is widely pursued in research and industry. Corner sites seem perfect but impractical because of the low content. Stubbornly reducing the size would risk the stability, and lose the merits of nanomaterials – easy recycle. Meanwhile the internal atoms have few chances to contact with substrate. Decreasing the amount of such unnecessary atoms would enhance the mass activity and reduce the employment of scarce metals. Then a structure with pure edges seem like a promising solution for reducing the amount of catalysts while maintain or enhance the activities. Thanks to the great development in nanomaterials synthesis, innumerable morphologies have been prepared in the past several decades, especially in the last 15 years. NFs with internal atoms etched out and edges conserved can be realized in many cases. These unique structures are ideal platform for edge studies comparing to integrated solid particles, because of simplified interference. However, the catalytic performance study is still in fancy because the challenge in synthesis is still the first obstacle for systematically research. Here we briefly introduce the fabrications of NFs, and some of their enhanced performances in catalytic reactions. A more comprehensive summary could be made only after vast researches have been carried out.
4.1.1. Conventional Nanoframe Synthesis Methods
From the concept of thermodynamic, NFs are not stable, like nanocrystal with high index facet.121 Fortunately, lots of crystals with high index facets have been generated by dynamic control, so have been in NF synthesis. There are lots of similarities both in synthesis and properties in these two areas, in brief, there are lots of atoms with low CN in both two cases. When we talk about edges in daily life, we usually refer to the ends of a flat surface. However, there is no flat surface in atom scale if we consider the atom as a sphere. What makes the edges of NPs so special is mainly the low CN. We may define the edges in nanoscale by CN changes in facets, because the CN is always lower at edges comparing with facet atoms. Here we consider a face center cubic package as an example (Table 3 ). For cubic structure enclosed by (100) facets, the nearest CN of atoms on (100) facet is 8, edge is 5 and corner is 3. While as for tetrahedron with only (111) facets encased, the nearest CN of atoms on surface is 9, edge is 6 and corner is 4. For (111) facets enclosed octahedron, the nearest CN of atoms on surface is 9, edge is 7 and corner is 3. It seems ok to define the edges as the atoms possess the second lowest CN on the surface. However, there would be great confusion in particles enclosed by high index facets. For instance, there are two kinds of atoms on (211) facets, one has nearest CN of 7, the other has 9, meanwhile edge is 7. Nearest CN of a series of atoms on surface could reach the nearest CN of edges of octahedron, and is the same with the edges of (211) facets. However, they are not atoms on the same environment because their CN of longer distance are different. Actually crystals with high index facets also represent high activity in many cases,118, 122, 123, 124, 125, 126 as well as edges. For accuracy and simplicity, we consider the edges as the turn of facets. Thus NFs could be seen as pure edges without other surface or interior atoms.
Table 3.
CN of nearest atoms in face center cubic packing particles enclosed by different facets
| Cube (100) | Tetrahedron (111) | Octahedron (111) | High index facet (211) | |
|---|---|---|---|---|
| Facet | 8 | 9 | 9 | 7 or 9 |
| Edge | 5 | 6 | 7 | 7 |
| Corner | 3 | 4 | 3 |
To get a NF constituted by edges, an etching process is always required. There are two kinds of typical conventional synthesis methods based on the etching methods. One is a template assist etching process, metal is firstly deposited on the edges of a well‐controlled solid template particle, then a proper etchant is introduced and the template is removed, leaving the frame alone. The other method is a kinetic controlled dissolution process, a stable or metastable particle is generate at first, then another etchant or nothing is added to move out the internal atoms, depending on the stabilities of NPs. Both of these two process are driven by kinetic control, the former one requires two motif to constitute an integral particles, the growth of frame on particle is controlled by kinetic effects. While the etching process of second method is controlled by kinetic effects. We briefly take a view at this two kinds of methods in the following sections.
Template Assist Etching Process: Seed mediated growth is powerful to prepare core‐shell structure. Then how to deposit the second material only on the template's edges in order to get frame structure? The solution relies on two aspects: surface chemistry and specific affinity on edges. Nanomaterials are usually fabricated in the presence of capping agents. The introduction of capping agents adsorbed on surface is to inhibit the further enlarge of the particles. Modifying the surface chemistry of nanoparticles is the first consideration of nanomaterial applications.127 By selecting proper capping to cap the facets, we can deposit other wanted materials on the active edges and corners. Subsequently etching the template would lead to a frame portraying the outline of the pristine NP. As for specific affinity on edges, this is usually realized in Au‐Ag system,128 they are easy to react in suitable position under controllable conditions. The specific affinity also exists in some other nonmetallic materials.129
We first look at an instance of surface chemistry controlled NF fabrication. Xia group successfully synthesized Rh cubic NF by etching the Pd template (Figure 8 a).130 They firstly synthesized Pd cube enclosed by Br− capped (100) facets, then reduced Rh salts at the edges of Pd cube. The Rh would selectively grow on the edges because of the passivated (100) facets, and preferentially grew along <111> direction at optimized precursors concentration because of a kinetic effect.122 When Br− was absent in the system, Rh could also grow on the surface. Due to the inherent different resistance to oxidative corrosion, Pd cube could be etched and Rh frame could be obtained (Figure 8b). In fact kinetic control here is vital, too, however, the surface control is the starting step of the heterogeneous growth of Rh, thus we categorize it into template assist etching process.
Figure 8.
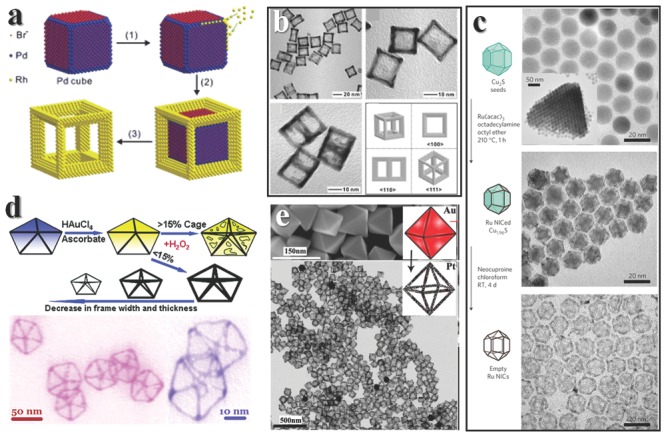
The growth of NFs. a) Formation of Rh NFs with the help of Pd cube. b) TEM images of Rh NFs. Reproduced with permission.130 c) Ru NFs can be fabricated via growth on edges of Cu2S seeds and then the etching of seeds. Reproduced with permission.135 Copyright 2010, Macmillan Publishers Ltd. d) Au atoms can deposit on the edges of Ag decahedra, thus lead to the formation of NFs. Reproduced with permission.134 Copyright 2011, American Chemical Society. e) Pt can grow on the edges of Au (up) and form NFs (down). Reproduced with permission.128 Copyright 2012, American Chemical Society.
We then take a view at NF fabrication driven by specific affinity on edges. Ag and Au are elements in the same group, their atom radius, lattice structures and parameters are quite similar, thus behave similar behaviors in many cases. However, the reduction potential of AuCl4 −/Au is 0.99 V vs SHE, and AgCl/Ag is 0.22 V vs SHE, thus AuCl4 − can etch Ag spontaneous, one AuCl4 − can react with three Ag atoms. The surface energies of different facets usually increase in the order γ(111) < γ(100) < γ(110).131 It is reasonable to hold the point that Au would preferentially etch (110) facets, then (100) facets, and the last are (111) facets. Au triangle frames can be generated by galvanic etching Ag triangular nanoprisms enclosed by (111) and (110) facets.132 The Au indeed etches Ag atoms on (110) facets and depose on (110) facets. If the etching is achieved only by AuCl4 −, the thickness and roughness cannot be well controlled. However, introducing the second etchant at proper time would be salutary for morphology control, because of the sudden depletion of Ag. Au triangle frames with thickness less than 2 nm or 6 nm could be synthesized in such way.133
There are several kinds of NPs that encased only by (111) facets. When using Ag decahedra as template, Au atoms would preferentially deposit on edge sites because of low CN. After H2O2 etches the Ag decahedra, an Au decahedral NF is generated.134 Controlling the amount of Au loaded would help to prepare frames with different thickness. While the gold deposition on (100) facets may lead to a porous mesh in similar reaction procedure (Figure 7d). Pt NFs could be generated by using Au as seeds, and the principles are similar with the Ag‐Au systems. When the edge sites of decahedra Au erode because of low CN, new facets are produced and Pt are preferentially deposited on the newly formed faces. After etching and annealing, smooth Pt NFs can be obtained (Figure 8e).128 Pt octahedral frames can be fabricated by using Au octahedron as templates.
Metal NPs are not the only choice for templates. When using Cu2S as template to grow Ru, hexagonal biprism Ru NF can be synthesized after etching the Cu2S (Figure 8c).135 This is a case of edge growth with no specific affinity between templates and metal, the details of driving force still need to be explored. Saturated NaCl aqueous solution can be used as cubic template, too. Adding AgNO3 into the solution would directly generate NaCl/AgCl core‐frame structure, then using water to wash the products would take the NaCl core away and leave a frame of cube. Soft reduction treatment would lead to the formation of Ag@AgCl NFs.136
The template assist etching process is benefit for structure well‐control. There are many tunable parameters to get ultrathin and ultrafine structure. The combinations of templates and frames are versatile and easy accessible. However, the common rules in this method is not well established yet. The design of new frames need to be ingeniously designed, and lots of trial‐error experiments are required. There is still plenty room in this areas. Well‐controlled synthesis of NFs would certainly lead to new insights both in material fabrications and catalytic processes.
Kinetic Controlled Dissolution Process: When something is not thermodynamically stable, an efficient procedure to achieve it is by kinetic control. All practical chemical reactions are less or more away from ideal thermodynamics. Kinetic effects even dominate in vast real processes. In the aforementioned template assist etching process, the deposition of frame material on template edges is usually controlled by kinetic effects. However, the specific character of such process is the present of template, so we name it by template. Herein we discuss kinetic controlled dissolution process with no template involved, the only driving force may be kinetic effects. There are also two forms in this process, based on the stability of the intermediates. In one case, metastable particles as intermediates are formed at beginning because of a fast reaction, after dissolution of internal atoms in the present of low concentration oxidant, finally frames are formed. In the other case, a stable particle is generate as the intermediates, then proper etchant is introduced. Based on the inherent difference of edges and surfaces, something would remain. If edges remain, NF is fabricated. If edges dissolve, the trial fails. Etching the nanosphere would lead to hollow structure or yolk‐shell structure because of the different migration rate, this is so called Kirkendall effect.137 Regular NFs would be formed under suitable environment driving by Kirkendall effect.
PtNi3 polyhedra is not a thermodynamic stable particle because of the active Ni atoms on surface. After the synthesis of PtNi3 NP, Ni atom on surface would easily leak out because of the existence of dissolved O2 in solvent even at room temperature. Finally the Pt3Ni NFs are generated in the solution (Figure 9 b).138 Pt4Ni NFs could be prepared via similar process.139 DFT calculations illustrate that after Ni atoms dissolved, Pt atoms on the surface aggregate to form a “porous Pt shell”. Then leaking the internal Ni atoms dominates the dissolution driving by Kirkendall effect and results in the NFs.140 PtCu3 NFs would be fabricated in similar way.141 However, the details still need to be further explored because similar PtCu3 can be generated in quite different conditions (Figure 9a).142 Ag‐Au octahedra NFs could be achieved in a two‐step process.143 Ag nanosphere is prepared at the beginning of the reaction, then galvanic etching by AuCl4 − could change the morphologies and compositions, and finally Ag‐Au NFs are produced (Figure 9c). Such alloying‐dealloying process could finely control the final morphologies, comparing to the direct etching process to get NFs, though both of them may form NFs as final products under suitable conditions.144, 145, 146
Figure 9.

Enhanced properties of NFs. a) CV of PtCu3 indicates a degration of NFs. Reproduced with permission.141 Copyright 2012, American Chemical Society. b) However, the Pt3Ni NFs are quite stable. Reproduced with permission.138 Copyright 2014, American Association for the Advancement of Science. c,d) NFs could serve as catalysts for organic synthesis. Reproduced with premission.140, 143 Copyright 2012, 2014, American Chemical Society. The TEM images inset in panels (a,b,c) are the pristine NF structures, and the cartoon image at the bottom in panel (d) depicts the fabrication method of Au on the corner of NF.
We have discussed the metal NF so far, oxides or other materials of NFs have also been synthesized by etching corresponding materials. Cu2O NFs can be synthesized by a rapidly etching process.147 Details studies reveal that the (100) faces of Cu2O are stable under HCl etching,148 thus different novel structures can be fabricate by using various substrate. However, the (100) faces are less stable in other oxidative etching conditions, and leave NFs with part of other facets remained.149 Ag2O NFs can be formed in a similar manner with Cu2O frames,150 the stability under etching is in the order (111) > (110) > (100). Taking advantages of this rule, various NFs structure is realized from diverse substrates.
Yet we have seen lots of NF synthesis, however the fabrication methods are still limited in several compounds. General and practical methods is still in fancy. The past several years have witnessed great success in material synthesis and applications, new materials hit headline in research almost every day. Thus, we are confident to predict that NFs would earn their status in the soon coming future.
4.1.2. Applications of Nanoframes
Due to the limitation in synthesis, only a few researches have focused on their chemical advantages, while some of the researches make use of their novel structures or physical properties to realize new phenomena. For example, Xia group capsuled the gold nanocages with thermal response polymer, then light induced phototermal effect of gold can manipulate the cages to open or close to release the chemicals inside.151 There are also many studies concerning the surface‐enhanced Raman spectroscopy (SERS) effects of Ag152 or Au153 NFs or their assemble structures.154 Meanwhile, the cage structure can also store small NPs inside and prepare new morphologies.135 There are also some researches mentioned the enhanced catalytic performances of NFs, we present some researches at this area in this section.
As we have mentioned in the previous discussion, the NF structures are not thermodynamically stable, especially for the alloy. This is indeed true in Pt‐Cu NF.141, 142 As depicted in Figure 9a, the pristine cyclic voltammogram (CV) curve shows no under potential H ad/desorption peaks (‐0.24–0.1V), due to the Cu coverage on the surface. As the bias potential applied, the intensity of Cu dissolution peak decreases further, and finally the shape of CV curve resembles to commercial Pt CV curve, meanwhile, shows better performance comparing to PtCu3 nanoparticles. The electrochemical dealloying has been proven to successfully synthesize highly active electrocatalysts.155 In another test, the Pt‐Cu NFs show higher performance than commercial Pt catalyst.
NFs are not always fragile in any cases. The aforementioned Pt3Ni NFs138 could maintain their morphologies and activities even after 10 000 cycles without loss for oxygen reduction reaction, while stay a factor of 22 enhancement in specific activity and 36 in mass activity (Figure 9b), which is essential for proton exchange membrane fuel cell.156 The secret of stability may be ascribed to the Pt‐skin coverage on the surface, preventing the aggregate of oxygenated intermediates. This hint enlightens stable NF design: cover the frame with something active but stable. Pt‐Ni NFs could also serve as hydrogenation catalyst.140 Considering the hydrogenation of 4‐nitrobenzaldehyde as a model reaction, different catalysts were tested. As drawn in Figure 8d, Pt3Ni NF shows high selectivity while the turn over frequency (TOF) is relatively low comparing with other truncated structure. A good way to enhance both the activity and selectivity is by sophisticated loading Au on the frames. A proper load content would help to both increase selectivity and activity (Figure 9d).
Several other reactions are employed in illustrating the enhanced performances. Au triangular NF with tailorable thickness range from 1.8 nm to 6 nm shows about 2.4 times reduction rate of 4‐nitrophenol, comparing to frames with thickness larger than 10 nm.133 In catalytic reaction, the Au NFs show a reaction rate about 2 times faster than Au nanoboxes, and much faster than solid particles.157 Oxidizing aniline to form aromatic azo compounds under mild condition could be achieved by Au‐Ag octahedral NFs, the yield is about 3 times higher than Au‐Ag NPs at 32 h (Figure 9c).143 The applications of NFs are not restricted in organic synthesis. The Ag@AgCl NF could degrade methyl orange under irradiation and show great efficient comparing to P25.136 A Cu2O‐Au complex NF was test to degrade methylene blue and results shown faster degradation than Cu2O.158
In summary, NFs have shed light on improving performances of catalysts. However, there are still many problems need to be solved besides obstacles in synthesis. Stability under reaction is the first concern for practical application. Though NFs possess innate instabilities, there are ways to avoid it while maintain high activities. The second thing lies on the way is that easy to scale up. Low harvest is accessible in lab, but unacceptable for industry. Exploring new reactions would also benefit for NFs applications.
4.2. Ultrathin Nanowires Resemble Edges?
In the view of quantum theory, size determines the boundary condition of wave functions of material. If “the underlying physical laws necessary for … the whole of chemistry are thus completely know”,159 then size would no doubt determine the structures and properties of materials. From the points of geometry, the content of atoms at edge becomes larger and larger when the particle get smaller and smaller. However, as we described before, stubbornly reducing the size would risk the stability, and lose the merits of nanomaterials – easy recycle, NFs could be a solution. On the other hand, UNW may be another solution, as we have seen in section 3.2, ultrathin Au nanowires which possess high edge atom content could improve the CO production.75 Ultrathin Pt nanowire with diameter about 3 nm also shows enhanced properties of electrochemical oxidation of formic acid or methanol comparing with commercial Pt/C catalysts.119 The results show promising future for application of UNW. Strictly speaking, “ultrathin” is not a scientific expression, as it does not refer to a specific size range. UNWs are usually considered to be less than 10 nm in diameters in many papers, while some researchers concern the size down to sub‐5 or sub‐2 nm. Here we have no intend to argue which definition of size is more accurate, we focus on the chemical aspects of UNW. The debate of “ultrathin” may be ended one day "when … approximate practical methods of applying quantum mechanics should be developed”.159
Synthesis is always the first step for material realization. There are mainly three categories of fabrication methods in general: template assist, ligand control and oriented attachment synthesis.160 The template assist synthesis involves the use of a hard or soft structure as template, which could limit the space of nanowire growth. For example, calix(4)hydroquinone could self‐assemble to nanotubes with diameter down to atomic scale, then reducing Ag salts in the space‐limited nanotube would produce ultrathin Ag nanowire with 0.4 nm width.161 Soft template may occur during the synthesis, oleylamine is a kind of amine with long alkyl chains and a C‐C double bond in the middle of the chain, which in turn would allow for the easier to self‐assemble in certain condition, and Au nanowires with diameter about 1.6 nm could be generate in the channel.162, 163 Introducing the triisopropylsilane into the synthesis could accelerate the reaction with slightly changing the diameter to 1.8 nm.164 The principle for ligand control synthesis lies on that one ligand binds very strongly on the sides of the nanostructure and greatly inhibit the growth along this direction, thus lead to the wire formation.165 Oriented attachment may result in UNWs with constant diameter, the process is similar with polymerization reactions. SnO2 ultrathin nanowire with diameter of ca. 0.5–2.5 and ca. 1.5–4.5 nm could be prepared via the oriented attachment of SnO2 quantum dots.166 An etching method of Te nanowires could also help to fabricate several kinds of metal telluride UNWs.167 There is no limitation of fabrication method, more and more efficient strategies may be achieved and the rationale of fabrication may be unraveled in the future.
The successful synthesis put the field forward to the novel phenomena related with the ultrathin size.168 The relative orientations and the volume fractions of the planar defect could determine the elastic modulus of nanowire.169 However when the sizes of nanowires narrow down to ultrathin, it would be hard to determine what is defect. Ultrathin nanowires could be flexible, and resemble the viscosity behavior of polymers.170 What is more, ultrathin inorganic nanocoils may mimic the self‐folding behavior of biomacromolecues (Figure 10 h,i).171 As for metal, the structure could be changed dramatically when size down to atomic level, helicity has been obtained in Au nanowires172 and Pt nanotubes.173
Figure 10.
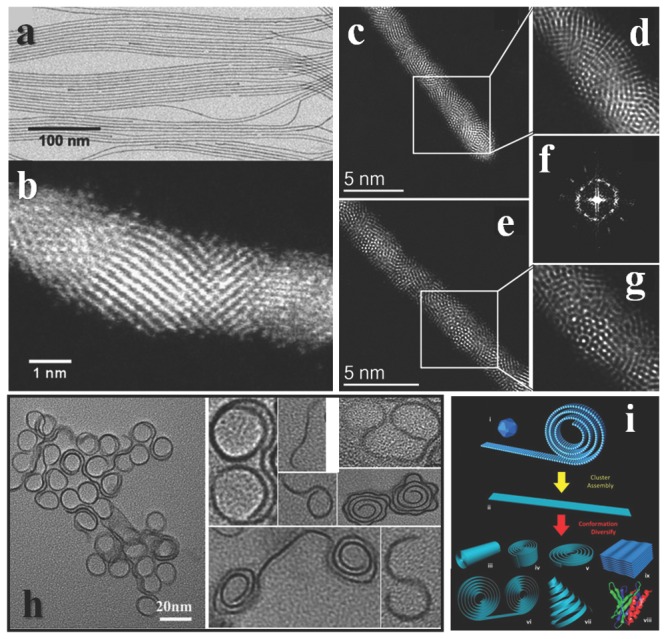
Structures of UNWs. a) TEM and b) STEM image of Au UNWs. Reproduced with permission.174 Copyright 2014, American Chemical Society. c,e) Aberration‐corrected STEM‐HAADF images of the Ag/Au nanowires with the helical icosahedral structure in different orientations. d,g) Corresponding high‐magnification areas of the icosahedral packing. f) FFT obtained along the axis of the nanowire of (c) showing the 5‐fold symmetry of Au nanowires. Reproduced with permission.175 Copyright 2011, American Chemical Society. h) The folding of ultrathin inorganic materials. Reproduced with permission.171 Copyright 2013, American Chemical Society. Schematic of 2D cluster‐assembly and possible variations in the ultrathin nanoribbon conformation. Reproduced with permission.170 Copyright 2013, American Chemical Society.
It is the structure that determines the properties. However, the details of ultrathin structure remain a challenge to study. Notwithstanding, TEM is a powerful tool to characterize the morphology, and with the help of energy disperse spectrum or other accessories, element mapping or even atomic element mapping could be realized. The higher resolution usually ties to high voltage electron beam, but ultrathin structures usually cannot withstand such high energy electron beam. Such contradiction greatly hinders the structure analysis of ultrathin nanowire. Nowadays, people are making progress. High‐angle annular dark‐field (HAADF) imaging at low voltage could reduce the damage of materials under characterization. Gold nanowire with diameter about 1.7 nm was successfully analyzed under such technique. The nanowire is not atomic uniform along the wire, and the surface is not flat (Figure 10a, b).174 The Ag‐Au alloy UNWs possess more complete structures with icosahedral and decahedral packing along the wire (Figure 10c–g).175 The atomic structure of metal oxide UNWs might be more intricate, because their constitutions are even more complicated.
One possible factor that edge sites are active centers is the unsaturated coordination as we have discussed before. UNW may expose vast unsaturated coordinated atoms according to geometry, however, the vast experiments on different types of UNW are still lack, thus the researches of UNW are hindered. When size extremely narrows down, the boundary between molecule and crystal blurs. It would be hard to distinguish defects and conformation in ultrathin nanowires. Maybe we could use some knowledge from the statistics of polymer science, and build up a practical statistic explanation of the conformation in ultrathin nanowires. One more thing we need to notice is that the UNW may be not stable without capping agent. The strong binding ligands may prevent the abilities of ultrathin nanowires. Moreover, the use of UNW as catalyst is seldom reported, thus the better understanding of performance is hampered. In short, there is still a long way to realize the ultrathin nanowire as high active catalyst.
5. Looking Beyond Catalysts: Edges of Graphene
C is probably the most intriguing element in the periodic table. Regardless of that life is based on C, materials made by C have been used since ancient time, such as the “lead” in a pencil, fuel for fire, doping in iron. Now C materials revive once again, in names of graphene, carbon nanotube, etc. Among all the allotropes of C, graphene have become a star since the preparation by repeated peeling at 2004,176 because of the potential in overcoming the bottleneck of silica materials in semiconductor industry. Graphene is a honeycomb two dimensional semimetal materials with zero bandgap. The zero bandgap results in the inability of switching off for logic applications,177 which is deadly for application. One of the solutions is to constrain graphene nanoribbons (GNRs) with regular edges. The theoretical explanations for opening the bandgap, in short, are the linear nature of the energy dispersion near the edges of the Brillouin zone, and different boundary conditions of wave functions imposed at the edges.178 Thus the edge structure would determine many physical properties, like optical,179, 180 magnetic,178 electronic181, 182, 183 properties.
Here we briefly review the types of edges, synthesis methods and electrochemical properties of edges in graphene. There are several types of edge structures, including achiral and chiral edges, stable, metastable and transition structures.184 Though there is no perfect method to synthesis high quality graphene with perfect edge structure yet, the fabrication technique is making progress.185 As for researches of chemical difference between edges and basal plane, electrochemical properties are powerful evidence, though there have been so few studies.186
In general, there are two types of achiral edges in GRNs, one is zigzag GNR (ZGNR), the other is armchair GNR (AGNR), based on the orientation of hexagons (Figure 11 a–c). These are two most seen edge structures after Joule heating of graphene.187 The chiral‐GNR (CGNR) is composed by a combination of armchair and zigzag sites. The zigzag edges are metastable and easy to reconstruction into pentagon‐heptagon configurations at room temperature based on theoretical calculations.188 The chemical natures of zigzag sites and armchair sites were long under debating that whether there are substituent groups. However, a complete study of theoretical and experimental studies from various standpoints shown that the zigzag sites are carbene‐like and armchair sites are carbyne‐like structure with neither H‐terminated nor unadulterated free σ radicals.189 Another type of edges consists of series of dangling C atoms protruding from a zigzag edges, is called extended Klein (EK) edge, which is assumed unstable.190 EK edges have been observed in recent studies under aberration‐corrected transmission electron microscopy (AC‐TEM), results illustrate that they would turn back to zigzag edge structures or reconstruction to pentagonal edge structures in short time.191
Figure 11.

Edge structures of graphene and the fabrication of GNR. a) The geometries of graphene edges: (from top to bottom) reconstructed zigzag [ZZ(57)], armchair, reconstructed armchair [AC(677)], zigzag, and pentagonal armchair [AC(56)]. In the right is structure of EK edges. b) Experimental evidence of zigzag reconstruction. c) Experimental evidence of armchair edge reconstruction. Reproduced with premission.185, 191 Copyright 2013, Royal Society of Chemistry; Copyright 2014, American Chemical Society. d) Schematic representation of the method used for unzipping carbon nanotubes to form graphene nanoribbons. Reproduced with permission.184 Copyright 2011, Royal Society of Chemistry.
One thing need to be really careful about the characterization with TEM is the reconstruction under high energy electron beams. For example, the maximum energy of an 80 keV incident electron that can be transferred to a carbon atom is 15.8 eV, and the knock‐on energy threshold for ejection of an in‐lattice carbon atom with three bonds is 17 eV. It seems safe to observe edge structure under such condition, however, the threshold drops below to 15 eV for sites with a neighboring vacancy.192 Such concern may expand to characterization in other nanomaterials. As for using STM and AFM to obtain edge structures, the low scan speed to get high resolution is really a risk for stability, the substrate and temperature would also influence the image of real atom arrangement. The spectrum method may be a good solution for mild and efficient characterization.193 All carbons show common features in their Raman spectra, the peaks lied at around 1580 and 1350 cm−1 are called G and D peaks respectively. For ideal zigzag edges, the D peak is zero, and large for armchair in principle. This implies the possibility to characterize the edge structure simply by Raman spectra, however, the real sample does not show such obvious feature.194
There are many fabrication method for GNR, including mechanical or chemical methods. Among all the methods, chemical vapor deposition (CVD) is benefit for large amount synthesis in a relative short time, and the deposited GNR can be easily transferred onto other substrate. The growth of graphene by CVD might follow the classic crystal growth theory.195 During CVD growth, C atoms are incorporated on the edges from the catalyst surface, however, the details at atomic scale are still needed to be explored. The shapes of graphene prepared by CVD are determined by the catalysts and other parameters, as well as the edge structure,196 since the graphene edge may be terminated by catalyst atoms. For example, the Cu terminated zigzag edge dramatically reduces the threshold barrier to add carbon atoms from 2.47 to 0.80 eV, thus lead to a fast growth of armchair edge.197
Unzipping CNTs along their longitudinal direction is an efficient method to prepare regular edge structure. Several kinds of unzipping methods are developed in various condition (Figure 11d). Multi‐wall CNTs can be unzipped in the oxidative condition with H2SO4 and KMnO4,198 or by intercalation and exfoliation with alkali‐metal and NH3.199 Cutting graphene with catalyst NPs like thermally activated nickel NPs can also produce smooth and orientated graphene edges.200 Other methods using electron or other beam could also be utilized to unzip the CNTs.201 The as prepared GNRs could be further modified using STM lithography,202 chemical etching.203 The etching strategy can extend to modify the structure of MoS2.204 Meanwhile, another “bottom‐up” method is emerging, the on‐surface polymerization from specific small molecules to GNR is a fascinating strategy to control GNR structure.205
Modifying the edge structure with different heteroatoms would also help to tune the bandgap.206 Both the covalent or noncovalent bonding at edges are available for decoration207, 208 via various methods.209 Readers could find a comprehensive review written by Georgakilas et al.210 Functionalization of graphene is of great interest in chemistry, and the well‐functionalized graphene expand their potential in various applications.211
The chemical environments of basal plane and edge are different, one can foresee that their electrochemical properties are not the same. For instance, metal nanowire212 or metal oxide nanowire213 can be deposited at the edges of graphite instead of on the basal plane. However, direct test of edge is of extremely difficult because of the obstacle to fabricate electrode on edges and nearly inevitable interference of basal plane. By applying CV for the oxidation of ferrocyanide and KCl solution at highly orient pyrolytic graphite (HOPG) electrode, on behalf of basal plane, and at an edge plane pyrolytic graphite electrode, the calculated electron transfer‐rate constant of edge is 7 orders of magnitude higher than the basal plane.214 In situ AFM215 and high resolution atomic force electrochemical microscopy216 illustrate that the basal plane is not totally inert, the electron can also transfer at pristine graphic basal plane. A recent electrochemical test using region‐specific cover with non‐conducting pinhole‐free thin film on single layer graphene sheets reveals that the edge is over 4 orders of magnitude higher specific capacitance, and has faster electron transfer rate than basal plane.217
6. Conclusion
In conclusion, edges are important parts of particles. The atoms on edge sites may be more active than atoms of facets. There are several reasons for this, including different chemical environments compared with atoms on inner surfaces, which may also lead to charge redistribution at interface, low CN. Research on MoS2 and related materials clearly shows that it is the edges that promote hydrogen evolution. Further studies to enhance the activity should be carried out focusing on the tuning of edge structures. Inverse catalysts represent another aspect of edge sites: the edge of an interface. The cooperation of various components around the interface can help to realize new reactions or to optimize low efficiency reactions. Edges of nanoparticles may also serve as highly active centers for reactions. Two kinds of novel structures, NFs and UNWs, are considered as atypical edges. It is not surprising that NFs have unique catalytic activities. However, the catalytic performances of UNWs are less studied yet. When the size of UNWs approach the molecular level, the boundary of the crystal and molecule should be reconsidered. Looking beyond the edges in catalysts, the edge structure of graphene is also essential for their physical or chemical properties. The band structures of zigzag or armchair edge sites in graphene are quite different, and this is also true for other 2D materials, such as MoS2.218 In short, the edges of two dimensional materials are pivotal for their applications.
Despite the great success in material fabrication, there are still many obstacles that hinder the understanding of nanoworld. As size decreases, the characterization of materials become difficult, mainly because of the inherent instability of materials. Our limited understanding of crystals or solids at sizes near the molecular level may also hamper the realization of applications. Furthermore, the identification of the active center in heterogeneous catalyst is still a challenge in many cases due to the lack of proper method to track the reactions. Although computer‐aided calculations have achieved great success in detailing many reactions, they usually require a long time to achieve suitable accuracy. Therefore further research, both experimental and theoretical, needs to be done to decipher the structures and properties of nanomaterials in the future.
Acknowledgements
This work was supported by NSFC (21431003, 91127040, 21221062), and the State Key Project of Fundamental Research for Nanoscience and Nanotechnology (2011CB932402).
Biographies
Bing Ni received his bachelors degree from the Department of Chemisty, Tsinghua University in 2014. He is currently pursuing his Ph.D. in Department of Chemisty, Tsinghua University, under the supervision of Prof. Xun Wang. His current interests include the synthesis of ultrathin structures and properties of monodisperse nanocrystals.

Xun Wang received his Ph.D. from the Department of Chemistry, Tsinghua University in 2004. He then joined the faculty of the Department of Chemistry, Tsinghua University in 2004, and was promoted to associate professor and to full professor in 2005 and 2007, respectively. His current research interests include the synthetic methodology, formation mechanism, and properties of monodisperse nanocrystals.

Ni B. Wang X. (2015). Face the Edges: Catalytic Active Sites of Nanomaterials. Adv. Sci., 2: 1500085. doi: 10.1002/advs.201500085
References
- 1. Tong H., Ouyang S., Bi Y., Umezawa N., Oshikiri M., Ye J., Adv. Mater. 2012, 24, 229. [DOI] [PubMed] [Google Scholar]
- 2. Narayanan R., El‐Sayed M. A., J. Am. Chem. Soc. 2003, 125, 8340. [DOI] [PubMed] [Google Scholar]
- 3. Kamat P. V., Chem. Rev. 1993, 93, 267. [Google Scholar]
- 4. Zhu W., Michalsky R., Metin Ö., Lv H., Guo S., Wright C. J., Sun X., Peterson A. A., Sun S., J. Am. Chem. Soc. 2013, 135, 16833. [DOI] [PubMed] [Google Scholar]
- 5. Cui C.‐H., Yu S.‐H., Acc. Chem. Res. 2013, 46, 1427. [DOI] [PubMed] [Google Scholar]
- 6. Burda C., Chen X., Narayanan R., El‐Sayed M. A., Chem. Rev. 2005, 105, 1025. [DOI] [PubMed] [Google Scholar]
- 7. Love J. C., Estroff L. A., Kriebel J. K., Nuzzo R. G., Whitesides G. M., Chem. Rev. 2005, 105, 1103. [DOI] [PubMed] [Google Scholar]
- 8. Ceyer S. T., Annu. Rev. Phys. Chem. 1988, 39, 479. [Google Scholar]
- 9. Raybaud P., Hafner J., Kresse G., Kasztelan S., Toulhoat H., J. Catal. 2000, 189, 129. [Google Scholar]
- 10. Castro Neto A. H., Guinea F., Peres N. M. R., Novoselov K. S., Geim A. K., Rev. Mod. Phys. 2009, 81, 109. [Google Scholar]
- 11. Phan N. T. S., Van Der Sluys M., Jones C. W., Adv. Synth. Catal. 2006, 348, 609. [Google Scholar]
- 12. Polshettiwar V., Varma R. S., Green Chem. 2010, 12, 743. [Google Scholar]
- 13. Kalidindi S. B., Jagirdar B. R., ChemSusChem 2012, 5, 65. [DOI] [PubMed] [Google Scholar]
- 14. Astruc D., Lu F., Aranzaes J. R., Angew. Chem. Int. Ed. 2005, 44, 7852. [DOI] [PubMed] [Google Scholar]
- 15. Li Y., Sci. China Chem. 2014, 924. [Google Scholar]
- 16. Guo X., Fang G., Li G., Ma H., Fan H., Yu L., Ma C., Wu X., Deng D., Wei M., Tan D., Si R., Zhang S., Li J., Sun L., Tang Z., Pan X., Bao X., Science 2014, 344, 616. [DOI] [PubMed] [Google Scholar]
- 17. Bell A. T., Sci. China Chem. 2014, 923. [Google Scholar]
- 18. Lukowski M. A., Daniel A. S., Meng F., Forticaux A., Li L., Jin S., J. Am. Chem. Soc. 2013, 135, 10274. [DOI] [PubMed] [Google Scholar]
- 19. Hao Y., Ji S., Liu X., Zhang D., Shi D., Sci. China Chem. 2014, 57, 866. [Google Scholar]
- 20. Ji Y., Wu L., Fan Q., Acta Chim. Sinica 2014, 72, 798. [Google Scholar]
- 21. Schlögl R., Abd Hamid S. B., Angew. Chem. Int. Ed. 2004, 43, 1628. [DOI] [PubMed] [Google Scholar]
- 22. Wang Y.‐G., Yoon Y., Glezakou V.‐A., Li J., Rousseau R., J. Am. Chem. Soc. 2013, 135, 10673. [DOI] [PubMed] [Google Scholar]
- 23. Lu Y., Jiang Y., Gao X., Wang X., Chen W., J. Am. Chem. Soc. 2014, 136, 11687. [DOI] [PubMed] [Google Scholar]
- 24. Zeuthen H., Kudernatsch W., Merte L. R., Ono L. K., Lammich L., Besenbacher F., Wendt S., ACS Nano 2015, 9, 573. [DOI] [PubMed] [Google Scholar]
- 25. Xi Z., Acc. Chem. Res. 2010, 43, 1342. [DOI] [PubMed] [Google Scholar]
- 26. Burgess B. K., Lowe D. J., Chem. Rev. 1996, 96, 2983. [DOI] [PubMed] [Google Scholar]
- 27. Semba K., Nakao Y., J. Am. Chem. Soc. 2014, 136, 7567. [DOI] [PubMed] [Google Scholar]
- 28. Xiao B., Niu Z., Wang Y.‐G., Jia W., Shang J., Zhang L., Wang D., Fu Y., Zeng J., He W., Wu K., Li J., Yang J., Liu L., Li Y., J. Am. Chem. Soc. 2015, 137, 3791. [DOI] [PubMed] [Google Scholar]
- 29. Liu Z.‐P., Hu P., J. Am. Chem. Soc. 2003, 125, 1958.12580623 [Google Scholar]
- 30. Yan Y., Xia B., Xu Z., Wang X., ACS Catal. 2014, 4, 1693. [Google Scholar]
- 31. Walter M. G., Warren E. L., McKone J. R., Boettcher S. W., Mi Q., Santori E. A., Lewis N. S., Chem. Rev. 2010, 110, 6446. [DOI] [PubMed] [Google Scholar]
- 32. Greeley J., Jaramillo T. F., Bonde J., Chorkendorff I., Norskov J. K., Nat. Mater. 2006, 5, 909. [DOI] [PubMed] [Google Scholar]
- 33. Rees D. C., Howard J. B., Science 2003, 300, 929. [DOI] [PubMed] [Google Scholar]
- 34. Evans D. J., Pickett C. J., Chem. Soc. Rev. 2003, 32, 268. [DOI] [PubMed] [Google Scholar]
- 35. Hinnemann B., Moses P. G., Bonde J., Jørgensen K. P., Nielsen J. H., Horch S., Chorkendorff I., Nørskov J. K., J. Am. Chem. Soc. 2005, 127, 5308. [DOI] [PubMed] [Google Scholar]
- 36. Jaramillo T. F., Jørgensen K. P., Bonde J., Nielsen J. H., Horch S., Chorkendorff I., Science 2007, 317, 100. [DOI] [PubMed] [Google Scholar]
- 37. Helveg S., Lauritsen J. V., Lægsgaard E., Stensgaard I., Nørskov J. K., Clausen B. S., Topsøe H., Besenbacher F., Phys. Rev. Lett. 2000, 84, 951. [DOI] [PubMed] [Google Scholar]
- 38. Xu M., Liang T., Shi M., Chen H., Chem. Rev. 2013, 113, 3766. [DOI] [PubMed] [Google Scholar]
- 39. Hansen L. P., Ramasse Q. M., Kisielowski C., Brorson M., Johnson E., Topsøe H., Helveg S., Angew. Chem. Int. Ed. 2011, 50, 10153. [DOI] [PubMed] [Google Scholar]
- 40. Bollinger M. V., Lauritsen J. V., Jacobsen K. W., Nørskov J. K., Helveg S., Besenbacher F., Phys. Rev. Lett. 2001, 87, 196803. [DOI] [PubMed] [Google Scholar]
- 41. Byskov L., Nørskov J., Clausen B., Topsøe H., Catal. Lett. 2000, 64, 95. [Google Scholar]
- 42. Lauritsen J. V., Kibsgaard J., Helveg S., Topsoe H., Clausen B. S., Laegsgaard E., Besenbacher F., Nat Nanotechnol. 2007, 2, 53. [DOI] [PubMed] [Google Scholar]
- 43. Chhowalla M., Shin H. S., Eda G., Li L.‐J., Loh K. P., Zhang H., Nat. Chem. 2013, 5, 263. [DOI] [PubMed] [Google Scholar]
- 44. Albu‐Yaron A., Levy M., Tenne R., Popovitz‐Biro R., Weidenbach M., Bar‐Sadan M., Houben L., Enyashin A. N., Seifert G., Feuermann D., Katz E. A., Gordon J. M., Angew. Chem. Int. Ed. 2011, 50, 1810. [DOI] [PubMed] [Google Scholar]
- 45. Laursen A. B., Kegnaes S., Dahl S., Chorkendorff I., Energy Environ. Sci. 2012, 5, 5577. [Google Scholar]
- 46. Karunadasa H. I., Montalvo E., Sun Y., Majda M., Long J. R., Chang C. J., Science 2012, 335, 698. [DOI] [PubMed] [Google Scholar]
- 47. Kibsgaard J., Chen Z., Reinecke B. N., Jaramillo T. F., Nat. Mater. 2012, 11, 963. [DOI] [PubMed] [Google Scholar]
- 48. Xie J., Zhang H., Li S., Wang R., Sun X., Zhou M., Zhou J., Lou X. W., Xie Y., Adv. Mater. 2013, 25, 5807. [DOI] [PubMed] [Google Scholar]
- 49. Li Y., Wang H., Xie L., Liang Y., Hong G., Dai H., J. Am. Chem. Soc. 2011, 133, 7296. [DOI] [PubMed] [Google Scholar]
- 50. Senthilkumar V., Tam L., Kim Y., Sim Y., Seong M.‐J., Jang J. I., Nano Res. 2014, 7, 1759. [Google Scholar]
- 51. Kong D., Wang H., Cha J. J., Pasta M., Koski K. J., Yao J., Cui Y., Nano Lett. 2013, 13, 1341. [DOI] [PubMed] [Google Scholar]
- 52. Wang H., Zhang Q., Yao H., Liang Z., Lee H.‐W., Hsu P.‐C., Zheng G., Cui Y., Nano Lett. 2014, 14, 7138. [DOI] [PubMed] [Google Scholar]
- 53. Wang H., Tsai C., Kong D., Chan K., Abild‐Pedersen F., Nørskov J., Cui Y., Nano Res. 2015, 8, 566. [Google Scholar]
- 54. Gao M.‐R., Liang J.‐X., Zheng Y.‐R., Xu Y.‐F., Jiang J., Gao Q., Li J., Yu S.‐H., Nat. Commun. 2015, 6, 5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang X., Ni B., Chem. Sci. 2015, doi: 10.1039/C5SC00836K. [Google Scholar]
- 56. Meng F., Audrey M. S. A. F., Song J., Acc. Chem. Res. 2013, 46, 1616. [DOI] [PubMed] [Google Scholar]
- 57. Chen L., Liu B., Abbas A. N., Ma Y., Fang X., Liu Y., Zhou C., ACS Nano 2014, 8, 11543. [DOI] [PubMed] [Google Scholar]
- 58. Zhang L., Liu K., Wong A. B., Kim J., Hong X., Liu C., Cao T., Louie S. G., Wang F., Yang P., Nano Lett. 2014, 14, 6418. [DOI] [PubMed] [Google Scholar]
- 59. Zhu Q., Wegener S. L., Xie C., Uche O., Neurock M., Marks T. J., Nat. Chem. 2013, 5, 104. [DOI] [PubMed] [Google Scholar]
- 60. Voiry D., Yamaguchi H., Li J., Silva R., Alves D. C. B., Fujita T., Chen M., Asefa T., Shenoy V. B., Eda G., Chhowalla M., Nat. Mater. 2013, 12, 850. [DOI] [PubMed] [Google Scholar]
- 61. Choi C., Feng J., Li Y., Wu J., Zak A., Tenne R., Dai H., Nano Res. 2013, 6, 921. [Google Scholar]
- 62. Tsai C., Chan K., Abild‐Pedersen F., Norskov J. K., PCCP 2014, 16, 13156. [DOI] [PubMed] [Google Scholar]
- 63. Hashmi A. S. K., Hutchings G. J., Angew. Chem. Int. Ed. 2006, 45, 7896. [DOI] [PubMed] [Google Scholar]
- 64. Haruta M., Kobayashi T., Sano H., Yamada N., Chem. Lett. 1987, 16, 405. [Google Scholar]
- 65. Goeppert A., Czaun M., May R. B., Prakash G. K. S., Olah G. A., Narayanan S. R., J. Am. Chem. Soc. 2011, 133, 20164. [DOI] [PubMed] [Google Scholar]
- 66. Bonin J., Robert M., Routier M., J. Am. Chem. Soc. 2014, 136, 16768. [DOI] [PubMed] [Google Scholar]
- 67. Yadav R. K., Oh G. H., Park N.‐J., Kumar A., Kong K.‐j., Baeg J.‐O., J. Am. Chem. Soc. 2014, 136, 16728. [DOI] [PubMed] [Google Scholar]
- 68. Li X., Wen J., Low J., Fang Y., Yu J., Sci. China Mater. 2014, 57, 70. [Google Scholar]
- 69. Peterson A. A., Nørskov J. K., J. Phys. Chem. Lett. 2012, 3, 251. [Google Scholar]
- 70. Peterson A. A., Abild‐Pedersen F., Studt F., Rossmeisl J., Norskov J. K., Energy Environ. Sci. 2010, 3, 1311. [Google Scholar]
- 71. Durand W. J., Peterson A. A., Studt F., Abild‐Pedersen F., Nørskov J. K., Surf. Sci. 2011, 605, 1354. [Google Scholar]
- 72. Hansen H. A., Varley J. B., Peterson A. A., Nørskov J. K., J. Phys. Chem. Lett. 2013, 4, 388. [DOI] [PubMed] [Google Scholar]
- 73. Kauffman D. R., Alfonso D., Matranga C., Qian H., Jin R., J. Am. Chem. Soc. 2012, 134, 10237. [DOI] [PubMed] [Google Scholar]
- 74. Mistry H., Reske R., Zeng Z., Zhao Z.‐J., Greeley J., Strasser P., Cuenya B. R., J. Am. Chem. Soc. 2014, 136, 16473. [DOI] [PubMed] [Google Scholar]
- 75. Zhu W., Zhang Y.‐J., Zhang H., Lv H., Li Q., Michalsky R., Peterson A. A., Sun S., J. Am. Chem. Soc. 2014, 136, 16132. [DOI] [PubMed] [Google Scholar]
- 76. Ertl G., Neumann M., Streit K. M., Surf. Sci. 1977, 64, 393. [Google Scholar]
- 77. Yeo Y. Y., Vattuone L., King D. A., J. Chem. Phys. 1997, 106, 392. [Google Scholar]
- 78. Fischer‐Wolfarth J.‐H., Hartmann J., Farmer J. A., Flores‐Camacho J. M., Campbell C. T., Schauermann S., Freund H.‐J., Rev. Sci. Instrum. 2011, 82, 024102. [DOI] [PubMed] [Google Scholar]
- 79. Golibrzuch K., Shirhatti P. R., Geweke J., Werdecker J., Kandratsenka A., Auerbach D. J., Wodtke A. M., Bartels C., J. Am. Chem. Soc. 2014, 137, 1465. [DOI] [PubMed] [Google Scholar]
- 80. Kuhl K. P., Hatsukade T., Cave E. R., Abram D. N., Kibsgaard J., Jaramillo T. F., J. Am. Chem. Soc. 2014, 136, 14107. [DOI] [PubMed] [Google Scholar]
- 81. Schouten K. J. P., Kwon Y., van der Ham C. J., Qin Z., Koper M. T. M., Chem. Sci. 2011, 2, 1902. [Google Scholar]
- 82. Schouten K. J. P., Qin Z., Gallent E. P., Koper M. T. M., J. Am. Chem. Soc. 2012, 134, 9864. [DOI] [PubMed] [Google Scholar]
- 83. Kim Y.‐G., Baricuatro J. H., Javier A., Gregoire J. M., Soriaga M. P., Langmuir 2014, 30, 15053. [DOI] [PubMed] [Google Scholar]
- 84. Lim H.‐K., Shin H., Goddard W. A., Hwang Y. J., Min B. K., Kim H., J. Am. Chem. Soc. 2014, 136, 11355. [DOI] [PubMed] [Google Scholar]
- 85. Manthiram K., Beberwyck B. J., Alivisatos A. P., J. Am. Chem. Soc. 2014, 136, 13319. [DOI] [PubMed] [Google Scholar]
- 86. Kim D., Resasco J., Yu Y., Asiri A. M., Yang P., Nat. Commun. 2014, 5, 4948. [DOI] [PubMed] [Google Scholar]
- 87. Tauster S. J., Fung S. C., Garten R. L., J. Am. Chem. Soc. 1978, 100, 170. [Google Scholar]
- 88. Tauster S. J., Acc. Chem. Res. 1987, 20, 389. [Google Scholar]
- 89. Bonanni S., Aït‐Mansour K., Brune H., Harbich W., ACS Catal. 2011, 1, 385. [Google Scholar]
- 90. Li L., Zeng X. C., J. Am. Chem. Soc. 2014, 136, 15857. [DOI] [PubMed] [Google Scholar]
- 91. Xia B. Y., Wu H. B., Yan Y., Wang H. B., Wang X., Small 2014, 10, 2336. [DOI] [PubMed] [Google Scholar]
- 92. Subbaraman R., Tripkovic D., Strmcnik D., Chang K.‐C., Uchimura M., Paulikas A. P., Stamenkovic V., Markovic N. M., Science 2011, 334, 1256. [DOI] [PubMed] [Google Scholar]
- 93. Fu Q., Yang F., Bao X., Acc. Chem. Res. 2013, 46, 1692. [DOI] [PubMed] [Google Scholar]
- 94. Fu Q., Li W.‐X., Yao Y., Liu H., Su H.‐Y., Ma D., Gu X.‐K., Chen L., Wang Z., Zhang H., Wang B., Bao X., Science 2010, 328, 1141. [DOI] [PubMed] [Google Scholar]
- 95. Mu R., Guo X., Fu Q., Bao X., J. Phys. Chem. C 2011, 115, 20590. [Google Scholar]
- 96. Mu R., Fu Q., Xu H., Zhang H., Huang Y., Jiang Z., Zhang S., Tan D., Bao X., J. Am. Chem. Soc. 2011, 133, 1978. [DOI] [PubMed] [Google Scholar]
- 97. Xu H., Fu Q., Guo X., Bao X., ChemCatChem 2012, 4, 1645. [Google Scholar]
- 98. Yao Y., Fu Q., Wang Z., Tan D., Bao X., J. Phys. Chem. C 2010, 114, 17069. [Google Scholar]
- 99. Wang Y., Zhang H., Yao X., Zhao H., J. Phys. Chem. C 2013, 117, 1672. [Google Scholar]
- 100. Gu X.‐K., Ouyang R., Sun D., Su H.‐Y., Li W.‐X., ChemSusChem 2012, 5, 871. [DOI] [PubMed] [Google Scholar]
- 101. Wang W., Zhang H., Wang W., Zhao A., Wang B., Hou J. G., Chem. Phys. Lett. 2010, 500, 76. [Google Scholar]
- 102. Merte L. R., Knudsen J., Grabow L. C., Vang R. T., Lægsgaard E., Mavrikakis M., Besenbacher F., Surf. Sci. 2009, 603, L15. [Google Scholar]
- 103. Huang W., Ranke W., Surf. Sci. 2006, 600, 793. [Google Scholar]
- 104. Xu L., Wu Z., Zhang Y., Chen B., Jiang Z., Ma Y., Huang W., J. Phys. Chem. C 2011, 115, 14290. [Google Scholar]
- 105. Rodriguez J. A., Ma S., Liu P., Hrbek J., Evans J., Pérez M., Science 2007, 318, 1757. [DOI] [PubMed] [Google Scholar]
- 106. Ketteler G., Ranke W., J. Phys. Chem. B 2003, 107, 4320. [Google Scholar]
- 107. Ebensperger C., Gubo M., Meyer W., Hammer L., Heinz K., Phys. Rev. B 2010, 81, 235405. [Google Scholar]
- 108. Franz T., Zabloudil J., Mittendorfer F., Gragnaniello L., Parteder G., Allegretti F., Surnev S., Netzer F. P., J. Phys. Chem. Lett. 2011, 3, 186. [Google Scholar]
- 109. Freund H.‐J., Pacchioni G., Chem. Soc. Rev. 2008, 37, 2224. [DOI] [PubMed] [Google Scholar]
- 110. Surnev S., Fortunelli A., Netzer F. P., Chem. Rev. 2012, 113, 4314. [DOI] [PubMed] [Google Scholar]
- 111. An K., Alayoglu S., Musselwhite N., Plamthottam S., Melaet G., Lindeman A. E., Somorjai G. A., J. Am. Chem. Soc. 2013, 135, 16689. [DOI] [PubMed] [Google Scholar]
- 112. Willinger M. G., Zhang W., Bondarchuk O., Shaikhutdinov S., Freund H.‐J., Schlögl R., Angew. Chem. Int. Ed. 2014, 53, 5998. [DOI] [PubMed] [Google Scholar]
- 113. Liu P., Ge X., Wang R., Ma H., Ding Y., Langmuir 2009, 25, 561. [DOI] [PubMed] [Google Scholar]
- 114. Bai Y., Zhang W., Zhang Z., Zhou J., Wang X., Wang C., Huang W., Jiang J., Xiong Y., J. Am. Chem. Soc. 2014, 136, 14650. [DOI] [PubMed] [Google Scholar]
- 115. Matthey D., Wang J. G., Wendt S., Matthiesen J., Schaub R., Lægsgaard E., Hammer B., Besenbacher F., Science 2007, 315, 1692. [DOI] [PubMed] [Google Scholar]
- 116. Green I. X., Tang W., Neurock M., Yates J. T., Science 2011, 333, 736. [DOI] [PubMed] [Google Scholar]
- 117. McEntee M., Tang W., Neurock M., Yates J. T., J. Am. Chem. Soc. 2014, 136, 5116. [DOI] [PubMed] [Google Scholar]
- 118. Xia B. Y., Wu H. B., Wang X., Lou X. W., Angew. Chem. 2013, 125, 12563. [Google Scholar]
- 119. Xia B. Y., Wu H. B., Yan Y., Lou X. W., Wang X., J. Am. Chem. Soc. 2013, 135, 9480. [DOI] [PubMed] [Google Scholar]
- 120. Xia B. Y., Wu H. B., Li N., Yan Y., Lou X. W., Wang X., Angew. Chem. 2015, 127, 3868. [DOI] [PubMed] [Google Scholar]
- 121. Zhang H., Jin M., Xia Y., Angew. Chem. Int. Ed. 2012, 51, 7656. [DOI] [PubMed] [Google Scholar]
- 122. Zhang H., Li W., Jin M., Zeng J., Yu T., Yang D., Xia Y., Nano Lett. 2010, 11, 898. [DOI] [PubMed] [Google Scholar]
- 123. Jin M., Zhang H., Xie Z., Xia Y., Angew. Chem. Int. Ed. 2011, 50, 7850. [DOI] [PubMed] [Google Scholar]
- 124. Xia X., Zeng J., McDearmon B., Zheng Y., Li Q., Xia Y., Angew. Chem. Int. Ed. 2011, 50, 12542. [DOI] [PubMed] [Google Scholar]
- 125. Zhang J., Langille M. R., Personick M. L., Zhang K., Li S., Mirkin C. A., J. Am. Chem. Soc. 2010, 132, 14012. [DOI] [PubMed] [Google Scholar]
- 126. Huang X., Zhao Z., Fan J., Tan Y., Zheng N., J. Am. Chem. Soc. 2011, 133, 4718. [DOI] [PubMed] [Google Scholar]
- 127. Kovalenko M. V., Manna L., Cabot A., Hens Z., Talapin D. V., Kagan C. R., Klimov V. I., Rogach A. L., Reiss P., Milliron D. J., Guyot‐Sionnnest P., Konstantatos G., Parak W. J., Hyeon T., Korgel B. A., Murray C. B., Heiss W., ACS Nano 2015. 9, 1012. [DOI] [PubMed] [Google Scholar]
- 128. Fan N., Yang Y., Wang W., Zhang L., Chen W., Zou C., Huang S., ACS Nano 2012, 6, 4072. [DOI] [PubMed] [Google Scholar]
- 129. Wu X., Yu Y., Liu Y., Xu Y., Liu C., Zhang B., Angew. Chem. Int. Ed. 2012, 51, 3211. [DOI] [PubMed] [Google Scholar]
- 130. Xie S., Lu N., Xie Z., Wang J., Kim M. J., Xia Y., Angew. Chem. Int. Ed. 2012, 51, 10266. [DOI] [PubMed] [Google Scholar]
- 131. Wang Z. L., J. Phys. Chem. B 2000, 104, 1153. [Google Scholar]
- 132. Métraux G. S., Cao Y. C., Jin R., Mirkin C. A., Nano Lett. 2003, 3, 519. [Google Scholar]
- 133. Shahjamali M. M., Bosman M., Cao S., Huang X., Cao X., Zhang H., Pramana S. S., Xue C., Small 2013, 9, 2880. [DOI] [PubMed] [Google Scholar]
- 134. McEachran M., Keogh D., Pietrobon B., Cathcart N., Gourevich I., Coombs N., Kitaev V., J. Am. Chem. Soc. 2011, 133, 8066. [DOI] [PubMed] [Google Scholar]
- 135. Macdonald J. E., Bar Sadan M., Houben L., Popov I., Banin U., Nat. Mater. 2010, 9, 810. [DOI] [PubMed] [Google Scholar]
- 136. Han C., Ge L., Chen C., Li Y., Zhao Z., Xiao X., Li Z., Zhang J., J. Mater. Chem. A 2014, 2, 12594. [Google Scholar]
- 137. Yin Y., Rioux R. M., Erdonmez C. K., Hughes S., Somorjai G. A., Alivisatos A. P., Science 2004, 304, 711. [DOI] [PubMed] [Google Scholar]
- 138. Chen C., Kang Y., Huo Z., Zhu Z., Huang W., Xin H. L., Snyder J. D., Li D., Herron J. A., Mavrikakis M., Chi M., More K. L., Li Y., Markovic N. M., Somorjai G. A., Yang P., Stamenkovic V. R., Science 2014, 343, 1339. [DOI] [PubMed] [Google Scholar]
- 139. Wang Y., Chen Y., Nan C., Li L., Wang D., Peng Q., Li Y., Nano Res. 2015, 8, 140. [Google Scholar]
- 140. Wu Y., Wang D., Zhou G., Yu R., Chen C., Li Y., J. Am. Chem. Soc. 2014, 136, 11594. [DOI] [PubMed] [Google Scholar]
- 141. Xia B. Y., Wu H. B., Wang X., Lou X. W., J. Am. Chem. Soc. 2012, 134, 13934. [DOI] [PubMed] [Google Scholar]
- 142. Nosheen F., Zhang Z.‐c., Zhuang J., Wang X., Nanoscale 2013, 5, 3660. [DOI] [PubMed] [Google Scholar]
- 143. Hong X., Wang D., Cai S., Rong H., Li Y., J. Am. Chem. Soc. 2012, 134, 18165. [DOI] [PubMed] [Google Scholar]
- 144. Lu X., Au L., McLellan J., Li Z.‐Y., Marquez M., Xia Y., Nano Lett. 2007, 7, 1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Zhang Q., Cobley C. M., Zeng J., Wen L.‐P., Chen J., Xia Y., J. Phys. Chem. C 2010, 114, 6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Skrabalak S. E., Chen J., Sun Y., Lu X., Au L., Cobley C. M., Xia Y., Acc. Chem. Res. 2008, 41, 1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kuo C.‐H., Huang M. H., J. Am. Chem. Soc. 2008, 130, 12815. [DOI] [PubMed] [Google Scholar]
- 148. Tsai Y.‐H., Chiu C.‐Y., Huang M. H., J. Phys. Chem. C 2013, 117, 24611. [Google Scholar]
- 149. Sui Y., Fu W., Zeng Y., Yang H., Zhang Y., Chen H., Li Y., Li M., Zou G., Angew. Chem. Int. Ed. 2010, 49, 4282. [DOI] [PubMed] [Google Scholar]
- 150. Lyu L.‐M., Huang M. H., J. Phys. Chem. C 2011, 115, 17768. [Google Scholar]
- 151. Yavuz M. S., Cheng Y., Chen J., Cobley C. M., Zhang Q., Rycenga M., Xie J., Kim C., Song K. H., Schwartz A. G., Wang L. V., Xia Y., Nat. Mater. 2009, 8, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Mulvihill M. J., Ling X. Y., Henzie J., Yang P., J. Am. Chem. Soc. 2009, 132, 268. [DOI] [PubMed] [Google Scholar]
- 153. Mahmoud M. A., El‐Sayed M. A., J. Phys. Chem. C 2008, 112, 14618. [Google Scholar]
- 154. Mahmoud M. A., El‐Sayed M. A., Nano Lett. 2009, 9, 3025. [DOI] [PubMed] [Google Scholar]
- 155. Oezaslan M., Heggen M., Strasser P., J. Am. Chem. Soc. 2011, 134, 514. [DOI] [PubMed] [Google Scholar]
- 156. Su L., Jia W., Li C.‐M., Lei Y., ChemSusChem 2014, 7, 361. [DOI] [PubMed] [Google Scholar]
- 157. Zeng J., Zhang Q., Chen J., Xia Y., Nano Lett. 2009, 10, 30. [DOI] [PubMed] [Google Scholar]
- 158. Mahmoud M. A., Qian W., El‐Sayed M. A., Nano Lett. 2011, 11, 3285. [DOI] [PubMed] [Google Scholar]
- 159. Dirac P. A. M., Proc. R. Soc. London 1929, 123, 714. [Google Scholar]
- 160. Cademartiri L., Ozin G. A., Adv. Mater. 2009, 21, 1013. [Google Scholar]
- 161. Hong B. H., Bae S. C., Lee C.‐W., Jeong S., Kim K. S., Science 2001, 294, 348. [DOI] [PubMed] [Google Scholar]
- 162. Huo Z., Tsung C.‐k., Huang W., Zhang X., Yang P., Nano Lett. 2008, 8, 2041. [DOI] [PubMed] [Google Scholar]
- 163. Pazos‐Pérez N., Baranov D., Irsen S., Hilgendorff M., Liz‐Marzán L. M., Giersig M., Langmuir 2008, 24, 9855. [DOI] [PubMed] [Google Scholar]
- 164. Feng H., Yang Y., You Y., Li G., Guo J., Yu T., Shen Z., Wu T., Xing B., Chem. Commun. 2009, 1984. [DOI] [PubMed] [Google Scholar]
- 165. Peng X., Adv. Mater. 2003, 15, 459. [Google Scholar]
- 166. Xu X., Zhuang J., Wang X., J. Am. Chem. Soc. 2008, 130, 12527. [DOI] [PubMed] [Google Scholar]
- 167. Yang H., Finefrock S. W., Albarracin Caballero J. D., Wu Y., J. Am. Chem. Soc. 2014, 136, 10242. [DOI] [PubMed] [Google Scholar]
- 168. Hu S., Wang X., Chem. Soc. Rev. 2013, 42, 5577. [DOI] [PubMed] [Google Scholar]
- 169. Dai S., Zhao J., He M.‐r., Wang X., Wan J., Shan Z., Zhu J., Nano Lett. 2014, 15, 8. [DOI] [PubMed] [Google Scholar]
- 170. Hu S., Liu H., Wang P., Wang X., J. Am. Chem. Soc. 2013, 135, 11115. [DOI] [PubMed] [Google Scholar]
- 171. Wang P.‐p., Yang Y., Zhuang J., Wang X., J. Am. Chem. Soc. 2013, 135, 6834. [DOI] [PubMed] [Google Scholar]
- 172. Kondo Y., Takayanagi K., Science 2000, 289, 606. [DOI] [PubMed] [Google Scholar]
- 173. Oshima Y., Koizumi H., Mouri K., Hirayama H., Takayanagi K., Kondo Y., Phys. Rev. B 2002, 65, 121401. [Google Scholar]
- 174. Lacroix L.‐M., Arenal R., Viau G., J. Am. Chem. Soc. 2014, 136, 13075. [DOI] [PubMed] [Google Scholar]
- 175. Velázquez‐Salazar J. J., Esparza R., Mejía‐Rosales S. J., Estrada‐Salas R., Ponce A., Deepak F. L., Castro‐Guerrero C., José‐Yacamán M., ACS Nano 2011, 5, 6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Novoselov K. S., Geim A. K., Morozov S. V., Jiang D., Zhang Y., Dubonos S. V., Grigorieva I. V., Firsov A. A., Science 2004, 306, 666. [DOI] [PubMed] [Google Scholar]
- 177. Schwierz F., Nat. Nanotechnol. 2010, 5, 487. [DOI] [PubMed] [Google Scholar]
- 178. Abergel D. S. L., Apalkov V., Berashevich J., Ziegler K., Chakraborty T., Adv. Phys. 2010, 59, 261. [Google Scholar]
- 179. Malola S., Häkkinen H., Koskinen P., Eur. Phys. J. D 2009, 52, 71. [Google Scholar]
- 180. Savin A. V., Kivshar Y. S., Phys. Rev. B 2010, 81, 165418. [Google Scholar]
- 181. Han M. Y., Özyilmaz B., Zhang Y., Kim P., Phys. Rev. Lett. 2007, 98, 206805. [DOI] [PubMed] [Google Scholar]
- 182. Dubois S. M. M., Lopez‐Bezanilla A., Cresti A., Triozon F., Biel B., Charlier J.‐C., Roche S., ACS Nano 2010, 4, 1971. [DOI] [PubMed] [Google Scholar]
- 183. Nika D. L., Pokatilov E. P., Askerov A. S., Balandin A. A., Phys. Rev. B 2009, 79, 155413. [Google Scholar]
- 184. Jia X., Campos‐Delgado J., Terrones M., Meunier V., Dresselhaus M. S., Nanoscale 2011, 3, 86. [DOI] [PubMed] [Google Scholar]
- 185. Zhang X., Xin J., Ding F., Nanoscale 2013, 5, 2556. [DOI] [PubMed] [Google Scholar]
- 186. Yang W., Ratinac K. R., Ringer S. P., Thordarson P., Gooding J. J., Braet F., Angew. Chem. Int. Ed. 2010, 49, 2114. [DOI] [PubMed] [Google Scholar]
- 187. Jia X., Hofmann M., Meunier V., Sumpter B. G., Campos‐Delgado J., Romo‐Herrera J. M., Son H., Hsieh Y.‐P., Reina A., Kong J., Terrones M., Dresselhaus M. S., Science 2009, 323, 1701. [DOI] [PubMed] [Google Scholar]
- 188. Koskinen P., Malola S., Häkkinen H., Phys. Rev. Lett. 2008, 101, 115502. [DOI] [PubMed] [Google Scholar]
- 189. Radovic L. R., Bockrath B., J. Am. Chem. Soc. 2005, 127, 5917. [DOI] [PubMed] [Google Scholar]
- 190. Klein D. J., Bytautas L., J. Phys. Chem. A 1999, 103, 5196. [Google Scholar]
- 191. He K., Robertson A. W., Lee S., Yoon E., Lee G.‐D., Warner J. H., ACS Nano 2014, 8, 12272. [DOI] [PubMed] [Google Scholar]
- 192. Girit Ç. Ö., Meyer J. C., Erni R., Rossell M. D., Kisielowski C., Yang L., Park C.‐H., Crommie M. F., Cohen M. L., Louie S. G., Zettl A., Science 2009, 323, 1705. [DOI] [PubMed] [Google Scholar]
- 193. Tao C., Jiao L., Yazyev O. V., Chen Y.‐C., Feng J., Zhang X., Capaz R. B., Tour J. M., Zettl A., Louie S. G., Dai H., Crommie M. F., Nat. Phys. 2011, 7, 616. [Google Scholar]
- 194. Casiraghi C., Hartschuh A., Qian H., Piscanec S., Georgi C., Fasoli A., Novoselov K. S., Basko D. M., Ferrari A. C., Nano Lett. 2009, 9, 1433. [DOI] [PubMed] [Google Scholar]
- 195. Zhou L., Zhang L., Liao L, Yang M., Xie Q., Peng H., Liu Z., Liu Z., Acta Chim. Sinica 2014, 72, 289. [Google Scholar]
- 196. Gao J., Zhao J., Ding F., J. Am. Chem. Soc. 2012, 134, 6204. [DOI] [PubMed] [Google Scholar]
- 197. Shu H., Chen X., Tao X., Ding F., ACS Nano 2012, 6, 3243. [DOI] [PubMed] [Google Scholar]
- 198. Kosynkin D. V., Higginbotham A. L., Sinitskii A., Lomeda J. R., Dimiev A., Price B. K., Tour J. M., Nature 2009, 458, 872. [DOI] [PubMed] [Google Scholar]
- 199. Cano‐Márquez A. G., Rodríguez‐Macías F. J., Campos‐Delgado J., Espinosa‐González C. G., Tristán‐López F., Ramírez‐González D., Cullen D. A., Smith D. J., Terrones M., Vega‐Cantú Y. I., Nano Lett. 2009, 9, 1527. [DOI] [PubMed] [Google Scholar]
- 200. Campos L. C., Manfrinato V. R., Sanchez‐Yamagishi J. D., Kong J., Jarillo‐Herrero P., Nano Lett. 2009, 9, 2600. [DOI] [PubMed] [Google Scholar]
- 201. Jiao L., Zhang L., Wang X., Diankov G., Dai H., Nature 2009, 458, 877. [DOI] [PubMed] [Google Scholar]
- 202. Tapaszto L., Dobrik G., Lambin P., Biro L. P., Nat. Nanotechnol. 2008, 3, 397. [DOI] [PubMed] [Google Scholar]
- 203. Wang X., Dai H., Nat. Chem. 2010, 2, 661. [DOI] [PubMed] [Google Scholar]
- 204. Huang Y., Wu J., Xu X., Ho Y., Ni G., Zou Q., Koon G., Zhao W., Castro Neto A. H., Eda G., Shen C., Özyilmaz B., Nano Res. 2013, 6, 200. [Google Scholar]
- 205. Basagni A., Sedona F., Pignedoli C. A., Cattelan M., Nicolas L., Casarin M., Sambi M., J. Am. Chem. Soc. 2015, 137, 1802. [DOI] [PubMed] [Google Scholar]
- 206. Li Z., Yang J., Hou J. G., J. Am. Chem. Soc. 2008, 130, 4224. [DOI] [PubMed] [Google Scholar]
- 207. Boukhvalov D. W., Katsnelson M. I., Nano Lett. 2008, 8, 4373. [DOI] [PubMed] [Google Scholar]
- 208. Chen C., Long M., Wu H., Cai W., Sci. China Chem. 2013, 56, 354. [Google Scholar]
- 209. Zhou L., Zhang L., Liao L, Yang M., Xie Q., Peng H., Liu Z., Liu Z., Acta Chim. Sinica 2014, 72, 289. [Google Scholar]
- 210. Georgakilas V., Otyepka M., Bourlinos A. B., Chandra V., Kim N., Kemp K. C., Hobza P., Zboril R., Kim K. S., Chem. Rev. 2012, 112, 6156. [DOI] [PubMed] [Google Scholar]
- 211. Muge A., Yves J. C., Jpn. J. Appl. Phys. 2011, 50, 070101. [Google Scholar]
- 212. Zach M. P., Ng K. H., Penner R. M., Science 2000, 290, 2120. [DOI] [PubMed] [Google Scholar]
- 213. Walter E. C., Zach M. P., Favier F., Murray B. J., Inazu K., Hemminger J. C., Penner R. M., ChemPhysChem 2003, 4, 131. [DOI] [PubMed] [Google Scholar]
- 214. Banks C. E., Davies T. J., Wildgoose G. G., Compton R. G., Chem. Commun. 2005, 829. [DOI] [PubMed] [Google Scholar]
- 215. Patel A. N., Collignon M. G., O'Connell M. A., Hung W. O. Y., McKelvey K., Macpherson J. V., Unwin P. R., J. Am. Chem. Soc. 2012, 134, 20117. [DOI] [PubMed] [Google Scholar]
- 216. Anne A., Cambril E., Chovin A., Demaille C., Goyer C., ACS Nano 2009, 3, 2927. [DOI] [PubMed] [Google Scholar]
- 217. Yuan W., Zhou Y., Li Y., Li C., Peng H., Zhang J., Liu Z., Dai L., Shi G., Sci. Rep. 2013, 3, 2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Li W., Zhang G., Guo M., Zhang Y.‐W., Nano Res. 2014, 7, 518. [Google Scholar]