

Abstract
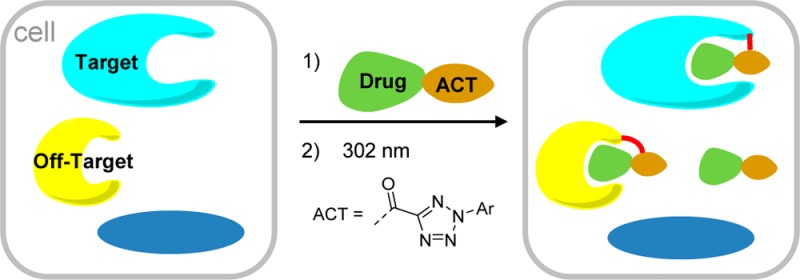
Photoaffinity labels are powerful tools for dissecting ligand–protein interactions, and they have a broad utility in medicinal chemistry and drug discovery. Traditional photoaffinity labels work through nonspecific C–H/X–H bond insertion reactions with the protein of interest by the highly reactive photogenerated intermediate. Herein, we report a new photoaffinity label, 2-aryl-5-carboxytetrazole (ACT), that interacts with the target protein via a unique mechanism in which the photogenerated carboxynitrile imine reacts with a proximal nucleophile near the target active site. In two distinct case studies, we demonstrate that the attachment of ACT to a ligand does not significantly alter the binding affinity and specificity of the parent drug. Compared with diazirine and benzophenone, two commonly used photoaffinity labels, in two case studies ACT showed higher photo-cross-linking yields toward their protein targets in vitro based on mass spectrometry analysis. In the in situ target identification studies, ACT successfully captured the desired targets with an efficiency comparable to the diazirine. We expect that further development of this class of photoaffinity labels will lead to a broad range of applications across target identification, and validation and elucidation of the binding site in drug discovery.
Introduction
In target-based approaches to drug discovery, linking the observed phenotypic response to a ligand of interest with on-target modulation is a critical step.1 To this end, both on- and off-targets of a drug candidate need to be identified and characterized prior to clinical development. Among many target identification methods, photoaffinity-labeling is particularly attractive, as the transient association of the molecular targets with a drug candidate becomes permanent after photo-cross-linking in the native cellular environment. In addition, for targets that are part of a fragile multiprotein complex, the in situ covalent capture prevents potential loss of the targets after cell lysis. Because photoaffinity probes are generally used in excess relative to their targets in order to drive the formation of the target–drug complexes, nonspecific targets can also be captured during photo-cross-linking. To overcome this problem, two strategies have been employed: (i) use ligands with higher affinity so that the photoaffinity probes can be used at lower concentrations; and (ii) use photoaffinity labels that generate reactive intermediates with high, yet selective reactivities toward the ligand-bound targets. To this end, only a few photoaffinity labels have been reported in the past 40 years (Chart 1), including phenyl azide,2 diazirine (DA),3 and benzophenone (BP).4 While these photoaffinity labels have shown tremendous versatility in biomedical research, nevertheless they have two major shortcomings:5 (i) the photogenerated nitrene, carbene, and diradical intermediates exhibit extremely short half-lives, leading to very low target capturing yields, and (ii) the nitrone, carbene, and diradical intermediates are prone to react nonselectively with any proximal C–H/X–H bonds (X = N, O, S), resulting in high background.
Chart 1. Structures of Photoaffinity Labels.

To balance reactivity with specificity, we envisioned that alternative photogenerated intermediates may exhibit longer half-lives and greater functional group selectivity. Indeed, Hamachi and co-workers have reported elegant ligand-directed chemistries based on the electrophilic tosyl6 and acyl imidazolyl7 groups and demonstrated their exquisite specificity in selective protein target labeling in situ.8 Inspired by this work, we hypothesized that an appropriately functionalized photoreactive tetrazole9 could serve as a highly selective, electrophilic photoaffinity label for in situ target capture.10 Here we report the development of 2-aryl-5-carboxytetrazole (ACT, Chart 1) as a robust photoaffinity label for identification of the targets and off-targets of Dasatinib and JQ-1−two drugs profiled extensively in the literature.11,12 Compared with DA and BP, ACT gave higher yields in the ligand-directed photo-cross-linking reactions with the recombinant target proteins. In addition, the ACT-based probes facilitated the in situ target identification in a manner similar to the DA-based ones.
Results and Discussion
Design, Synthesis, and Biological Evaluation of the Photoaffinity Probes
Whereas the tosyl and acyl imidazolyl groups were successfully employed in labeling endogenous targets in living cells, they are not ideal affinity labels for general target identification because of concerns about their stability in cellular milieu.13 We hypothesized that ACT should serve as an ideal photoaffinity label based on the following considerations: (i) 5-carboxy-substituted 2-aryltetrazoles are photoactive;14 (ii) placement of a carboxyl group at the C5-position of 2H-tetrazole increases the electrophilicity of the photogenerated carboxy-nitrile imine intermediate; (iii) nucleophilic thiol-addition of 2-mercaptobenzoic acid15 and 3-mercaptopropionic acid16 to the base-generated carboxy-nitrile imine was reported in the literature; and (iv) the photogenerated carboxy-nitrile imine intermediate should undergo rapid medium quenching when a proximal nucleophile is not available (Figure S1),9,17 minimizing the undesired reactions with nonspecific targets. Thus, two series of photoaffinity probes were prepared (see Supporting Information for synthetic schemes): one series is based on Dasatinib, a potent inhibitor of Bcr-Abl kinase,18 the Src family kinases, as well as BTK (Bruton’s tyrosine kinase);19 and the other is based on JQ-1, a potent inhibitor of the BET family of bromodomain proteins20 (Figure 1a). Specifically, three photoaffinity labels, ACT, DA, and BP, were attached to Dasatinib or JQ-1 via the previously reported linkage sites.11,12 The linker length was varied to adjust the distance of the reactive intermediate from the target binding site (Figure 1a). An alkyne tag was placed on the photoaffinity labels to enable the click chemistry-mediated detection and enrichment of the targets from cell lysates.21 To determine how the attachment of photoaffinity label affects the inhibitory activity and specificity, kinome profiling was carried out for Dasatinib-derived probes (Figure 1b and Table S1), while the in vitro binding assay was performed for JQ-1-derived probes (Figure 1c and Table S2). We found that DA probes 2a and 2b retained most of their inhibitory activities while ACT probes 1a and 1b showed modest reduction (2–20-fold), particularly for 1a with the shorter linker. In comparison, the BP probes exhibited the largest reduction in inhibitory activity (25–400-fold), presumably due to the positioning of a large, flat aromatic structure in the solvent-exposed hinge region. For the JQ-1 series, almost all the photoaffinity probes demonstrated greater inhibitory activities than JQ-1, indicating that the hydrophobic photoaffinity labels form additional interactions with BRD2-4 outside the shallow and flat canonical binding pocket.20 In the cell proliferation assays, the photoaffinity label-linked JQ-1 probes showed potencies similar to the parent JQ-1 against leukemic cell line SKM-1, but reduced activities against breast carcinoma MX-1 as well as nonsmall cell lung carcinoma NCI-H1299 (Table S2), likely due to the permeability difference of the photoaffinity probes and/or the disparate dependency of these cell lines on BRD proteins for proliferation.
Figure 1.

Dasatinib and JQ-1-derived photoaffinity probes containing 2-aryl-5-carboxy-tetrazole (ACT), diazirine (DA), or benzophenone (BP) photoaffinity label (PAL) and their biological activities. (a) Structures of the photoaffinity probes. (b) Kinome profiling of Dasatinib-derived photoaffinity probes. A panel of 82 protein kinases were surveyed in this assay, and inhibition constants, Ki, are given in micromolar. See Table S1 in the Supporting Information for Ki values. The nonreceptor tyrosine kinase targets are indicated by blue arrows. NV, no value. (c) Plots of the inhibition of BRD-2, -3, and -4 by JQ-1-derived photoaffinity probes. See Table S2 in the Supporting Information for Ki values.
Evaluation of Photo-Cross-Linking Efficiency in Vitro
To compare the efficiency of these three photoaffinity labels in covalently labeling their targets, we treated recombinant BTK and BRD4 proteins with appropriate probes, and we detected the photo-cross-linked adducts using in-gel fluorescence analysis after the copper-catalyzed click chemistry with rhodamine-azide.22 For BTK, all probes showed irradiation and ligand-dependent labeling, with 1a and 3a giving the strongest fluorescence (Figure 2a and Figure S2), suggesting other factors, e.g., click chemistry yield, may also affect the overall labeling efficiency. These probes also selectively labeled BTK in the K562 cell lysate spiked with recombinant BTK protein (Figure S3). For BRD4 protein, both ACT- and DA-based probes showed UV light- and ligand-dependent labeling, while BP-based probes 6a/6b exhibited strong background labeling, evidenced by lack of signal attenuation in the presence of JQ-1 (Figure 2b) as well as labeling of BSA which was added to the reaction mixture to prevent nonspecific binding of BRD4 to the plastic surface (Figure S4). In the K562 cell lysate spiked with recombinant BRD4, ACT-based probe 4a showed stronger labeling of BRD4 than DA-based probe 5a, while BP-based probe 6a showed no labeling (Figure S5), presumably due to its nonspecific associations with many cellular proteins.
Figure 2.

Evaluating the efficiency and selectivity of photoaffinity-labeling of recombinant proteins by the small-molecule probes. (a) Evaluating the BTK labeling efficiency using in-gel fluorescence (top panels). For reaction setup, 0.5 μg of BTK (final concentration ≈0.1 μM), 0.2 μM small-molecule probe, 10 μM Dasatinib (for competition only) in 50 μL of PBS were used. For photoirradiation, a hand-held UV lamp with a wavelength of 302 nm for ACT (5 min) and BP (20 min) or 365 nm for DA (10 min) was used. (b) Evaluating the BRD4 labeling efficiency using in-gel fluorescence (top panels). For reaction setup, 0.4 μg of BRD4 (final concentration ≈0.4 μM), 0.2 μM small-molecule probe, 5 μg of BSA, and 10 μM JQ-1 (for competition only) in 50 μL of PBS were used. The equal loading of proteins was verified by SYPRO Ruby staining of the same gels (bottom panels). See Supporting Information for procedures of click chemistry with TAMRA-azide and polyacrylamide gel electrophoresis.
Because the photoaffinity probes with the short linker in general exhibited higher labeling efficiency (Figure 2), we decided to focus on these probes in the following comparison studies. To quantify the photo-cross-linking yield, we incubated recombinant protein targets with appropriate Dasatinib or JQ-1 probes, subjected the mixture to a brief UV irradiation, and analyzed the mixtures by LC-MS. Gratifyingly, ACT-based probes 1a and 4a showed robust photo-cross-linking with their targets, reaching 60% cross-linking yield for 1a (Figure 3a) and 95% cross-linking yield for 4a (Figure 3d). In contrast, DA-based probes 2a and 5a gave the desired photo-cross-linked products in much lower yields (Figure 3b, 3e). The control experiment showed that the photoactivation efficiencies are similar between the ACT and DA probes (Figure S6). Surprisingly, BP-based probes 3a and 6a did not yield any detectable photo-cross-linked adducts; instead, the recombinant target proteins showed significant broadening of their mass peaks, suggesting the initial photoadducts, if they are formed, may have undergone fragmentation to generate less than expected lower-molecular weight adducts (Figure 3c, 3f). An alternative explanation is that the benzophenone serves as a photosensitizer to cause nonspecific oxidative damage to the proteins.23 Importantly, the ACT-mediated photo-cross-linking with the target protein is ligand-dependent, as addition of Dasatinib or JQ-1 into the reaction mixture abolished the photoadducts (Figure S7). In addition, the photo-cross-linking yield showed probe-concentration dependency as increasing amount of ACT-probe 4a used in the reaction led to a higher photo-cross-linking yield (Figure S8).
Figure 3.

Quantifying the cross-linking efficiency of the photoaffinity labels with recombinant target proteins by LC-MS. (a–c) Deconvoluted masses of the product mixture after incubating 2.5 μM human BTK387–659 with 25 μM Dasatinib probe 1a, 2a, or 3a in 100 μL of PBS for 15 min followed by photoirradiation with a hand-held UV lamp for 5 min (302 nm for ACT and BP, 365 nm for DA) on ice. (d–f) Deconvoluted masses of the product mixture after incubating 2.5 μM BRD444–168 with 5 μM JQ-1 probe 4a, 5a, or 6a in 100 μL of PBS followed by photoirradiation with a hand-held UV lamp for 5 min (302 nm for ACT and BP, 365 nm for DA) on ice. The cross-linking yield was calculated using the following equation: yield% = Iphotoadduct/(Itarget protein + Iphotoadduct), where Itarget protein and Iphotoadduct represent the ion counts of the target protein and the photoadduct, respectively, and marked at the upper-right of the spectra.
Identification of the ACT Cross-Linking Site by Tandem Mass Spectrometry
To identify cross-linking sites on the target protein, we digested the probe 1a-treated recombinant BTK protein with trypsin, and analyzed the product mixture by LC-MS/MS. A tripeptide fragment corresponding to BTK488–490 with the carboxy-nitrile imine linked with the Glu-488 side chain was identified (Figure 4a). It is noted that recombinant BTK387–659 protein contains 25 Glu and 14 Asp residues and only Glu-488 was detected as labeled, indicating that the photo-cross-linking is ligand-dependent. This ligand-directed proximity-driven reactivity is consistent with the probe docking model (Figure 4b) in which the binding of probe 1a to the kinase active site brings the C5 of the ACT ca. 6.9 Å away from the carboxylate of Glu-488; indeed, it is the only nucleophilic side chain within 9.0 Å from the electrophilic site. Certainly, because the ACT is completely solvent exposed and highly mobile, these distances may vary as the ACT orients itself dynamically relative to the BTK protein. We propose that the photoadduct is formed via nucleophilic addition of the Glu-488 carboxylate to the carboxy-nitrile imine intermediate followed by a 1,4-acyl shift (Figure 4c). This mechanism is consistent with a literature report in which quenching of the in situ generated diaryl nitrile imine by an excess carboxylic acid produced the N′-acyl-N′-aryl-benzohydrazide product in good yield.24 It is conceivable that other nucleophiles such as Cys (Figure S9),12 if they are in close proximity, may also participate in the cross-linking reactions with ACT for other targets. Since a recent report25 suggested that the photoreactivity of diaryltetrazole can be harnessed for photo-cross-linking with target proteins through their acidic side chains, we compared the intrinsic reactivity of the carboxy-nitrile imine to that of the diaryl-nitrile imine toward glutamic acid (10 mM) in mixed PBS/acetonitrile (1:1) solution. In the model study, the glutamate-quenching product was clearly detected for the diphenyl-nitrile imine (Figure S10). In contrast, the carboxy-nitrile imine underwent predominant chloride quenching when a weak nucleophile such as glutamic acid is present in solution (Figure S9), suggesting that the observed photo-cross-linking of 1a with Glu-488 of the BTK enzyme is not merely the result of elevated local concentration of the glutamate near the in situ generated carboxy-nitrile imine. Indeed, because of the rapid chloride quenching of the reactive carboxy-nitrile imine, ACT should be more suitable as a photoaffinity label than the diaryltetrazoles, as the background cross-linking reactions with the nucleophilic side chains present on protein surfaces would be minimal.
Figure 4.

Determination of the cross-linking site on BTK protein and the proposed ligand-directed cross-linking mechanism. (a) Sequence of the full-length His6-(TEV)-G-hBTK387–659. The MS/MS spectrum for probe 1a-modified tripeptide fragment, EMR, is shown with the fragment ions annotated on the structure. (b) A docking model of probe 1a bound to BTK (PDB code: 3K54) showing a proximal Glu-488 residue located on a loop 6.9 Å away from the C5 of the tetrazole ring. (c) Proposed mechanism of the ligand-dependent nucleophilic addition to the carboxy-nitrile imine followed by the O → N acyl shift to generate the specific photoadduct.
ACT-Enabled in Situ Target Identifications
Encouraged by high in vitro photo-cross-linking efficiency, we sought to assess the efficiency and selectivity of ACT as a new photoaffinity label for in situ target identification. For comparison, we included the DA-based probes 2a and 5a, as they exhibited excellent biological activities (Figure 1) and moderate photo-cross-linking reactivity (Figures 2 and 3). In brief, suspended K562 cells were treated with 1 μM probe 1a, 2a, 4a, or 5a for 5 h before UV irradiation (5 min for ACT probe-treated cells at 302 nm; 10 min for DA probe-treated cells at 365 nm). The cells were lysed, and the lysates were reacted with biotin azide prior to pulldown with the streptavidin agarose beads.20 Western blot analyses revealed that the Dasatinib targets, BTK, Src, and Csk, and the JQ-1 target, BRD4, were successfully captured by their respective photoaffinity probes, and pretreating the cells with 50 μM parent drug, Dasatinib or JQ-1, abolished the capture (Figure S11). In-gel digestion of the streptavidin captured proteins on SDS-PAGE gel followed by LC-MS/MS analyses produced lists of potential targets. To ensure that the captured proteins are derived from the ligand-dependent photo-cross-linking, high-confidence targets were compiled based on the following two criteria: (1) at least two unique peptides were identified in the MS, and (2) the area under the curve (AUC)−a measurement of MS signal intensity and reliability−for the parent drug-pretreated sample is not detectable. Using these criteria, six kinases were identified by probe 1a, five of which also appeared in probe 2a-treated cells, indicating ACT works similarly to DA (Figure 5a, Table S3). However, probe 1a failed to identify Abl protein, presumably due to a lack of proximal nucleophilic side chains near the kinase active site. For JQ-1 targets, probes 4a and 5a successfully captured the bromodomain proteins BRD-2, -3, and -4 with minimum off-targets (Figure 5b, Table S4), suggesting both ACT and DA are efficient in the in situ target identification. Comparison of our data with other literature-reported MS-based target identification studies revealed that these ACT- and DA-based photoaffinity probes performed exceptionally well (Tables S5–S6).
Figure 5.
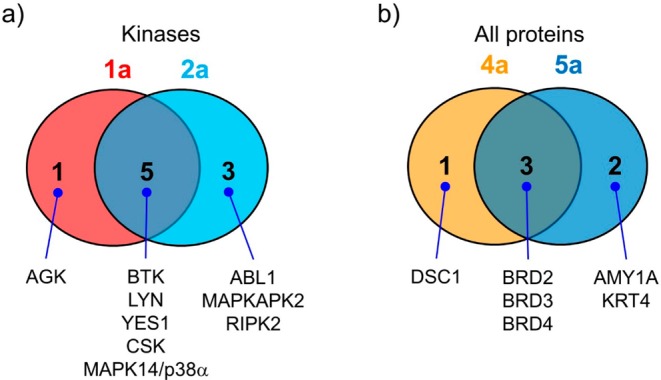
Venn diagrams of the identified protein targets by (a) Dasatinib-derived photoaffinity probes 1a and 2a; and (b) JQ-1-derived photoaffinity probes 4a and 5a.
Taken together, we show that ACT can serve as an effective photoaffinity label for target identification both in vitro and in live cells. Compared to the existing photoaffinity labels such as BP and DA, the main advantage of ACT lies in its unique photo-cross-linking mechanism, which in principle should lead to reduced background reactions with nonspecific targets as well as a facile mapping of the ligand-binding site. Structurally, ACT is comparable in size to BP and the electronically stabilized DA derivatives such as trifluoromethylaryl diazirine, and it features a modular design with the carboxy group at the C5-position of 2H-tetrazole, providing the conjugation handle for a drug molecule and the aryl group responsible for the photoreactivity. Compared to DA and BP, ACT showed higher cross-linking yields with the desired targets in vitro (Figure 3), but it produced similar efficiency in target capture in situ in a two-step cross-linking/capture procedure (Figure S11), suggesting additional optimization of the capture step may be necessary in order to achieve higher overall target capture yield. At present, an alkyne tag was appended onto the aryl ring to enable the click chemistry-mediated target capture. However, alternative chemical moieties that are captured covalently by engineered enzymes, e.g., the haloalkane moiety for HaloTag26 and the benzoguanine moiety for SNAP tag,27 will be explored in the future for more efficient target capture. Because of its unique cross-linking mechanism, a potential drawback of ACT is that a suitable nucleophile needs to be present near the ligand-binding site for a target to be captured and identified, which may result in false negative; for example, Abl kinase was not identified by 1a in this study. In principle, this limitation can be potentially overcome by increasing the linker length between ACT and the ligand to allow the survey of a larger area surrounding the ligand-binding pocket.
In summary, we have developed a new photoaffinity label, 2-aryl-5-carboxytetrazole (ACT), for efficient in situ target capture and subsequent identification. The attachment of ACT to two drug molecules was generally well tolerated without significantly altering the binding affinity and specificity. Compared with DA and BP, ACT provides a unique mechanism of target capture through which the photogenerated carboxy-nitrile imine intermediate reacts with a proximal nucleophile near the target active site. As a result, ACT displayed the cleanest and most efficient cross-linking with the recombinant target proteins in vitro among the three photoaffinity labels tested. In the in situ target identification studies with two previously profiled drugs, Dasatinib and JQ-1, ACT successfully captured the desired targets in both cases with an efficiency comparable to DA. While aniline was used as the aryl group in the present study, a wide range of heterocycles will be explored in the future with a goal to identify ACTs with enhanced solubility and photo-cross-linking reactivity.
Experimental Section
In-Gel Fluorescence Analysis of BTK Labeling by Dasatinib Probes in Vitro
One microliter of 0.5 mM Dasatinib in DMSO (for competition experiments) or DMSO (without Dasatinib competition) was added to 0.5 μg of BTK in 50 μL of PBS. After incubation at r.t. for 15 min, 1 μL of 10 μM photoaffinity probe in DMSO was added. After additional incubation at r.t. for 30 min, the mixture was irradiated with a hand-held 302 nm UV lamp, ca. 2–3 cm from the top of the sample. A premixed click reaction cocktail (6 μL, 1:3:1:1 of 50 mM CuSO4 in water/1.7 mM TBTA in 1:4 DMSO-tBuOH/50 mM TCEP in water/1.25 mM TAMRA-azide in DMSO) was added, and the reaction mixture was incubated at r.t. for 1 h. After 1 h, 500 μL of cold acetone was added, and the mixture was left at −20 °C overnight. The mixture was then centrifuged at 17,200g at 4 °C for 20 min and the pellet was collected. To the pellet was added 30 μL of 1 × SDS sample buffer, and the mixture was boiled at 95 °C for 10 min before SDS-PAGE with 4–20% Bis-Tris gel using MOPS as running buffer.
In-Gel Fluorescence Analysis of BTK Labeling by Dasatinib Probes in K562 Cell Lysate
One microliter of 0.5 mM Dasatinib in DMSO (for competition experiments) or DMSO (without Dasatinib competition) was added to 0.5 μg of BTK in 50 μL of 2 mg/mL K562 cell lysate in PBS. After incubation at r.t. for 15 min, 1 μL of 10 μM photoaffinity probe in DMSO was added. After additional incubation at r.t. for 30 min, the mixture was irradiated with a hand-held 302 nm UV lamp, ca. 2–3 cm from the top of the sample. A premixed click reaction cocktail (6 μL, 1:3:1:1 of 50 mM CuSO4 in water/1.7 mM TBTA in 1:4 DMSO-tBuOH/50 mM TCEP in water/1.25 mM TAMRA-azide in DMSO) was added, and the reaction mixture was incubated at r.t. for 1 h. After 1 h, 500 μL of cold acetone was added and the mixture was left at −20 °C overnight. The mixture was then centrifuged at 17,200g at 4 °C for 20 min, and the pellet was collected. To the pellet was added 30 μL of 1 × SDS sample buffer, and the mixture was boiled at 95 °C for 10 min before SDS-PAGE with 4–20% Bis-Tris gel using MOPS as running buffer.
In-Gel Fluorescence Analysis of BRD4 Labeling by JQ-1 Probes in Vitro
One microliter of 0.5 mM (+)-JQ-1 in DMSO (for competition experiments) or DMSO (without competition) was added to 0.4 μg of BRD4 and 5 μg of BSA (added to reduce nonspecific binding to the vial surface) in 50 μL of PBS. After incubation at r.t. for 15 min, 1 μL of 10 μM photoaffinity probe in DMSO was added. After additional incubation at r.t. for 30 min, the mixture was irradiated with a hand-held 302 nm UV lamp, ca. 2–3 cm from the top of the sample. A premixed click reaction cocktail (6 μL, 1:3:1:1 of 50 mM CuSO4 in water/1.7 mM TBTA in 1:4 DMSO-tBuOH/50 mM TCEP in water/1.25 mM TAMRA-azide in DMSO) was added, and the reaction mixture was incubated at r.t. for 1 h. After 1 h, 500 μL of cold acetone was added and the mixture was left at −20 °C overnight. The mixture was then centrifuged at 17,200g at 4 °C for 20 min and the pellet was collected. To the pellet was added 30 μL of 1 × SDS sample buffer, and the mixture was boiled at 95 °C for 10 min before SDS-PAGE with 4–20% Bis-Tris gel using MOPS as running buffer.
In-Gel Fluorescence Analysis of BRD4 Labeling by JQ-1 Probes in K562 Cell Lysate
One microliter of 0.5 mM (+)-JQ-1 in DMSO (for competition experiments) or DMSO (without competition) was added to 0.1 μg of BRD4 in 50 μL of 2 mg/mL K562 lysate in PBS. After incubation at r.t. for 15 min, 1 μL of 10 μM photoaffinity probe in DMSO was added. After additional incubation at r.t. for 30 min, the mixture was irradiated with a hand-held 302 nm UV lamp, ca. 2–3 cm from the top of the sample. A premixed click reaction cocktail (6 μL, 1:3:1:1 of 50 mM CuSO4 in water/1.7 mM TBTA in 1:4 DMSO-tBuOH/50 mM TCEP in water/1.25 mM TAMRA-azide in DMSO) was added, and the reaction mixture was incubated at r.t. for 1 h. After 1 h, 500 μL of cold acetone was added and the mixture was left at −20 °C overnight. The mixture was then centrifuged at 17,200g at 4 °C for 20 min and the pellet was collected. To the pellet was added 30 μL of 1 × SDS sample buffer, and the mixture was boiled at 95 °C for 10 min before SDS-PAGE with 4–20% Bis-Tris gel using MOPS as running buffer.
Kinome Profiling
Plates were stamped with 5 μL of kinase buffer (Life Technologies #PR4940D) containing recombinant kinase (2.5–10 nM final concentration), Eu or Tb labeled antibodies (His or GST; 0.5–2 nM final concentration), and fluorescently tagged probe (3–200 nM final concentration). Appropriate probes were diluted in kinase buffer, and 120 μL of compound was added to the plate using Biomex FX. Plates were incubated at r.t. for 2 h and read on an Envision plate reader (PerkinElmer). The Ki values were calculated using the Assay Explorer software (Accelrys). Example for BTK kinase: BTK (Invitrogen, PV3363) was added to 5 μL of kinase buffer (#PR4940D) to a final concentration of 10 nM, supplemented with 2 nM Tb labeled anti-His antibody and 200 nM Oregon Green labeled probe. Afterward, 120 μL of diluted compound in kinase buffer was added and the plate was incubated at r.t. for 2 h. The plate was read on an Envision plate reader, and the Ki values were calculated using the Assay Explorer software.
In Situ Target Identification
Cell Treatment
One hundred million K562 cells were plated in 20 mL of DMEM media (∼5 million cells/mL) without FBS and antibiotics. Twenty μL of 50 mM unmodified ligand (Dasatinib or (+)-JQ-1) in DMSO (competition experiments) or DMSO (without competition, control) was added to the cells, and the mixture was incubated for 30 min (37 °C, 5% CO2, gentle shaking). Afterward, 20 μL of 1 mM probe in DMSO (all experiments except the control) or DMSO (control) was added (final competitor concentration = 50 μM, final probe concentration = 1 μM), and the sample was kept in the incubator for 5 h (37 °C, 5% CO2, gentle shaking).
Photoirradiation and Cell Lysis
After 5 h of incubation, cells were washed twice with 2 mL of PBS and then resuspended in 2 mL of PBS in 35 mm Petri dishes. The mixture was irradiated with a hand-held 302 nm UV lamp, ca. 2–3 cm from the top of the sample placed on ice. PBS was changed to 2 mL of 0.02% Tween-20 in PBS, and a protease inhibitor cocktail was added (Amresco, #M250). The suspended cells were lysed with sonication (10 × 10 s with 10 s breaks, 40% power) on ice. The lysate was centrifuged (20 min, 17,200g, 4 °C) and filtered through a 0.2 μm membrane. The protein concentration was measured to be 8–12 mg/mL using the BCA assay.
Click Reaction and Target Affinity Capture
Click reaction was performed by following a published procedure.22 In brief, 10 mg of cell lysate was diluted with PBS to 5 mL to obtain a final concentration of 2 mg/mL. To the above solution, 113 μL of 5 mM azide-PEG3-biotin (Aldrich, #762024) in DMSO, 113 μL of 50 mM TCEP in PBS, 340 μL of 1.7 mM TBTA in 1:4 DMSO-tBuOH, and 113 μL of 50 mM CuSO4 were added. The mixture was gently mixed at r.t. for 1 h before 45 mL of acetone was added. The reaction mixture was kept at −20 °C overnight. After centrifugation, the protein pellet was collected, washed with 2 × 10 mL of cold methanol, and redissolved in 14 mL of 0.1% SDS in PBS. Prewashed streptavidin agarose beads (60 μL, Thermo Scientific, #20347) were added, and the mixture was rocked at 4 °C overnight. The beads were washed with 3 × 1 mL of 0.1% SDS in PBS followed by 5 × 1 mL of PBS. Then, 60 μL of 2 × SDS sample buffer was added and the mixture was boiled at 95 °C for 12 min before samples were loaded onto SDS-PAGE gel.
Determination of Intact Masses of Protein Substrates
RPLC-MS was performed using an Agilent 1100 HPLC coupled to an Agilent LC/MSD TOF running MassHunter Workstation Acquisition B.04.00. Data was deconvoluted in MassHunter Qualitative Analysis B.07.00 using the maximum entropy algorithm with a 0.5 Da mass step, proton mass adduct, and baseline subtract factor 7.0.
Peptide Mapping of the Cross-Linking Site
The site of modification of BTK by probe 1a was determined by in-gel trypsin digestion of the band corresponding to the protein after labeling with 1a as described in the following reference: Shevchenko, A. Evaluation of the efficiency of in-gel digestion of proteins by peptide isotopic labeling and MALDI mass spectrometry.28 LC-MS/MS analysis was performed using a Waters NanoAcquity HPLC system coupled to a Thermo Fisher Scientific Fusion mass spectrometer. Separation of the peptides was achieved using a Thermo EasySpray PepMap column (ES802; C18, 2 μm, 100 Å, 75 μm × 25 cm) at a flow of 0.25 μL/min, with a gradient starting at 5% B (B = 0.1% formic acid in acetonitrile, A = 0.1% formic acid in water), ramping to 15% B at 2 min, 15–35% B over 20 min, followed by a 5 min ramp to 80% B, washing for 6 min at elevated flow (0.4 μL/min), before returning to the starting conditions. The Fusion source was operated at 1.9 kV in positive ion mode with MS detection in the Orbitrap using 120 K resolution. The modified tripeptide was identified by its fragmentation spectrum that resulted from quadrupole isolation of the triply charged ion using an isolation window of 1.8 m/z, and fragmentation via HCD at 26% collision energy, with fragment ion detection in the Orbitrap at 15K resolution. Data was interpreted manually.
Proteomics by LC-MS/MS
Protein from in situ enriched samples was eluted from beads with 100 μL of 2 × LDS-PAGE sample buffer (Invitrogen; 141 mM Tris base, 106 mM Tris·HCl, 2% LDS, 10% glycerol, 0.51 mM EDTA, 0.22 mM SERVA Blue G, 0.175 mM Phenol Red, pH 8.5), and the mixture was heated to 80 °C for 10 min. A 20-μL sample was applied to SDS-PAGE running with 4–12% Bis-Tris gel and MOPS running buffer to remove the detergent. In-gel digestion was performed on 6 gel sections as described.28 Five microliters, representing 10% of each sample, was loaded via Waters NanoAcquity autosampler onto an Acclaim pep map precolumn (P/N 164535) with online trapping and salt removal (trapping flow rate at 5 μL/min for 3.5 min). Analytical separation was performed over a 90 min run using an Easy Spray column (ES802) heated to 45 °C. Reverse phase gradient was delivered at a flow rate of 0.225 μL/min by Waters NanoAcquity HPLC as follows: 0 min 10% B, 55 min 25% B, 60 min 40% B, 60.1 min 98.0% B, 65.1 min 10% B, 89.0% B, where B is 0.1% formic acid in acetonitrile. Spectra were collected on a Thermo Fisher Scientific Fusion mass spectrometer using the following parameters: 2.1 kV spray voltage, 275 °C transfer tube temperature, 350–1500 m/z scan range with a quadrupole isolation window of 1.6 m/z, MS1 in the Orbitrap at 120 K resolution, MS2 by CID in the ion trap with rapid speed, MS2 scans collected with top speed 3 s cycle, dynamic exclusion with repeat count 1 if occurs within 30 s and exclude for 60 s. MIPS on with charge states 2–7 allowed with 4E5 AGC orbitrap and 2E3 ion trap settings.
Proteomics Bioinformatics
Raw files were processed by Proteome Discoverer (v 2.1.081) and searched by Mascot (v 2.4.0) using the Uniprot human database (downloaded 08-10-2015). MS1 tolerance was set to 20 ppm, and MS2 tolerances were set to 0.8 Da. Static cysteine carbamidomethyl and variable oxidized methionine were included as modifications. FDR was set to 1% via Percolator. Criteria for acceptance were based on designation as a “Master” Protein with greater than 1 unique peptide and high protein confidence. Label-free quantitation was performed with the Precursor Ions Area Detector function. Areas under the curve (AUCs) less than 2.0E5 were determined below LOQ based on previous studies on the performance of the instrument using proteomic reagent standards. Thresholds per experiment were set for significant differences dependent upon the determination of potential sample loading bias by comparing total ion chromatogram (TIC) intensity between paired injections (± competition) and average signal from nonspecifically binding protein background.
Acknowledgments
We gratefully acknowledge the NIH (GM085092 to Q.L.), AbbVie (to Q.L.), and the Rosztoczy Foundation (to A.H.) for financial support. Maricel Torrent, Paul Richardson, Jan Waters, Shaun McLoughlin, Hua Tang, Matt Kurnick, Peter Kovar, and Nirupama Soni, all AbbVie employees, and Marco Jacinto from the Lin lab are acknowledged for their contributions to various portions of this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06645.
Author Contributions
§ A.H. and J.M. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Bunnage M. E.; Gilbert A. M.; Jones L. H.; Hett E. C. Nat. Chem. Biol. 2015, 11, 368–372. 10.1038/nchembio.1813. [DOI] [PubMed] [Google Scholar]
- Fleet G. W. J.; Porter R. R.; Knowles J. R. Nature 1969, 224, 511–512. 10.1038/224511a0. [DOI] [Google Scholar]
- Smith R. A. G.; Knowles J. R. J. Am. Chem. Soc. 1973, 95, 5072–5073. 10.1021/ja00796a062. [DOI] [PubMed] [Google Scholar]
- Galardy R. E.; Craig L. C.; Jamieson J. D.; Printz M. P. J. Biol. Chem. 1974, 249, 3510–3518. [PubMed] [Google Scholar]
- Fleming S. A. Tetrahedron 1995, 51, 12479–12520. 10.1016/0040-4020(95)00598-3. [DOI] [Google Scholar]
- Tsukiji S.; Miyagawa M.; Takaoka Y.; Tamura T.; Hamachi I. Nat. Chem. Biol. 2009, 5, 341–343. 10.1038/nchembio.157. [DOI] [PubMed] [Google Scholar]
- Fujishima S. H.; Yasui R.; Miki T.; Ojida A.; Hamachi I. J. Am. Chem. Soc. 2012, 134, 3961–3964. 10.1021/ja2108855. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Hamachi I. Acc. Chem. Res. 2012, 45, 1460–1469. 10.1021/ar200334r. [DOI] [PubMed] [Google Scholar]
- Herner A.; Lin Q. Topics Curr. Chem. 2016, 374, 77–107. 10.1007/s41061-015-0002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battenberg O. A.; Nodwell M. B.; Sieber S. A. J. Org. Chem. 2011, 76, 6075–6087. 10.1021/jo201281c. [DOI] [PubMed] [Google Scholar]
- Rix U.; Hantschel O.; Dürnberger G.; Remsing Rix L. L.; Planyavsky M.; Fernbach N. V.; Kaupe I.; Bennett K. L.; Valent P.; Colinge J.; Köcher T.; Superti-Furga G. Blood 2007, 110, 4055–4063. 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- Anders L.; Guenther M. G.; Qi J.; Fan Z. P.; Marineau J. J.; Rahl P. B.; Loven J.; Sigova A. A.; Smith W. B.; Lee T. I.; Bradner J. E.; Young R. A. Nat. Biotechnol. 2014, 32, 92–96. 10.1038/nbt.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka Y.; Sun Y.; Tsukiji S.; Hamachi I. Chem. Sci. 2011, 2, 511–520. 10.1039/C0SC00513D. [DOI] [Google Scholar]
- Wang Y.; Lin Q. Org. Lett. 2009, 11, 3570–3573. 10.1021/ol901300h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahra J. A.; Abu Thaher B. A.; El-Abadelah M. M.; Boese R. Org. Biomol. Chem. 2003, 1, 822–825. 10.1039/b211458p. [DOI] [PubMed] [Google Scholar]
- Zahra J. A.; Abu Thaher B. A.; El-Abadelah M. M.; Boese R. Org. Biomol. Chem. 2005, 3, 2599–2603. 10.1039/b505010c. [DOI] [PubMed] [Google Scholar]
- Zheng S.-L.; Wang Y.; Yu Z.; Lin Q.; Coppens P. J. Am. Chem. Soc. 2009, 131, 18036–18037. 10.1021/ja9094523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N. P.; Tran C.; Lee F. Y.; Chen P.; Norris D.; Sawyers C. L. Science 2004, 305, 399–401. 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. Nat. Biotechnol. 2007, 25, 1035–1044. 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Nature 2010, 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallana E.; Riguera R.; Fernandez-Megia E. Angew. Chem., Int. Ed. 2011, 50, 8794–8804. 10.1002/anie.201101019. [DOI] [PubMed] [Google Scholar]
- Speers A. E.; Cravatt B. F. Curr. Protoc Chem. Biol. 2009, 1, 29–41. 10.1002/9780470559277.ch090138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin B.; Cadet J. J. Am. Chem. Soc. 1995, 117, 12408–12415. 10.1021/ja00155a005. [DOI] [Google Scholar]
- Meier H.; Heimgartner H. Helv. Chim. Acta 1985, 68, 1283–1300. 10.1002/hlca.19850680524. [DOI] [Google Scholar]
- Li Z.; Qian L.; Li L.; Bernhammer J. C.; Huynh H. V.; Lee J. S.; Yao S. Q. Angew. Chem., Int. Ed. 2016, 55, 2002–2006. 10.1002/anie.201508104. [DOI] [PubMed] [Google Scholar]
- Los G. V.; Encell L. P.; McDougall M. G.; Hartzell D. D.; Karassina N.; Zimprich C.; Wood M. G.; Learish R.; Ohana R. F.; Urh M.; Simpson D.; Mendez J.; Zimmerman K.; Otto P.; Vidugiris G.; Zhu J.; Darzins A.; Klaubert D. H.; Bulleit R. F.; Wood K. V. ACS Chem. Biol. 2008, 3, 373–382. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Keppler A.; Gendreizig S.; Gronemeyer T.; Pick H.; Vogel H.; Johnsson K. Nat. Biotechnol. 2002, 21, 86–89. 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Shevchenko A. Anal. Biochem. 2001, 296, 279–283. 10.1006/abio.2001.5321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.