

Abstract
Vesicular stomatitis virus, a single-stranded RNA virus, triggers activation of the serine-threonine kinases RIP1 and RIP3, which damages mitochondria by activating the GTPase DRP1. This results in excessive production of reactive oxygen species and subsequent activation of the NLRP3 inflammasome.
Cell death and inflammation are important aspects of the host response to pathogens and cellular damage. In this issue of Nature Immunology, Wang et al. demonstrate that vesicular stomatitis virus (VSV), a single-stranded RNA virus, activates the NLPR3 inflammasome through a signaling cascade of the serine-threonine kinases RIP1 and RIP3 (ref. 1). The RIP1-RIP3 complex is known to mediate the form of programmed lytic cell death called ‘necroptosis’2. However, Wang et al. show that the necroptotic activity of the RIP1-RIP3 complex and its ability to trigger the NLRP3 inflammasome are mediated by two distinct pathways, one that involves the necrosis effector protein MLKL and another that involves the mitochondrial fission protein DRP1 (ref. 1). This is an elegant example of how both cell-death pathways and inflammatory pathways are interconnected in the context of the response to complex pathogens such as VSV.
Specific pattern-recognition receptors detect viral nucleic acids in different cellular compartments. In the vacuolar space, the Toll-like receptor TLR9 detects DNA viruses, and TLR3 and TLR7 detect RNA viruses. The detection of viruses through these TLRs results in the secretion of type I interferon that is critical for the clearance of viruses. Type I interferon is also secreted downstream of cGAS, Mda5 and RIG-I, which are cytosolic sensors for DNA and RNA. In contrast to the cytosolic sensors involved in the production of type I interferon, another cytosolic sensor, AIM2, activates caspase-1 in response to DNA, but how RNA activates caspase-1 has remained unclear. The NLRP3 inflammasome can be activated by numerous structurally and functionally diverse ligands, including several DNA and RNA viruses. How the NLRP3 inflammasome detects these viruses is not known. NLRP3 probably detects its various agonists by sensing cellular damage, such as excessive generation of reactive oxygen species (ROS)3, disruption of lysosomal compartment4 and lowering of the cytosolic potassium (K+) concentration5. Here, Wang et al. demonstrate a novel mechanism for the activation of NLRP3 downstream of RIP1 and RIP3 (ref. 1).
Necroptosis is a type of programmed cell death induced by the activation of signaling via death receptors, such as the receptor for tumor-necrosis factor, TLR signaling via the adaptor TRIF or receptors for interferons2. Stimulation of these receptors leads to the formation of a RIP1-RIP3–containing necrosis-signaling complex (the ‘necrosome’) that signals downstream, which results in necroptosis2. In addition to mediating necroptosis, the RIP1-RIP3 complex has also been linked to the activation of inflammasomes2. In the current study, Wang et al. first investigate whether the RIP1-RIP3 complex is required for the activation of NLRP3. They establish that RIP3 is critical for the production of interleukin 1β (IL-1β) and IL-18 during infection with RNA viruses (VSV, Sendai virus or influenza virus) but not during infection with DNA viruses (adenovirus or herpes simplex virus). Furthermore, IL-1β production is inhibited by the RIP1 inhibitors Nec-1 and Nec-1s, which suggests that infection with VSV promotes the induction of IL-1β through the formation of a RIP1-RIP3 complex. Notably, this complex is not required for the activation of NLRP3 by other activators of NLRP3, such as ATP and nigericin. The RIP1-RIP3 complex initiates the phosphorylation of MLKL and the mitochondrial phosphatase PGAM5, which eventually results in necroptosis. Through the use of Mlkl−/− and Pgam5−/− macrophages, the authors confirm that MLKL is required for VSV-induced necroptosis but not for the activation of NLRP3. These findings also exclude the possibility that the process of pore formation by MLKL during necroptosis could have triggered the activation of NLRP3. Thus, the signaling cascade that leads to necroptotic cell death following infection with VSV is distinct from the cascade that leads to the activation of NLRP3.
Because NLRP3 is known to respond to mitochondrial dynamics, the authors next examine the state of the mitochondria in VSV-infected macrophages. They observe aberrant mitochondrial fission, perinuclear aggregation of damaged mitochondria and increased ROS production following infection1. Taking those findings a step further, the authors elegantly show that the mitochondrial damage results from the activation of DRP1. DRP1 is a GTPase that mediates mitochondrial fission and is normally found in the cytosol. It can localize to the mitochondrial outer membrane during cellular stress, where it is known to oligomerize and constrict the two mitochondrial membranes in the process of fission6. By in vitro phosphorylation assays, the authors determine that RIP1 phosphorylates DRP1 at Ser616 (ref. 1). However, RIP1 is precipitated with DRP1 only in the presence of RIP3, which suggests the formation of a ternary complex. Phosphorylated DRP1 then localizes to mitochondrial outer membrane, where it engages the mitochondrial fission protein Fis1 and induces mitochondrial fission and thus generates ROS and activates NLRP3. In addition, poly(I:C), a synthetic analog of double-stranded RNA (dsRNA), also induces phosphorylation of DRP1 and activation of NLRP3 similarly to infection with VSV. The induction of mitochondrial fission with the chemical compound CCCP, on the other hand, causes the activation of NLRP3 in a RIP3-independent manner. Thus, mitochondrial fission is probably the cellular event that NLRP3 detects downstream of VSV infection.
Although Wang et al. convincingly demonstrate distinct roles for DRP1 and MLKL downstream of the activation of RIP1-RIP3 (ref. 1), the sensor that detects RNA upstream of RIP1-RIP3 remains unclear. Published reports have suggested that DHX33 (a member of the DExD/H-box family of helicases) activates NLRP3 in response to dsRNA7. However, the authors eliminate this possibility in their VSV-infection model. They also rule out the possibility that TLR3, RIG-I and Mda5 are involved through the use of macrophages deficient in each of these molecules. Therefore, what activates RIP1-RIP3 in response to VSV or other RNA viruses remains to be clarified.
Cytosolic dsRNA has been shown to trigger the activation of NLRP3 through membrane permeabilization dependent on the signaling adaptor MAVS, which results in K+ efflux8. In that study, mitochondrial ROS caused damage to the plasma membrane and K+ efflux, which then triggered NLRP3 (ref. 8). It is possible that the RIP1-RIP3-DRP1 complex is upstream of the mitochondrial production of ROS induced by cytosolic dsRNA, but that was not examined in that study8. Similarly, Wang et al. do not investigate whether VSV-induced production of ROS leads to a decrease in the cytosolic concentration of K+ (ref. 1). The main difficulty in reconciling the data from the two studies is that the former shows that MAVS is essential8, while in the latter, RIG-I is not involved1. Because both studies use poly(I:C), the difference in dependence on MAVS could arise from experimental variations or the highly complex nature of poly(I:C) itself. Despite discrepancies in the sensors ‘opted’ by cytosolic RNA in these two studies, it is still plausible that they converge on one common cellular event. Notably, another study has suggested a direct link between NLRP3 and MAVS9. Whether there is a plausible hypothesis to explain these diverse mechanisms for the activation of NLRP3 remains to be seen. In vivo, viruses can undoubtedly trigger RIP1-RIP3 activity through a variety of different pathways. The two distinct outcomes of formation of the RIP1-RIP3 complex presented here may be used at different stages of infection. For VSV, it appears that the virus has evolved to evade the inflammasomes, because wild-type mice and mice deficient in caspase-1 and caspase-11 have similar susceptibility to VSV10. However, another RNA virus, influenza A virus, appears to be sensitive to activation with caspase-1 (refs. 11,12). Nevertheless, as researchers continue to consider the physiological roles of RIP1-RIP3, whether the phenotype of RIP3-deficient mice in a particular infection is due to necroptosis or activation with caspase-1 should now routinely be considered.
Figure.
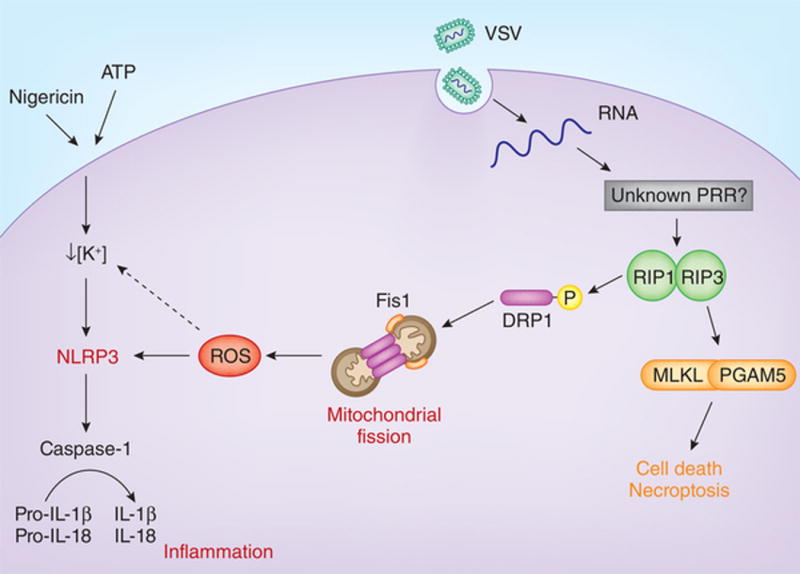
Once activated, NLRP3 leads to the processing of pro-caspase-1 into its active form. Active caspase-1 then cleaves the pro-IL-1β and pro-IL-18 into their secreted, mature forms. Wang et al. show that during infection with RNA viruses such as VSV, the activation of NLRP3 is closely tied to necroptosis. Virus-derived cytosolic RNA induces formation of the RIP1-RIP3 complex, in which RIP1 phosphorylates DRP1. DRP1 then engages Fis1 on the mitochondrial outer membrane, where DRP1 is known to oligomerize to induce mitochondrial fission. Mitochondrial fission leads to increased cytosolic levels of ROS and activated NLRP3. Notably, the authors demonstrate that this arm of RIP1-RIP3 signaling cascade is distinct from the necroptotic arm that is executed by MLKL and PGAM5. Additionally, the mechanisms presented here are also distinct from the activation of NLRP3 by other well-known NLRP3 agonists, such as ATP and nigericin. PRR, pattern-recognition receptor.
References
- 1.Wang, et al. Nat Immunol. 2014;15:1126–1133. doi: 10.1038/ni.3015. [DOI] [PubMed] [Google Scholar]
- 2.Kaczmarek A, Vandenabeele P, Krysko DV. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Zhou R, Yazdi AS, Menu P, Tschopp J. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 4.Hornung V, et al. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muñoz-Planillo R, et al. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitoma H, et al. Immunity. 2013;39:123–135. doi: 10.1016/j.immuni.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franchi L, et al. J Immunol. 2014;193:4214–4222. doi: 10.4049/jimmunol.1400582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. Cell. 2013;153:348–361. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajan JV, Rodriguez D, Miao EA, Aderem A. J Virol. 2011;85:4167–4172. doi: 10.1128/JVI.01687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen IC, et al. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas PG, et al. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]