

Abstract
The dentate gyrus plays critical roles both in cognitive processing, and in regulation of the induction and propagation of pathological activity. The cellular and circuit mechanisms underlying these diverse functions overlap extensively. At the cellular level, the intrinsic properties of dentate granule cells combine to endow these neurons with a fundamental reluctance to activate, one of their hallmark traits. At the circuit level, the dentate gyrus constitutes one of the more heavily inhibited regions of the brain, with strong, fast feedforward and feedback GABAergic inhibition dominating responses to afferent activation. In pathologic states such as epilepsy, a number of alterations within the dentate gyrus combine to compromise the regulatory properties of this circuit, culminating in a collapse of its normal function. This epilepsy-associated transformation in the fundamental properties of this critical regulatory hippocampal circuit may contribute both to seizure propensity, and cognitive and emotional comorbidities characteristic of this disease state.
Keywords: Epilepsy, Dentate gyrus, Hippocampus, GABA, Patch clamp, Functional imaging, Calcium imaging, Dentate granule cells
1 INTRODUCTION
Situated as the initial component of the canonical trisynaptic circuit, the dentate gyrus (DG) is a critical entry point to the hippocampus, functioning as a key regulator of cortical input to the limbic system. The DG is involved in the performance of hippocampal-dependent tasks and is postulated to accomplish these cognitive functions through a transformation of highly active and multimodal afferent cortical inputs into a sparse neural code in which very few of its principal cells activate. In this chapter, we will discuss how the sparse, selective activation properties of the DG’s principal cells, dentate granule cells (DGCs), are critical to hippocampal cognitive function and how this characteristic firing property plays a key role in the regulation of seizure activity in diseases such as epilepsy. Finally, we will consider mechanisms that may erode appropriate DG circuit activation and play a pivotal role in epileptogenesis, seizure propensity in epilepsy, as well as the cognitive comorbidities associated with the disease.
In vivo recording studies have demonstrated that DGCs exhibit spatially selective firing in extremely small populations (Chawla et al., 2005; Jung and McNaughton, 1993). This characteristic sparse activation is thought to enable DGCs to participate in the execution of cognitive functions such as pattern separation and novelty detection (Leutgeb et al., 2007). The propensity for DGCs to generate action potentials (APs) is normally tightly constrained by a combination of cell-intrinsic properties and powerful local inhibitory control, culminating in a population of neurons that are extremely reluctant to activate. However, when DGCs do activate, they exert strong excitatory influence on their downstream targets. A secondary consequence of the DG’s low excitability is its ability to restrict relay of pathological, synchronous cortical activity into the hippocampus and regulate seizure activity in diseases such as epilepsy: a phenomenon termed “dentate gating” (Lothman et al., 1992). However, if this regulatory ability becomes compromised, the DG’s powerful excitatory influence on its downstream targets in the hippocampus can allow it to relay and amplify synchronous pathological activity through the limbic system, potentially fomenting seizure activity.
These characteristic DG circuit properties are significantly disrupted both in humans with epilepsy and in animal models of the disorder. A large aggregation of cellular and circuit alterations occurs in the DG during epilepsy development, including sprouting of pathologic, recurrent excitatory networks, molecular and cellular alterations of local inhibitory circuits, aberrant neurogenesis, astrocytic gliosis, and changes in the intrinsic properties of DGCs. These pathology-associated alterations have generated a prevalent hypothesis that the DG’s normal gating function is compromised during epilepsy and its development and contributes to increased seizure propensity. However, this DG gate failure hypothesis has yet to receive adequate experimental support, and if found to exist, the exact mechanisms mediating such a circuit collapse are likely mechanistically complex.
2 ACTIVITY IN THE DG IS SPARSE
Among the most unusual properties of the DG is the uniquely low level of neuronal activity among its principal cell population. Assays measuring DGC activity over the last several decades have consistently described DGC activation as unusually low compared to many other brain areas. In vivo unit recordings during spatial navigation tasks have demonstrated exceedingly low levels of DGC activity; the vast majority of recorded DGCs display extremely low mean firing rates (typically <0.5 Hz) and activate only in tightly restricted spatial and directional receptive fields (Jung and McNaughton, 1993; Leutgeb et al., 2007; Neunuebel and Knierim, 2012). Moreover, studies examining neuronal expression of activity-dependent immediate-early genes such as c-Fos, Zif286, and Arc as surrogate measures of DGC AP firing have found exceptionally sparse labeling in DGCs, with only ~2–5% of DGCs expressing these genes, even after exposure to multiple environments or spatial cognitive tasks (Alme et al., 2010; Chawla et al., 2005; Tashiro et al., 2007). In comparison, similar spatial navigation sessions activate immediate-early gene expression in ~40% of hippocampal pyramidal cells in downstream CA subfields (Guzowski et al., 1999), highlighting the comparative paucity of DGC activation.
The extremely sparse activation of DGCs is particularly puzzling given the anatomy of their primary afferents from the entorhinal cortex (EC). DGCs receive massively convergent and divergent inputs from the medial and lateral entorhinal cortices. Singly labeled EC stellate cells projecting to the DG can project to as much as 1/3 of the rodent DG, making en passant synapses with DGCs throughout this extensive axonal arbor (Tamamaki and Nojyo, 1993). Conversely, retrograde tracing studies have shown that discrete regions of the DG can receive inputs from substantial portions of the EC, with a single DGC receiving inputs from 3600 to 5600 neurons widely distributed across the EC (Patton and McNaughton, 1995; van Groen et al., 2003). In addition to the massive convergence and divergence of DG afferents, the originating EC stellate cells of this pathway, particularly “grid cells” in the medial EC, are promiscuously active. These neurons exhibit large-scale activation and demonstrate tessellated grid-like receptive fields across spatial environments (Fyhn et al., 2004, 2007). Given the repeating nature of grid cell activity throughout spatial environments, it appears that a significant portion of DG afferents are almost always active, given that the vertices of grid cells receptive fields comprise approximately 20% of the entire environment (Lisman, 2011). Thus, the convergence, divergence, and nearly continuous activity of the EC perforant path projection provide substantial excitatory input to the DG.
Given this robust and constant excitatory drive impinging onto DGCs from the EC, why are so few of these cells active during behavioral tasks? It is likely that cell-intrinsic biophysical properties of DGCs contribute in part to their relative inexcitability. DGCs exhibit hyperpolarized membrane potentials compared to most hippocampal neurons, typically resting near −85 to −90 mV, and their high membrane resistance endows them with a long time constant for integrating synaptic inputs. Further, they lack regenerative calcium conductances to permit phasic or “burst mode” firing and they exhibit remarkable spike frequency adaptation during sustained depolarizations (Fricke and Prince, 1984; Spruston and Johnston, 1992; Staley et al., 1992). Finally, DGC dendrites display significant attenuation of synaptic input owing to the lack of active conductances that would allow dendritic spiking or input amplification (Krueppel et al., 2011). This combination of properties endows DGCs with an innate reluctance to fire APs.
In addition to these relatively hypoexcitable cell-intrinsic properties, DGCs reside in a strongly inhibitory local network, which provides rich feedforward, feedback, and tonic inhibitory input. Feedforward inhibitory drive onto DGCs is particularly strong due to a number of factors. First, several classes of feedforward inhibitory interneurons, including both somatically targeting fast-spiking interneurons on the granule cell layer’s periphery as well as dendritically targeting hilar interneurons, are far more easily recruited by perforant path activation than are DGCs (Ewell and Jones, 2010; Scharfman, 1991). Second, the fast-spiking basket cell interneurons of the DG are particularly specialized to integrate afferent inputs, generate APs, and powerfully inhibit DGCs upon perforant path activation with extraordinary rapidity. Among the many specializations of these basket cells are high K+–Na+ conductance ratios in their dendrites which allow for rapid and temporally precise integration and activation (Hu et al., 2010), nonuniform cable properties that accelerate the time course of fast somatic synaptic potentials and elevate the efficacy of slower distal inputs (Nörenberg et al., 2010), and highly efficient calcium-buffering abilities that allow them to rapidly and repeatedly couple synchronous transmitter release with AP firing (Aponte et al., 2008; see Hu et al., 2014; for review of fast-spiking interneurons). This fast, strong feedforward inhibition would function to constrain DGC firing, and limit firing rates of DGCs when spiking occurs, because this inhibitory input primarily targets DGC somata and axon initial segments, the cellular domains of synaptic integration of excitatory and inhibitory inputs.
Third, feedback inhibition in the DG also appears particularly specialized. DGC axons are normally not present in the molecular layer or granule cell layer, but are instead restricted exclusively to the hilus (Claiborne et al., 1986), where 50% of their target cell population is comprised of GABAergic interneurons (Houser, 2007; Houser and Esclapez, 1994), suggesting that many of the DGCs’ postsynaptic targets may be inhibitory neurons. DGCs are unique among cortical principal cells in that they are endowed with a repertoire of differing synaptic terminal types in their axons. These axons give rise to 7–12 mossy fiber boutons which synapse with excitatory hilar mossy cells and onto CA3 pyramidal cells, but they have a far larger number (100–150) of smaller terminals, including filopodial extensions and en passant synapses, which primarily target interneurons (Acsády et al., 1998). Due to this target selectivity, the vast majority of DGCs’ postsynaptic targets may be inhibitory interneurons. These targets include both hilar feedback interneurons (a majority of which are somatostatin positive) that provide both dendritic feedback inhibition onto DGCs, which limits the strength of cortical afferent inputs, as well as fast-spiking interneurons with somatically targeted inhibition (typically also performing feedforward functions). This biases synaptic integration toward restriction of DGC activation (Freund and Buzsáki, 1996). This feedback inhibition is hypothesized to contribute to making the DG a competitive network, or a “winner-take-all” scheme, in which the activation of a sparse population of DGCs effectively silences the remaining majority of the population (de Almeida et al., 2009a,b; Rolls, 2010).
Given the high levels of activity of its upstream cortical afferents in relation to the surprisingly low activity levels of its principal cells, the information transformation within the DG has been described as a “sparsification” operation or as a form of “sparse coding” in which the DG’s output becomes a more sparse representation of its incoming neuronal activity pattern, both in terms of number of DGCs activating, as well as their limited firing rates (Acsády and Káli, 2007; O’Reilly and McClelland, 1994; Treves and Rolls, 1994). The sheer paucity of DGC activation raises an important question with regard to cognitive processing. If so few DGCs are active, how can their extremely sparse activity contribute in any significant way toward neural processing of hippocampal-dependent cognitive tasks? In short, the DG could only effectively transmit information through an amplifying mechanism by which DGCs could strongly excite their downstream targets in the hilus, and perhaps more critically, the pyramidal cells of CA3, without relying on synaptic convergence (since DGCs only target a few CA3 pyramidal cells). Without such a mechanism, the sparse coding in the DG would be a synaptic “dead end” and little, if any, information content could be relayed to downstream hippocampal structures. In vivo recordings have shown that single DGCs are indeed capable of reliably activating CA3 pyramidal cells (Henze et al., 2002). Unique properties of DGC axon terminals appear to be specialized to achieve this end. Mossy fiber boutons synapse with excitatory hilar mossy cells and CA3 pyramidal cells. These synapses are unique in the mammalian brain in that they are exceptionally large, contain multiple release sites, and display extraordinarily robust frequency facilitation, even at very low frequencies (Salin et al., 1996). Furthermore, even though the majority of DGCs’ synaptic targets are feedback (onto DGCs) and feedforward (onto CA3) inhibitory interneurons, these connections are mediated by filopodial synapses, which, in contrast to mossy fiber boutons, exhibit rapid depression upon repetitive activation (Mori et al., 2004). These specialized synaptic properties allow DGCs to act as conditional “detonators” (McNaughton and Morris, 1987) of their postsynaptic CA3 targets during periods of elevated firing rates.
3 WHAT FUNCTIONS ARE SERVED BY SPARSELY ACTIVATING DGCs?
What are the implications of a sparsely coding network of cells that rarely activate, yet when they do, exert massive excitatory influence on their postsynaptic targets? The answer varies depending on whether one is considering the DG’s role in cognition and memory, or epilepsy. The DG’s position at the entrance to the hippocampus combined with the sparse activation of DGCs led to the theory that DG circuitry transforms promiscuous and highly active cortical input into a sparse neural code suitable for representing novel aspects of episodic memories (Treves and Rolls, 1994; Treves et al., 2008). This DG-mediated transformation is critical to many aspects of hippocampal-dependent neural processing, most notably pattern separation and pattern completion, which are fundamental components of episodic memory encoding (Leutgeb et al., 2007; McHugh et al., 2007; Nakashiba et al., 2012; Neunuebel and Knierim, 2014). Behaviorally, pattern separation is the ability to discriminate subtle differences between similar episodes (eg, “where one parked one’s car today is not necessarily the same place as yesterday”; O’Reilly and McClelland, 1994). From a computational perspective, this translates to the "process of transforming similar inputs into more dissimilar outputs" (Piatti et al., 2013), thereby detecting novelty and isolating salient differences between inputs. Computational models suggest that the DG could separate similar patterns through the output of different, small subsets of DGCs with distinct DGC ensembles activating to each unique stimulus (de Almeida et al., 2009b, O’Reilly and McClelland, 1994). However, tetrode recording and immediate-early gene reporter studies have both reported that the same sparse population of DGCs may activate in multiple environments (Alme et al., 2010; Leutgeb et al., 2007) and that the rest of the population may remain essentially dormant during a given behavioral epoch. This conflicts with the expectation of sparse firing of multiple, distinct DGC ensembles in different environments (Chawla et al., 2005; Jung and McNaughton, 1993) predicted by computational models (de Almeida et al., 2009b; O’Reilly and McClelland, 1994). In this case, rate coding within the active population of cells could instead differentiate inputs. Other models hypothesize that newborn and mature DGC’s contribute differentially to the active population of DGCs involved in pattern separation (Aimone et al., 2011; Lisman, 2011; see Piatti et al., 2013 for review of neurogenesis and sparse coding). It is not yet known which of these coding schemes is utilized by the DG in execution of DG-dependent tasks such as pattern separation. What remains abundantly clear, however, is that information coding is achieved through sparse activation of principal cells within the DG and that this is critical to normal execution of DG-dependent cognitive function.
4 DENTATE GATING: A SECONDARY CONSEQUENCE OF THE DG’s SPARSE CODE
A secondary consequence of the DG’s sparse activation and low excitability has received considerable attention with regard to epilepsy: the DG is capable of preventing and restricting the relay of pathological, synchronous activity in the EC into the hippocampus and limbic system, regulating seizure activity in diseases such as epilepsy. This phenomenon has been termed “dentate gating” (Heinemann et al., 1992; Lothman et al., 1991). The term, “dentate gating,” and others, such as “the dentate gate,” “gatekeeper,” “regulatory checkpoint,” “critical checkpoint,” and “filter function,” have all been used extensively by epilepsy researchers to describe the phenomena in which the dentate effectively limits (or sometimes fails to limit) cortical input from entering the hippocampus proper. These terms do not semantically imply that this behavior is the function of the DG, but rather a role that emerges as a consequence of the DG’s transformation of cortical inputs into a sparse, neural code during execution of its cognitive functions.
Some of the first evidence suggesting a “gatekeeper” role for the DG appeared in early in vivo field recordings from the DG molecular layer in rabbits during electrical stimulation of the EC and perforant path. In these studies, the DG was capable of filtering out higher frequency inputs (>10 Hz) via EPSP habituation due to large, slow poststimulus IPSPs as long as the stimulus train was brief; higher frequency stimulation for longer durations (several seconds) appeared to break this filter down and facilitate DG EPSPs (Andersen et al., 1966). Later, Collins et al. (1983) more directly demonstrated the DG’s role in regulating seizure propagation in behaving rats. Seizure-like activity was initiated in the EC by focal application of chemoconvulsants, and then activation of hippocampal structures was measured post hoc using a metabolic deoxyglucose autoradiography assay in sectioned hippocampal slices. When convulsant injection failed to induce epileptic activity, 10 or fewer spike events were observed per minute in the EC and there were no apparent changes in deoxyglucose autoradiography or behavior. When mild convulsive activity was initiated (10–30 interictal spikes per minute), animals exhibited slight to no signs of behavioral seizures, which included intermittent staring and sniffing. In these animals, deoxyglucose uptake was restricted to the EC and DG molecular layer and there was no indication of propagation of seizure activity into the DG granule cell layer or CA fields of the hippocampus proper. However, when greater than 40 interictal spikes per minute were induced (by either convulsants alone or in combination with electrical stimulation), animals displayed clear behavioral seizures, including shaking, sniffing, head nodding, and freezing. In these animals, metabolic changes were observed to spread through the EC, the DG, and beyond into CA3, CA1, and other extrahippocampal structures in both the ipsilateral and contralateral hemispheres. Since enhanced deoxyglucose uptake was initially restricted to only the DG molecular layer in mild to moderate convulsive states, and only propagated beyond the DG through the entire hippocampus and beyond after strong convulsive states, the authors concluded that this sequential metabolic activation suggests that “the [DG] acts as a restrictive gateway for seizure spread from EC to the rest of the limbic system.”
Stringer and Lothman further developed this gating theory by introducing a concept termed “maximal dentate activation” (MDA; Lothman et al., 1992; Stringer et al., 1989). In these studies, either the perforant path or CA3 was electrically stimulated in urethane-anesthetized rats. MDA was defined as the state in which stimulation elicited a saturating response in the DG. Stimulation frequencies between 10 and 40 Hz most easily elicited MDA, and once MDA occurred, electrical recordings revealed a marked, negative DC shift indicating depolarization of DGCs. This was accompanied by a rapid and substantial elevation of extracellular K+ concentrations (~6–8 mM increase) and the emergence of large-amplitude DGC population spikes and bursting. Stimuli above the MDA threshold also elicited prominent after-discharges as well as synchronous epileptiform discharges in CA3, CA1, subiculum, and the EC. When stimulation was applied to the angular bundle of the perforant path, MDA occurred first in the DG, before subsequent propagation to CA1, ruling out the possibility of direct EC of the temporoammonic pathway to CA1 being responsible for CA1’s activation. Moreover, when the DGCs were selectively lesioned with colchicine, stimulation that would normally generate MDA failed to elicit it, and there was no concomitant activation of CA1. These results showed clearly that maximal activation of the DG, even unilaterally, can both initiate and sustain seizure activity throughout the hippocampus and extrahippocampal structures bilaterally. In total, these results indicated that when the DG fails to filter and control cortical excitatory inputs as a gatekeeper or regulatory checkpoint, it acts instead as an ictal amplifier or promoter of seizure activity (further reviewed in Heinemann et al., 1992). This duplicitous role of the DG as both a filter and amplifier likely emerges from a combination of DGCs’ reluctance to fire, the rich inhibitory network they reside in, and the unique properties of the large mossy fiber boutons of their axons endowing with them the ability to “detonate” area CA3 when upon DGC activation.
Recent studies have further supported the DG’s role as a gatekeeper. Among the most visually demonstrative of these are studies utilizing dynamic imaging techniques. In particular, use of voltage-sensitive dyes, when combined with state-of-the-art cameras, can resolve propagation of synaptic potentials through multiple sites within a brain slice, at an extremely high temporal resolution. Voltage-sensitive dye imaging can therefore facilitate recording of synaptic integration and propagation of activity across multiple circuits. Specifically within hippocampal and parahippocampal structures, this technique allows simultaneous monitoring of afferent activation of the DG via the EC, processing within the DG, and propagation of DG signals to efferent structures in CA3 and CA1.
One early voltage-sensitive dye imaging study strikingly illustrated the gating dynamics of the DG (Iijima et al., 1996). In this study, rat entorhinal cortical–hippocampal slices were perfused with low concentrations of GABA-A receptor antagonists, the EC was stimulated, and activation of the slices imaged. This study demonstrated that the disinhibited DG more easily allowed cortical activity to enter the hippocampus and activate the entire hippocampal loop, later reentering the EC. Further experiments in the absence of inhibitory blockade showed that repetitive stimulation of the EC would initially activate only cortical areas. Each successive stimulus would increase sustained activity in the EC, and only after multiple successive stimuli would the DG “gate” collapse, allowing activity to penetrate through DG and enter the hippocampus proper. This study clearly demonstrated that activation of the EC is usually restricted by the DG, but that successive stimulation could, within a short time frame, erode gating and elicit propagation activity into the hippocampus.
We (Ang et al., 2006; Yu et al., 2013; Fig. 1) have further utilized voltage-sensitive dye imaging techniques to monitor activation of the DG and downstream structures in control and epileptic animals, in response to stimulation of the perforant path. Our recordings clearly illustrate the “gatekeeper” function of the DG (Fig. 2A–C), as well as the important finding that activation of GABA-A receptors on DGCs is required for this filtering of EC inputs. Even modest levels of disinhibition (pharmacologic block of 20–25% of IPSC amplitudes with picrotoxin) resulted in DG gating collapse and activation of downstream CA3 (Fig. 1D–F).
FIG. 1.

“Gatekeeper” function of the DG is maintained by GABAergic inhibition. Simultaneous voltage-sensitive dye (A), snapshot taken at the peak of the response, (B) trace illustrating the VSD response over time, (C) patch clamp (top) and field potential recording (bottom) of DG response to perforant path activation in control ACSF. (A) Corresponding to a 10–15 mV EPSP in (B), which does not result in activation of downstream structures (note lack of response in area CA3 in (A) and (B)). This lack of CA3 activation is because DGCs do not fire APs in response to perforant path activation under these conditions. This is evident in both the patch (C, top trace, the neuron depolarized to Vm of −50 mV) and field potential recording (C, bottom trace), due to powerful feedforward inhibition activated by perforant path stimulation (C, note large IPSP in patch recording). The importance of inhibition in mediating this “gatekeeper” function is illustrated in responses in (D), (E), and (F), following perfusion with 5 μM picrotoxin, a noncompetitive GABA-A receptor antagonist. This concentration blocks 20–25% of inhibition (see inset [located above (E)] depicting an averaged spontaneous IPSC [sIPSC] before and after perfusion with 5 μM picrotoxin). During 25% GABAergic blockade, perforant path activation resulted in powerful activation of both the DG and downstream structures (CA3 and hilus; D, E). It also triggered AP firing in DGCs (see patch and field potential recordings in (F), both of which exhibit AP firing). A grayscale image of the slice, with patch and field potential recording electrode location is depicted in the inset above (A).
From Coulter, D.A., Carlson, G.C., 2007. Functional regulation of the dentate gyrus by GABA-mediated inhibition. Prog. Brain Res. 163, 235–243.
FIG. 2.

Postnatal development of DG gating behavior. (A) Top: A schematic illustration depicting subregions of the hippocampus. Bottom: The DG (gray box) is expanded in a VSD image with an overlay of the ROI delineating subregions used to measure DG responses elicited by perforant path stimulation. (B) VSDI time-resolved fluorescence plots for the subregions depicted in (A) for P12 (top) and P60 (bottom) animals. PP stimulation elicits comparable depolarizations in the DGC, hilus, and CA3 at P12, but little depolarization of hilus and CA3 at P60, despite robust responses in DGC. (C) DG response amplitude (ΔF/F) is comparable at all developmental ages (elicited by a 400 μA PP stimulus). (D) P12, P22, and P60 mice (n=8 slices in 3 animals, n=7 of 2 animals, and n=6 of 2 animals, respectively) show progressively less propagation of synaptic responses through DGC (green, light gray in the print version) to hilus (blue, dark gray in the print version) and CA3 (red, gray in the print version). All data points are normalized to DGC layer response at 400 μA, which is equivalent across groups (see C). (E) Plots of DG gating function, the ratio of DGC to CA3 activation intensity, depict the significant increase in the DG gating property as postnatal development progresses, at several stimulus intensities 200 μA (circle), 300 μA (square), and 400 μA (triangle). p=0.001 for the animal age factor affecting gating (two-way ANOVA). p=0.16 for stimulus intensity affecting gating (two-way ANOVA).
From Yu, E.P., Dengler, C.G., Frausto, S.F., Putt, M.E., Yue, C., Takano, H., Coulter, D.A., 2013. Protracted postnatal development of sparse, specific dentate granule cell activation in the mouse hippocampus. J. Neurosci. 33, 2947–2960.
In Yu et al. (2013), we further demonstrated the emergence of this gating function during postnatal development using a novel, combined dynamic imaging approach capable of resolving sequentially both synaptic potentials (voltage-sensitive dye imaging) and AP firing (multicellular calcium imaging in large populations of DGCs) in response to perforant path afferent activation in mouse hippocampal–entorhinal cortical slices. During postnatal development, DG gating function was expressed only as animals matured. Neonatal animals (P6) showed robust activation of both the DG and CA3 upon perforant path activation, whereas older animals (P22, P60) displayed robust gating in which the DG showed marked depolarization, but activation did not propagate into area CA3 (Fig. 2). Development of this gating property was primarily mediated by changes in local circuit inhibition, as inhibitory blockade (with picrotoxin) normalized responses at all developmental time points. During development, GABA responses are often depolarizing due to differential expression of the chloride accumulator, NKCC1, and chloride extruder, KCC2, which invert their relative expression levels during development to produce hyperpolarizing or shunting inhibitory currents at GABAergic synapses in adulthood (Hollrigel et al., 1998; Liu et al., 1996). These changes as well as further inhibitory synaptic innervation of DGCs and electrophysiological maturation of DGCs with development likely combine to establish the gating function of the DG.
The sequential imaging approach in this study allowed us not only to resolve the spatiotemporal properties of afferent activation in the DG and downstream structures using voltage-sensitive dye imaging but also to resolve cellular activation in large numbers of DGCs with single-cell resolution using multicellular calcium imaging. This technique can transduce AP firing in individual neurons into changes in fluorescence intensity through the use of calcium-chelating dyes, or genetically encoded calcium indicators. Using this imaging approach, we found that the population activity of DGCs displayed a progressive sparsification with postnatal development (Fig. 3) and that differences in proportional activation of DGCs were also normalized by inhibitory blockade, further demonstrating the critical role of inhibition in restricting DGC activation. An interesting side note of the study was that dentate gating and sparse firing of DGCs develop synchronously with the protracted representation of space within the hippocampus and EC, which become evident 3.5–4 weeks after birth (Langston et al., 2010) as well as with competency in hippocampal-dependent spatial memory tasks (Ainge and Langston, 2012; Rudy et al., 1987; Schenk, 1985). Given that appropriate neuronal activity within the DG is likely critical to completion of these tasks, the delays in development of adult levels of DG filtering and sparse DGC activation may contribute to the delayed development of both neuronal representations of space and competence in spatial memory tasks.
FIG. 3.
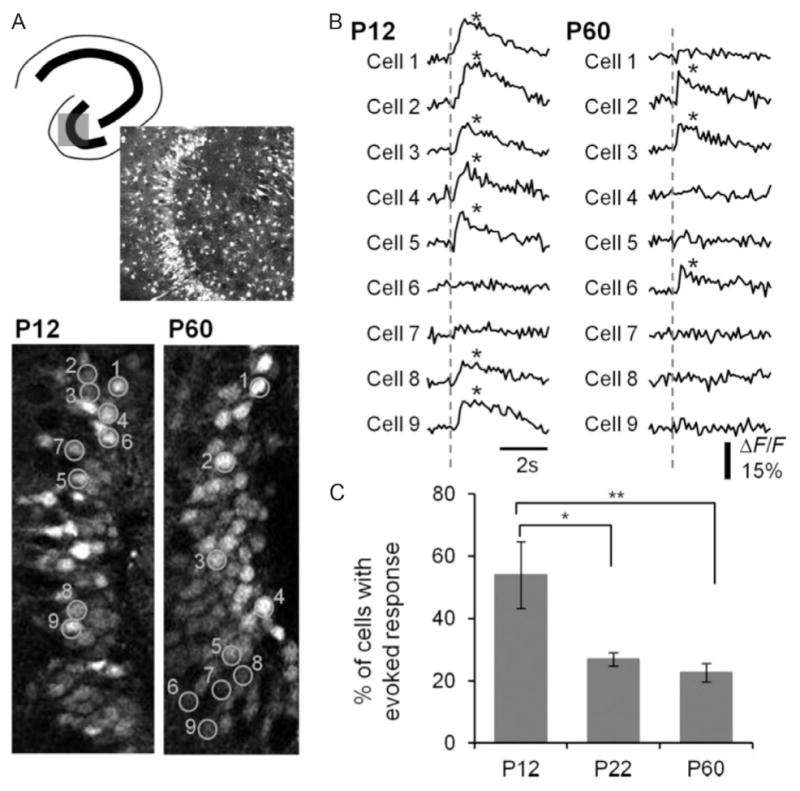
Decreased DGC activation during postnatal development. (A) Top: Schematic of a hippocampal slice depicting the imaged area in the DG and a Fura2-loaded DG of a P12 mouse (370×370 μm). Bottom: Image of P12 and P60 DGCs, with ROI created on a random sample of cells (90×200 μm). Numbers denote cell identification with time-resolved fluorescence responses depicted in (B). (B) Representative traces of time-resolved calcium imaging response for the ROI in the P12 and P60 images in (A). The dotted line indicates the time when PP stimulation (400 μA) occurred. An asterisk indicates detection of a calcium transient. (C) Plot of the percentage DGC activation by PP stimulation for P12, P22, and P60 animals. Note the decrease in cell activation with postnatal development. p=<0.0001 for the animal age factor (ANOVA). *p=<0.05, significant differences between P12 and P22 (Tukey’s multiple comparison post hoc testing). **p=<0.01, significant differences between P12 and P60 groups (Tukey’s multiple comparison post hoc testing). P12: (n=322 PTX-active cells in 10 imaged regions), P22: (n=198 in 10 imaged regions), and P60: (n=239 of 12 imaged regions).
From Yu, E.P., Dengler, C.G., Frausto, S.F., Putt, M.E., Yue, C., Takano, H., Coulter, D.A., 2013. Protracted postnatal development of sparse, specific dentate granule cell activation in the mouse hippocampus. J. Neurosci. 33, 2947–2960.
Recent studies have used optogenetic techniques to modulate dentate gating function directly in a mouse model of temporal lobe epilepsy. In Krook-Magnuson et al. (2015), investigators used a closed-loop system to detect seizures and selectively modulate the activity of DGCs by triggering either optical activation of these neurons with channelrhodopsin or inhibition with halorhodopsin. They found that optogenetic hyperpolarization of DGCs efficiently stopped spontaneous seizures (gate restoration). In contrast, optogenetic activation of DGCs exacerbated spontaneous seizures. Additionally, this study revealed that optogenetic activation of DGCs in healthy, nonepileptic animals elicits acute seizures and that the intensity and duration of these seizures increase with repeated DGC photostimulation. These results clearly support the concept that the DG is a critical node within the hippocampus and provides direct in vivo evidence consistent with the dentate “gate” hypothesis. Additional experiments have elaborated on this finding and demonstrated that optogenetic activation of DG inhibitory interneurons can immediately stop the spread of seizures in the hippocampus and EC, further highlighting the important role of inhibition within dentate circuitry in regulating cortical inputs (Lu et al., 2016).
The studies mentioned above all demonstrate ways in which the DG is a critical node within the hippocampal circuit, both for restricting entrance of EC activity into the hippocampus and, when sufficiently hyperactivated, allowing propagation of pathological activity to reverberate through the hippocampus. Given that the DG functions as a key node within the limbic system, a critical question is whether the DG’s gating function is compromised during epilepsy development and expression in both animal models and humans with epilepsy. Several lines of evidence have suggested that this may be the case. In one such study, Behr et al. (1998) demonstrated enhanced propagation of epileptiform activity through the DG in a rat kindling model of epilepsy. In this study, investigators prepared hippocampal–EC slices from control and kindled animals, evoked isolated epileptiform activity in the EC via local perfusion of GABA antagonists and high K+, and recorded activation of downstream DG and CA3 extracellularly. Slices from kindled animals displayed markedly enhanced propagation of EC bursting activity through the DG. Another study by Patrylo et al. (1999) showed a similar pattern of abnormal DG responses in a systemic kainate model of epilepsy in rats. In this investigation, slices from kainate-treated rats (both acutely 2–4 days following kainate administration, as wells as from chronically epileptic rats >3 months after kainate treatment) showed multiple population spikes following perforant path stimulation, while control slices consistently produced only a single spike in response to the same stimulus. An additional investigation in this kainate-induced epilepsy model also showed that slices from epileptic rats would produce epileptiform bursts, when stimulated repetitively at 5–10 Hz (in the range of hippocampal theta rhythms), while the same stimulation elicited only single spikes in control slices (Shao and Dudek, 2011). Further, in a slice voltage-sensitive dye imaging study in our laboratory (Pathak et al., 2007), we showed that in a rat pilocarpine status-epilepticus model, DG throughput onto downstream structures was also dysregulated for several days following status epilepticus. In this study (Fig. 1), control slices exhibit little downstream CA3 activation following perforant path stimulation. However, immediately following status epilepticus (and until 2 weeks post-SE), perforant path stimulation reliably activated CA3 showing a collapse of normal DG gating function. Interestingly in this model, normal gating function appeared to be restored to control levels in chronically epileptic animals. This caveat aside, these studies collectively show that the DG’s gatekeeper function can be corrupted in multiple models of epilepsy, including kindling, as well as kainate- and pilocarpine-induced status-epilepticus models. Further, these data are congruent with a number of electrophysiological studies demonstrating varying degrees of DGC hyperexcitability in hippocampal slices prepared from tissue resected from patients with intractable temporal lobe epilepsy (Isokawa and Fried, 1996; Masukawa et al., 1989, 1996; Williamson, 1994).
5 WHICH MECHANISMS CONTRIBUTE TO THE DG GATE BREAKDOWN IN EPILEPSY?
Failure of dentate gating and its hypothesized relationship to both icto- and epileptogenesis encouraged many studies detailed earlier. The common theme described in these investigations is that the DG is normally resistant to allowing EC activity to propagate to downstream structures of the hippocampus proper. In epilepsy, the gatekeeping function of the DG is compromised, allowing relay of cortical activity from the EC into the hippocampus, and propagation in a reverberatory cycle back to EC (Lothman et al., 1992). This hypothesis has prompted numerous studies to characterize cellular and circuit alterations in DG circuitry which may contribute toward failure of the DG’s gatekeeper function.
Among the earliest discovered and most striking of these alterations, and a classic hallmark of temporal lobe epilepsy, is the de novo sprouting of recurrent mossy fiber synapses. First described by Tauck and Nadler (1985), mossy fiber sprouting is the generation of aberrant mossy fiber collateral which forms recurrent, autoexcitatory synapses from DGCs to other DGCs via synapses located in the inner third of the molecular layer. This phenomenon, which is thought to be a form of reactive plasticity in response to death of many of the DGC population’s synaptic targets, is particularly well described in the DG in both human epilepsies, as well as in animal models of injury-induced epilepsy (de Lanerolle et al., 1989; Houser et al., 1990; Sutula et al., 1989; Tauck and Nadler, 1985), and is hypothesized to contribute to DG hyperexcitability in epilepsy. Initial electrophysiological experiments involved electrical antidromic stimulation of the hilus (to activate mossy fibers directly) and demonstrated the role of mossy fiber sprouting in elevating DGC excitability (Cronin et al., 1992; Tauck and Nadler, 1985), but these approaches were indirect because such stimulation might also activate other afferent fibers or excitatory mossy cells. Later however, direct evidence showing monosynaptic excitatory transmission between DGCs following mossy fiber sprouting was obtained through paired whole-cell recordings (Scharfman et al., 2003b). In this study, the amplitudes of these EPSPs between DGCs were small, and the percentage of paired connections was relatively low, suggesting that the mossy fiber recurrent excitatory network would be relatively weak at promoting hyperexcitability in the DG. Supporting this conclusion, use of rapamycin to suppress mossy fiber sprouting in a pilocarpine model of temporal lobe epilepsy did not reduce seizure frequency (Heng et al., 2013). However, in slices prepared from chronically epileptic rats, aberrant kainate receptors present at these sprouted synapses can interact with persistent sodium currents to generate abnormal, sustained, and rhythmic firing among DGCs (Artinian et al., 2011). Thus, the extent to which this mossy fiber sprouting contributes to DGC excitability in epilepsy is unclear. Interestingly, further work has shown that currents mediated through these aberrant kainate receptor-containing mossy fiber synapses can be selectively blocked using pharmacologic agents (Pinheiro et al., 2013); such tools may provide great utility in further clarifying this issue.
While increased recurrent excitatory drive may play a role in breakdown of the dentate gate, reduced inhibition of DGCs could also play a role in this process. GABAergic inhibition is a critical mediator of DG gating (Fig. 1), and compromised filter function of the DG is a major contributor to seizure propagation in models of epilepsy. One could easily hypothesize that the DG in these models undergoes disinhibition. However, in both kindling and post-status-epilepticus models of temporal lobe epilepsy, numerous studies have shown a surprising upregulation of GABA-A receptor expression, both synaptically (Buhl et al., 1996; Cohen et al., 2003; Nusser et al., 1998; Otis et al., 1994) and in whole-cell recordings (Brooks-Kayal et al., 1998; Gibbs et al., 1997; Leroy et al., 2004; Mtchedlishvili et al., 2001) from chronically epileptic rats. Some studies do, nevertheless, describe a transient decrease in GABAergic currents onto DGCs immediately following epileptogenic injuries that persists for weeks following initial injury, but this reduction recovers toward or beyond control levels over time (Cohen et al., 2003; Thind et al., 2010). However, in a recent study in a mouse model of temporal lobe epilepsy, phasic GABAergic inhibition was reduced, likely as a result of relocalization of γ2 GABA receptor-containing subunits away from the center of synaptic contacts (Zhang et al., 2007). While this result in mice differs from many of the results earlier (all conducted in rats), it may, in fact, highlight an important species difference between rat and mouse models of temporal lobe epilepsy.
While the amplitude of GABAergic inhibitory currents in DGCs may be increased in many chronically epileptic animals, several studies have demonstrated that inhibitory connectivity within the DG is compromised in epileptic animals following epileptogenic injuries and remain low in chronically epileptic animals (Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005; Sun et al., 2007). This decreased inhibitory synaptic connectivity is often presumed to be caused by a loss of hilar somatostatin-positive interneurons following epileptogenic insults (Sloviter, 1987). Further experiments have also demonstrated that deficits in inhibitory basket cell circuit function may also contribute to reduced inhibitory efficacy as a result of less excitatory synaptic drive onto basket cells, diminished pools of readily releasable vesicles, and frequent synaptic transmission failure between basket cells to DGCs (Zhang and Buckmaster, 2009).
While results implicating alterations in phasic DG disinhibition have been mixed, studies have shown that tonic inhibition within the DG is compromised as a result of epileptogenic injuries and epilepsy development. Tonic inhibition in the DG is typically mediated by δ subunit-containing GABA-A receptors, which are located primarily at nonsynaptic sites on DGCs. These receptors play a critical role in mediating controlled DG excitability via responding to GABA spillover from GABAergic synapses because of their perisynaptic localization (Wei et al., 2003), as well as their high affinity for GABA and their relatively slow desensitization rates (Haas and Macdonald, 1999; Mtchedlishvili and Kapur, 2006; Saxena and Macdonald, 1994). It is also likely that these receptors can be activated by neurogliaform interneurons in the DG’s molecular layer, which have dense local axonal plexuses that are thought to provide inhibition largely through “bulk transmission” of GABA through activation of extrasynaptic receptors by release of a local “cloud” of GABA (Armstrong et al., 2011, 2012). In models of temporal lobe epilepsy, studies have demonstrated a reduction of these extrasynaptic, tonic GABA-A receptors with a corresponding downregulation in expression of δ subunits (Peng et al., 2004; Zhang et al., 2007). As of now, the role of this reduction in tonic inhibition onto DGCs with regard to DG excitability and gating in epileptic animals is unknown. It is possible that repetitive activation of EC afferents, like those occurring during seizure initiation, will elevate extrasynaptic GABA concentrations and enhance tonic current in normal animals (helping to suppress ictal propagation), and loss of these receptors will compromise this check on excitability in animals with epilepsy.
However, the efficacy of ionotropic GABAergic synapses depends not only on the pre- and postsynaptic function of synaptic machinery or number of inhibitory synaptic inputs onto DGCs but also on the driving force of chloride ions across the neuronal membrane at these synapses. The chloride reversal potential is a potent determinant of ionotropic GABAergic inhibition. Typically, EGABA is around −70 to −80 mV in DGCs under normal conditions. However, immediately following status epilepticus, this reversal potential shifts to markedly less hyperpolarized levels of −55 to −60 mV, which greatly diminishes inhibitory efficacy. During this time period, DGC excitability is markedly enhanced via reduced inhibitory influence in DGC synaptic integration and further, the DG’s gating ability is consequently compromised (Pathak et al., 2007). This transient depolarizing shift in EGABA manifests as a result of reduced expression of the chloride-extruding potassium/chloride cotransporter, KCC2, which normally functions to maintain EGABA at a hyperpolarized levels. Interestingly, this phenomenon of reduced KCC2 expression in animals immediately following status epilepticus closely mirrors the developing brain in which GABA is also depolarizing. During development, the expression of the chloride extruder, KCC2, is low and the chloride accumulator, NKCC1, is relatively high before the “GABA switch” (around the second postnatal week), which involves reversed relative expression of these chloride transporters, and decreased DG gating efficacy (Ben-Ari, 2002; Owens and Kriegstein, 2002; Yu et al., 2013; Figs. 2 and 3). Interestingly, in Pathak et al. (2007), chloride reversal potentials return to near control levels as animals become chronically epileptic, and DG excitability appears to return to normal levels. However, even though transient, the failure of the DG gate immediately following epileptogenic injury may have critical implications in epileptogenesis, since the likelihood of synchronous, epileptiform activity reaching downstream structures is greatly enhanced. Bursting activity in the DG can potentially damage neurons in the hilus, and areas CA3 and CA1, leading to further neuronal loss, aberrant plasticity, and hippocampal sclerosis, in the period following the initial epileptogenic injury, thus furthering the initial insult and potentially promoting further epileptogenesis.
While GABAergic disinhibition and altered chloride regulation are potential mechanisms for enhancing DG excitability in epilepsy and its development, another possible contributor to aberrant DG circuit activation is astrocytic gliosis. Reactive astrocytosis is a prominent pathology in both human (Eid et al., 2004) and animal models of epilepsy (do Nascimento et al., 2012; Estrada et al., 2012; Xu et al., 2011). Studies from our laboratory have demonstrated that astrocytic gliosis can reduce the efficacy of inhibitory neurotransmission onto hippocampal principal cells via downregulation of glutamine synthetase. Loss of glutamine starves inhibitory synapses of necessary local precursors to regenerate GABA and results in disinhibition (Coulter and Eid, 2012; Ortinski et al., 2010). While it is currently known that astrogliosis is present in the hippocampus, and specifically, in the DG of patients with temporal lobe epilepsy and in animal models of the disease, the exact contributions of this pathology in altering circuit function within the DG are currently unknown. However, in one recent study, Dhaher et al. (2015) pharmacologically mimicked the glutamine synthetase-reducing effects of astrogliosis, by infusing the glutamine synthetase inhibitor, methionine sulfoximine into different hippocampal substructures, including the angular bundle of the perforant path, deep layers of the EC, the DG, CA1, subiculum, and the lateral ventricle and monitored animals for seizures via video and EEG recordings. Among all tested structures, methionine sulfoximine infusion into the DG produced the highest number of seizures over the recording period, highlighting both the importance of the DG in seizure generation, and its potential susceptibility for gliosis-mediated disinhibition in epilepsy.
Yet another circuit alteration in the epileptogenic DG is aberrant neurogenesis. The DG is a unique brain structure in that it continuously generates new, functional neurons throughout life (van Praag et al., 2002). These newly generated DGCs have electrophysiological properties distinct from their mature counterpart and some studies suggest that these newborn cells may be activated preferentially compared to mature DGCs (Alme et al., 2010; Marin-Burgin et al., 2012). However, following SE, there is a marked increase in neurogenesis of DGCs, many of which integrate aberrantly into the local DG network (Kron et al., 2010; Parent, 2007; Parent et al., 1997). Several studies have posited both pathophysiological and compensatory roles for these aberrantly integrated neurons following epileptogenic injuries. Transgenic ablation of DGC neurogenesis prior to the inciting injury has recently been demonstrated to decrease epilepsy severity (Cho et al., 2015). Many DGCs born following epileptogenic injuries migrate to ectopic locations within the hilus and integrate abnormally into hippocampal circuitry (Scharfman et al., 2003a). The accumulation of these ectopic, but not normotopic, adult-born DGCs correlates with epilepsy severity (Hester and Danzer, 2013) as normotopic adult-born DGCs in epileptic animals do not exhibit aberrant excitability (Jakubs et al., 2006). While these studies have shown that these newly generated DGC (whether normotopic or ectopic) can play both pathologic and apparently compensatory roles, it is currently unclear how these newborn neurons participate in aggregate DG function, or the degree to which they compromise activation and output of the DG.
6 CONCLUSIONS
The DG is a structure characterized by the sparse activity of its principal neurons, DGCs. This low excitability is important in the cognitive function of the hippocampus and results predominantly from the high degree of inhibitory synaptic regulation, as well as intrinsic properties of DGCs themselves. This sparse activation serves an additional role in the context of epilepsy, where the DG’s inherent ability to restrict pathological activation of the hippocampus and limbic system is of critical importance. A combination of circuit changes occurs in the DG during epilepsy development and expression, including mossy fiber sprouting, alterations to inhibitory circuits and their function, transmembrane chloride regulation, astrogliosis, aberrant neurogenesis, and alterations to the intrinsic properties of DGCs themselves. These epilepsy-associated modifications to the DG circuit may, to varying degrees, compromise the DG’s ability to fulfill its regulatory gatekeeper role. Given the abundance of alterations, the mechanisms mediating this circuit collapse are likely not mutually exclusive, but instead mechanistically complex. Given that the DG functions as both a sparsifying transformer of cortical information in spatial cognitive processes and a regulator of cortical hyperactivity, inappropriate or pathological excitability within this watershed circuit may contribute to seizure propagation, both primary and secondary epileptogenic processes, as well as the many cognitive comorbidities associated with epilepsy. Understanding how epilepsy development alters the basic circuit properties of hippocampal structures may be important not only in targeting new therapies for seizure amelioration but also in developing new treatments to reduce comorbidities accompanying epilepsy.
References
- Acsády L, Káli S. Models, structure, function: the transformation of cortical signals in the dentate gyrus. Prog Brain Res. 2007;163:577–599. doi: 10.1016/S0079-6123(07)63031-3. [DOI] [PubMed] [Google Scholar]
- Acsády L, Kamondi A, Sík A, Freund T, Buzsáki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 1998;18:3386–3403. doi: 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimone JB, Deng W, Gage FH. Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron. 2011;70:589–596. doi: 10.1016/j.neuron.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainge JA, Langston RF. Ontogeny of neural circuits underlying spatial memory in the rat. Front Neural Circuits. 2012;6:1–10. doi: 10.3389/fncir.2012.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alme CB, Buzzetti RA, Marrone DF, Leutgeb JK, Chawla MK, Schaner MJ, Bohanick JD, Khoboko T, Leutgeb S, Moser EI, Moser MB, McNaughton BL, Barnes CA. Hippocampal granule cells opt for early retirement. Hippocampus. 2010;20:1109–1123. doi: 10.1002/hipo.20810. [DOI] [PubMed] [Google Scholar]
- Andersen P, Holmqvist B, Voorhoeve PE. Entorhinal activation of dentate granule cells. Acta Physiol Scand. 1966;66:448–460. doi: 10.1111/j.1748-1716.1966.tb03223.x. [DOI] [PubMed] [Google Scholar]
- Ang CW, Carlson GC, Coulter DA. Massive and specific dysregulation of direct cortical input to the hippocampus in temporal lobe epilepsy. J Neurosci. 2006;26:11850–11856. doi: 10.1523/JNEUROSCI.2354-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte Y, Bischofberger J, Jonas P. Efficient Ca2+ buffering in fast-spiking basket cells of rat hippocampus. J Physiol. 2008;586:2061–2075. doi: 10.1113/jphysiol.2007.147298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C, Szabadics J, Tamás G, Soltesz I. Neurogliaform cells in the molecular layer of the dentate gyrus as feed-forward γ-aminobutyric acidergic modulators of entorhinal-hippocampal interplay. J Comp Neurol. 2011;519:1476–1491. doi: 10.1002/cne.22577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C, Krook-Magnuson E, Soltesz I. Neurogliaform and Ivy cells: a major family of nNOS expressing GABAergic neurons. Front Neural Circuits. 2012;6:23. doi: 10.3389/fncir.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artinian J, Peret A, Marti G, Epsztein J, Crépel V. Synaptic kainate receptors in interplay with INaP shift the sparse firing of dentate granule cells to a sustained rhythmic mode in temporal lobe epilepsy. J Neurosci. 2011;31:10811–10818. doi: 10.1523/JNEUROSCI.0388-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr J, Lyson KJ, Mody I. Enhanced propagation of epileptiform activity through the kindled dentate gyrus. J Neurophysiol. 1998;79:1726–1732. doi: 10.1152/jn.1998.79.4.1726. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- Chawla MK, Guzowski JF, Ramirez-Amaya V, Lipa P, Hoffman KL, Marriott LK, Worley PF, McNaughton BL, Barnes CA. Sparse, environmentally selective expression of Arc RNA in the upper blade of the rodent fascia dentata by brief spatial experience. Hippocampus. 2005;15:579–586. doi: 10.1002/hipo.20091. [DOI] [PubMed] [Google Scholar]
- Cho KO, Lybrand ZR, Ito N, Brulet R, Tafacory F, Zhang L, Good L, Ure K, Kernie SG, Birnbaum SG, Scharfman HE, Eisch AJ, Hsieh J. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat Commun. 2015;6:6606. doi: 10.1038/ncomms7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claiborne BJ, Amaral DG, Cowan WM. A light and electron microscopic analysis of the mossy fibers of the rat dentate gyrus. J Comp Neurol. 1986;246:435–458. doi: 10.1002/cne.902460403. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Lin DD, Quirk GL, Coulter DA. Dentate granule cell GABA(A) receptors in epileptic hippocampus: enhanced synaptic efficacy and altered pharmacology. Eur J Neurosci. 2003;17:1607–1616. doi: 10.1046/j.1460-9568.2003.02597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RC, Tearse RG, Lothman EW. Functional anatomy of limbic seizures: focal discharges from medial entorhinal cortex in rat. Brain Res. 1983;280:25–40. doi: 10.1016/0006-8993(83)91170-8. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60:1215–1226. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin J, Obenaus A, Houser CR, Edward Dudek F. Electrophysiology of dentate granule cells after kainate-induced synaptic reorganization of the mossy fibers. Brain Res. 1992;573:305–310. doi: 10.1016/0006-8993(92)90777-7. [DOI] [PubMed] [Google Scholar]
- de Almeida L, Idiart M, Lisman JE. A second function of gamma frequency oscillations: an E%-max winner-take-all mechanism selects which cells fire. J Neurosci. 2009a;29:7497–7503. doi: 10.1523/JNEUROSCI.6044-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida L, Idiart M, Lisman JE. The input-output transformation of the hippocampal granule cells: from grid cells to place fields. J Neurosci. 2009b;29:7504–7512. doi: 10.1523/JNEUROSCI.6048-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- Dhaher R, Wang H, Gruenbaum SE, Tu N, Lee TSW, Zaveri HP, Eid T. Effects of site-specific infusions of methionine sulfoximine on the temporal progression of seizures in a rat model of mesial temporal lobe epilepsy. Epilepsy Res. 2015;115:45–54. doi: 10.1016/j.eplepsyres.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Nascimento AL, dos Santos NF, Campos Pelágio F, Aparecida Teixeira S, de Moraes Ferrari EA, Langone F. Neuronal degeneration and gliosis time-course in the mouse hippocampal formation after pilocarpine-induced status epilepticus. Brain Res. 2012;1470:98–110. doi: 10.1016/j.brainres.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Eid T, Thomas MJ, Spencer DD, Rundén-Pran E, Lai JCK, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, De Lanerolle NC. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. 2004;363:28–37. doi: 10.1016/s0140-6736(03)15166-5. [DOI] [PubMed] [Google Scholar]
- Estrada FS, Hernández VS, López-Hernández E, Corona-Morales AA, Solís H, Escobar A, Zhang L. Glial activation in a pilocarpine rat model for epileptogenesis: a morphometric and quantitative analysis. Neurosci Lett. 2012;514:51–56. doi: 10.1016/j.neulet.2012.02.055. [DOI] [PubMed] [Google Scholar]
- Ewell LA, Jones MV. Frequency-tuned distribution of inhibition in the dentate gyrus. J Neurosci. 2010;30:12597–12607. doi: 10.1523/JNEUROSCI.1854-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fricke R, Prince D. Electrophysiology of dentate gyrus granule cells. J Neurophysiol. 1984;51:195–209. doi: 10.1152/jn.1984.51.2.195. [DOI] [PubMed] [Google Scholar]
- Fyhn M, Molden S, Witter MP, Moser EI, Moser MB. Spatial representation in the entorhinal cortex. Science. 2004;305:1258–1264. doi: 10.1126/science.1099901. [DOI] [PubMed] [Google Scholar]
- Fyhn M, Hafting T, Treves A, Moser MB, Moser EI. Hippocampal remapping and grid realignment in entorhinal cortex. Nature. 2007;446:190–194. doi: 10.1038/nature05601. [DOI] [PubMed] [Google Scholar]
- Gibbs JW, Shumate MD, Coulter DA. Differential epilepsy-associated alterations in postsynaptic GABA(A) receptor function in dentate granule and CA1 neurons. J Neurophysiol. 1997;77:1924–1938. doi: 10.1152/jn.1997.77.4.1924. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, McNaughton BL, Barnes CA, Worley PF. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci. 1999;2:1120–1124. doi: 10.1038/16046. [DOI] [PubMed] [Google Scholar]
- Haas KF, Macdonald RL. GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514(Pt. 1):27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, Zhang CL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl. 1992;7:273–280. [PubMed] [Google Scholar]
- Heng K, Haney MM, Buckmaster PS. High-dose rapamycin blocks mossy fiber sprouting but not seizures in a mouse model of temporal lobe epilepsy. Epilepsia. 2013;54:1535–1541. doi: 10.1111/epi.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze DA, Wittner L, Buzsáki G. Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci. 2002;5:790–795. doi: 10.1038/nn887. [DOI] [PubMed] [Google Scholar]
- Hester MS, Danzer SC. Accumulation of abnormal adult-generated hippocampal granule cells predicts seizure frequency and severity. J Neurosci. 2013;33:8926–8936. doi: 10.1523/JNEUROSCI.5161-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollrigel GS, Ross ST, Soltesz I. Temporal patterns and depolarizing actions of spontaneous GABAA receptor activation in granule cells of the early postnatal dentate gyrus. J Neurophysiol. 1998;80:2340–2351. doi: 10.1152/jn.1998.80.5.2340. [DOI] [PubMed] [Google Scholar]
- Houser CR. Interneurons of the dentate gyrus: an overview of cell types, terminal fields and neurochemical identity. Prog Brain Res. 2007;163:217–232. doi: 10.1016/S0079-6123(07)63013-1. [DOI] [PubMed] [Google Scholar]
- Houser CR, Esclapez M. Localization of mRNAs encoding two forms of glutamic acid decarboxylase in the rat hippocampal formation. Hippocampus. 1994;4:530–545. doi: 10.1002/hipo.450040503. [DOI] [PubMed] [Google Scholar]
- Houser CR, Miyashiro JE, Swartz BE, Walsh GO, Rich JR, Delgado-Escueta AV. Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J Neurosci. 1990;10:267–282. doi: 10.1523/JNEUROSCI.10-01-00267.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Martina M, Jonas P. Dendritic mechanisms underlying rapid synaptic activation of fast-spiking hippocampal interneurons. Science. 2010;327:52–58. doi: 10.1126/science.1177876. [DOI] [PubMed] [Google Scholar]
- Hu H, Gan J, Jonas P. Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science. 2014;345:1255263-1–13. doi: 10.1126/science.1255263. [DOI] [PubMed] [Google Scholar]
- Iijima T, Witter MP, Ichikawa M, Tominaga T, Kajiwara R, Matsumoto G. Entorhinal-hippocampal interactions revealed by real-time imaging. Science. 1996;272:1176–1179. doi: 10.1126/science.272.5265.1176. [DOI] [PubMed] [Google Scholar]
- Isokawa M, Fried I. Extracellular slow negative transient in the dentate gyrus of human epileptic hippocampus in vitro. Neuroscience. 1996;72:31–37. doi: 10.1016/0306-4522(95)00544-7. [DOI] [PubMed] [Google Scholar]
- Jakubs K, Nanobashvili A, Bonde S, Ekdahl CT, Kokaia Z, Kokaia M, Lindvall O. Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron. 2006;52:1047–1059. doi: 10.1016/j.neuron.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Jung MW, McNaughton BL. Spatial selectivity of unit activity in the hippocampal granular layer. Hippocampus. 1993;3:165–182. doi: 10.1002/hipo.450030209. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron MM, Zhang H, Parent JM. The developmental stage of dentate granule cells dictates their contribution to seizure-induced plasticity. J Neurosci. 2010;30:2051–2059. doi: 10.1523/JNEUROSCI.5655-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Bui A, Lew S, Oijala M, Soltesz I. In vivo evaluation of the dentate gate theory in epilepsy. J Physiol. 2015;593:2379–2388. doi: 10.1113/JP270056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueppel R, Remy S, Beck H. Dendritic integration in hippocampal dentate granule cells. Neuron. 2011;71:512–528. doi: 10.1016/j.neuron.2011.05.043. [DOI] [PubMed] [Google Scholar]
- Langston RF, Ainge JA, Couey JJ, Canto CB, Bjerknes TL, Witter MP, Moser EI, Moser MB. Development of the spatial representation system in the rat. Science. 2010;328:1576–1580. doi: 10.1126/science.1188210. [DOI] [PubMed] [Google Scholar]
- Leroy C, Poisbeau P, Keller AF, Nehlig A. Pharmacological plasticity of GABA(A) receptors at dentate gyrus synapses in a rat model of temporal lobe epilepsy. J Physiol. 2004;557:473–487. doi: 10.1113/jphysiol.2003.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315:961–966. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Lisman J. Formation of the non-functional and functional pools of granule cells in the dentate gyrus: role of neurogenesis, LTP and LTD. J Physiol. 2011;589:1905–1909. doi: 10.1113/jphysiol.2010.201137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YB, Lio PA, Pasternak JF, Trommer BL. Developmental changes in membrane properties and postsynaptic currents of granule cells in rat dentate gyrus. J Neurophysiol. 1996;76:1074–1088. doi: 10.1152/jn.1996.76.2.1074. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, Stringer JL. Functional anatomy of hippocampal seizures. Prog Neurobiol. 1991;37:1–82. doi: 10.1016/0301-0082(91)90011-o. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 1992;7:301–313. [PubMed] [Google Scholar]
- Lu Y, Zhong C, Wang L, Wei P, He W, Huang K, Zhang Y, Zhan Y, Feng G, Wang L. Optogenetic dissection of ictal propagation in the hippocampal–entorhinal cortex structures. Nat Commun. 2016;7:10962. doi: 10.1038/ncomms10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Burgin A, Mongiat LA, Pardi MB, Schinder AF. Unique processing during a period of high excitation/inhibition balance in adult-born neurons. Science. 2012;335:1238–1242. doi: 10.1126/science.1214956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masukawa LM, Higashima M, Kim JH, Spencer DD. Epileptiform discharges evoked in hippocampal brain slices from epileptic patients. Brain Res. 1989;493:168–174. doi: 10.1016/0006-8993(89)91012-3. [DOI] [PubMed] [Google Scholar]
- Masukawa LM, Wang H, O’Connor MJ, Uruno K. Prolonged field potentials evoked by 1 Hz stimulation in the dentate gyrus of temporal lobe epileptic human brain slices. Brain Res. 1996;721:132–139. doi: 10.1016/0006-8993(96)00153-9. [DOI] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, Lowell BB, Fanselow MS, Wilson MA, Tonegawa S. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science. 2007;317:94–99. doi: 10.1126/science.1140263. [DOI] [PubMed] [Google Scholar]
- McNaughton BL, Morris RGM. Hippocampal synaptic enhancement and information storage within a distributed memory system. Trends Neurosci. 1987;10:408–415. [Google Scholar]
- Mori M, Abegg MH, Gähwiler BH, Gerber U. A frequency-dependent switch from inhibition to excitation in a hippocampal unitary circuit. Nature. 2004;431:453–456. doi: 10.1038/nature02854. [DOI] [PubMed] [Google Scholar]
- Mtchedlishvili Z, Kapur J. High-affinity, slowly desensitizing GABA A receptors mediate tonic inhibition in hippocampal dentate granule cells. Mol Pharmacol. 2006;69:564–575. doi: 10.1124/mol.105.016683. [DOI] [PubMed] [Google Scholar]
- Mtchedlishvili Z, Bertram EH, Kapur J. Diminished allopregnanolone enhancement of GABA(A) receptor currents in a rat model of chronic temporal lobe epilepsy. J Physiol. 2001;537:453–465. doi: 10.1111/j.1469-7793.2001.00453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashiba T, Cushman JD, Pelkey KA, Renaudineau S, Buhl DL, McHugh TJ, Barrera VR, Chittajallu R, Iwamoto KS, McBain CJ, Fanselow MS, Tonegawa S. Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell. 2012;149:188–201. doi: 10.1016/j.cell.2012.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neunuebel JP, Knierim JJ. Spatial firing correlates of physiologically distinct cell types of the rat dentate gyrus. J Neurosci. 2012;32:3848–3858. doi: 10.1523/JNEUROSCI.6038-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neunuebel JP, Knierim JJ. CA3 retrieves coherent representations from degraded input: direct evidence for CA3 pattern completion and dentate gyrus pattern separation. Neuron. 2014;81:416–427. doi: 10.1016/j.neuron.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nörenberg A, Hu H, Vida I, Bartos M, Jonas P. Distinct nonuniform cable properties optimize rapid and efficient activation of fast-spiking GABAergic interneurons. Proc Natl Acad Sci USA. 2010;107:894–899. doi: 10.1073/pnas.0910716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Hajos N, Somogyi P, Mody I. Increased number of synaptic GABA(A) receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998;395:172–177. doi: 10.1038/25999. [DOI] [PubMed] [Google Scholar]
- O’Reilly RC, McClelland JL. Hippocampal conjunctive encoding, storage, and recall: avoiding a trade-off. Hippocampus. 1994;4:661–682. doi: 10.1002/hipo.450040605. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, De Koninck Y, Mody I. Lasting potentiation of inhibition is associated with an increased number of gamma-aminobutyric acid type A receptors activated during miniature inhibitory postsynaptic currents. Proc Natl Acad Sci USA. 1994;91:7698–7702. doi: 10.1073/pnas.91.16.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Parent JM. Adult neurogenesis in the intact and epileptic dentate gyrus. Prog Brain Res. 2007;163:529–540. doi: 10.1016/S0079-6123(07)63028-3. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrylo PR, Schweitzer JS, Dudek FE. Abnormal responses to perforant path stimulation in the dentate gyrus of slices from rats with kainate-induced epilepsy and mossy fiber reorganization. Epilepsy Res. 1999;36:31–42. doi: 10.1016/s0920-1211(99)00022-4. [DOI] [PubMed] [Google Scholar]
- Patton PE, McNaughton B. Connection matrix of the hippocampal formation. 1 The dentate gyrus. Hippocampus. 1995;5:245–286. doi: 10.1002/hipo.450050402. [DOI] [PubMed] [Google Scholar]
- Peng Z, Huang CS, Stell BM, Mody I, Houser CR. Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. 2004;24:8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti VC, Ewell LA, Leutgeb JK. Neurogenesis in the dentate gyrus: carrying the message or dictating the tone. Front Neurosci. 2013;7:50. doi: 10.3389/fnins.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro PS, Lanore F, Veran J, Artinian J, Blanchet C, Crépel V, Perrais D, Mulle C. Selective block of postsynaptic kainate receptors reveals their function at hippocampal mossy fiber synapses. Cereb Cortex. 2013;23:323–331. doi: 10.1093/cercor/bhs022. [DOI] [PubMed] [Google Scholar]
- Rolls ET. A computational theory of episodic memory formation in the hippocampus. Behav Brain Res. 2010;215:180–196. doi: 10.1016/j.bbr.2010.03.027. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Stadler-Morris S, Albert P. Ontogeny of spatial navigation behaviors in the rat: dissociation of “proximal”- and “distal”-cue-based behaviors. Behav Neurosci. 1987;101:62–73. doi: 10.1037//0735-7044.101.1.62. [DOI] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci USA. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NC, Macdonald RL. Assembly of GABAA receptor subunits: role of the delta subunit. J Neurosci. 1994;14:7077–7086. doi: 10.1523/JNEUROSCI.14-11-07077.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE. Dentate hilar cells with dendrites in the molecular layer have lower thresholds for synaptic activation by perforant path than granule cells. J Neurosci. 1991;11:1660–1673. doi: 10.1523/JNEUROSCI.11-06-01660.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AE, Berger RE, Goodman JH, Pierce JP. Perforant path activation of ectopic granule cells that are born after pilocarpine-induced seizures. Neuroscience. 2003a;121:1017–1029. doi: 10.1016/s0306-4522(03)00481-0. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AL, Berger RE, Goodman JH. Electrophysiological evidence of monosynaptic excitatory transmission between granule cells after seizure-induced mossy fiber sprouting. J Neurophysiol. 2003b;90:2536–2547. doi: 10.1152/jn.00251.2003. [DOI] [PubMed] [Google Scholar]
- Schenk F. Development of place navigation in rats from weaning to puberty. Behav Neural Biol. 1985;43:69–85. doi: 10.1016/s0163-1047(85)91510-9. [DOI] [PubMed] [Google Scholar]
- Shao LR, Dudek FE. Changes in mIPSCs and sIPSCs after kainate treatment: evidence for loss of inhibitory input to dentate granule cells and possible compensatory responses. J Neurophysiol. 2005;94:952–960. doi: 10.1152/jn.01342.2004. [DOI] [PubMed] [Google Scholar]
- Shao LR, Dudek FE. Repetitive perforant-path stimulation induces epileptiform bursts in minislices of dentate gyrus from rats with kainate-induced epilepsy. J Neurophysiol. 2011;105:522–527. doi: 10.1152/jn.00456.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- Spruston N, Johnston D. Perforated patch-clamp analysis of the passive membrane properties of three classes of hippocampal neurons. J Neurophysiol. 1992;67:508–529. doi: 10.1152/jn.1992.67.3.508. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Otis TS, Mody I. Membrane properties of dentate gyrus granule cells: comparison of sharp microelectrode and whole-cell recordings. J Neurophysiol. 1992;67:1346–1358. doi: 10.1152/jn.1992.67.5.1346. [DOI] [PubMed] [Google Scholar]
- Stringer JL, Williamson JM, Lothman EW. Induction of paroxysmal discharges in the dentate gyrus: frequency dependence and relationship to afterdischarge production. J Neurophysiol. 1989;62:126–135. doi: 10.1152/jn.1989.62.1.126. [DOI] [PubMed] [Google Scholar]
- Sun C, Mtchedlishvili Z, Bertram EH, Erisir A, Kapur J. Selective loss of dentate hilar interneurons contributes to reduced synaptic inhibition of granule cells in an electrical stimulation-based animal model of temporal lobe epilepsy. J Comp Neurol. 2007;500:876–893. doi: 10.1002/cne.21207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Nojyo Y. Projection of the entorhinal layer II neurons in the rat as revealed by intracellular pressure-injection of neurobiotin. Hippocampus. 1993;3:471–480. doi: 10.1002/hipo.450030408. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Makino H, Gage FH. Experience-specific functional modification of the dentate gyrus through adult neurogenesis: a critical period during an immature stage. J Neurosci. 2007;27:3252–3259. doi: 10.1523/JNEUROSCI.4941-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thind KK, Yamawaki R, Phanwar I, Zhang G, Wen X, Buckmaster PS. Initial loss but later excess of GABAergic synapses with dentate granule cells in a rat model of temporal lobe epilepsy. J Comp Neurol. 2010;518:647–667. doi: 10.1002/cne.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treves A, Rolls ET. Computational analysis of the role of the hippocampus in memory. Hippocampus. 1994;4:374–391. doi: 10.1002/hipo.450040319. [DOI] [PubMed] [Google Scholar]
- Treves A, Tashiro A, Witter MP, Moser EI. What is the mammalian dentate gyrus good for? Neuroscience. 2008;154:1155–1172. doi: 10.1016/j.neuroscience.2008.04.073. [DOI] [PubMed] [Google Scholar]
- van Groen T, Miettinen P, Kadish I. The entorhinal cortex of the mouse: organization of the projection to the hippocampal formation. Hippocampus. 2003;13:133–149. doi: 10.1002/hipo.10037. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Zhang N, Peng Z, Houser CR, Mody I. Perisynaptic localization of delta subunit-containing GABA(A) receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci. 2003;23:10650–10661. doi: 10.1523/JNEUROSCI.23-33-10650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A. Electrophysiology of epileptic human neocortical and hippocampal neurons maintained in vitro. Clin Neurosci. 1994;2:47–52. [Google Scholar]
- Xu Z, Xue T, Zhang Z, Wang X, Xu P, Zhang J, Lei X, Li Y, Xie Y, Wang L, Fang M, Chen Y. Role of signal transducer and activator of transcription-3 in up-regulation of GFAP after epilepsy. Neurochem Res. 2011;36:2208–2215. doi: 10.1007/s11064-011-0576-1. [DOI] [PubMed] [Google Scholar]
- Yu EP, Dengler CG, Frausto SF, Putt ME, Yue C, Takano H, Coulter DA. Protracted postnatal development of sparse, specific dentate granule cell activation in the mouse hippocampus. J Neurosci. 2013;33:2947–2960. doi: 10.1523/JNEUROSCI.1868-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Buckmaster PS. Dysfunction of the dentate basket cell circuit in a rat model of temporal lobe epilepsy. J Neurosci. 2009;29:7846–7856. doi: 10.1523/JNEUROSCI.6199-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wei W, Mody I, Houser CR. Altered localization of GABA(A) receptor subunits on dentate granule cell dendrites influences tonic and phasic inhibition in a mouse model of epilepsy. J Neurosci. 2007;27:7520–7531. doi: 10.1523/JNEUROSCI.1555-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]