

INTRODUCTION AND BIOMEDICAL SIGNIFICANCE
The medicinal value of Amaryllidaceae plant extracts has been recognized for a long time. It dates back to at least the fourth century B.C., when physician Hippocrates of Cos used oil from the daffodil Narciclasus poeticus L. for the treatment of uterine cancer.1 From the currently known 860 – 1100 species of plants belonging to the Amaryllidaceae family, at least 30 species have been used in ancient folk medicine to treat cancer. In more recent times, more than 100 structurally diverse alkaloids, possessing a wide spectrum of activities have been isolated from the Amaryllidaceae species.2 Lycorine (Fig. 1), shown to possess antitumor and antiviral activities,3 was the first member of the family isolated in 1877.4 Later, the phenanthridones narciclasine and 7-deoxynarciclasine (lycoricidine) attracted considerable interest due to their antineoplastic properties, including activity against Ehrlich carcinoma.5,6 The latter natural products have been proposed to inhibit the growth of eukaryotic cells by disrupting protein biosynthesis.7–9 Additional members of the Amaryllidaceae anticancer constituents include trans-dihydronarciclasine and 7-deoxy-trans-dihydronarciclasine, which were first synthetically prepared by hydrogenation of the double bond in their corresponding congeners10 and then isolated from natural sources.11,12
Fig 1.

The most promising spectrum of biological activities, however, resides with the isocarbostyril (+)-pancratistatin, isolated by Pettit and coworkers in 1984 from the bulbs of Hawaiian Hymenocallis littoralis (originally Pancratium littorale) in a low yield of 0.028%.13,14 Additional sources of this natural product were found later and include Haemanthus Kalbreyeri (India),15 Hymenocallis Pedalis (Seychelles), Hymenocallis speciosa (Singapore) and Hymenocallis sonoranensis (Mexico).16 Also, frozen extracts of the Texas grasshopper Brachystola magna from 1967, which were only analyzed recently, were found to have 0.041% pancratistatin.17 The structure of pancratistatin was determined by NMR spectral analysis and confirmed by the determination of an X-ray crystal structure of pancratistatin monomethyl ether.13 The X-ray structure of the natural product itself was obtained later.17 Similarly to narciclasine/7-deoxynarciclasine pair, pancratistatin was found to have its naturally occurring 7-deoxy congener, isolated by Ghosal and coworkers in 1989 from Haemanthus Kalbreyeri.15
The interest in pancratistatin stems from its strong in vitro cancer cell growth inhibitory activities against the US National Cancer Institute (NCI) panel of cancer cell lines, showing a highly characteristic differential cytotoxicity profile with a pronounced activity towards the melanoma panel of cell lines as well as a number of in vivo experimental cancer systems.12,14 Powerful antiviral18 and antiparasitic19 activities of pancratistatin constitute a related area of promise. Importantly, it has been noted that pancratistatin and 7-deoxypancratistatin are the only known agents (other than an interferon inducer) to show a significant chemotherapeutic efficacy in a Japanese encephalitis virus-infected mouse model.18 Based on its structural similarity to narciclasine, pancratistatin has been surmised to exert its antitumor effect through the disruption of protein biosynthesis.7–9 Notwithstanding, some recent intriguing data have indicated the involvement of an antiangiogenesis/vascular targeting mechanism for in vivo neoplastic disease.20,21 Additional promising data were recently reported by Pandey and coworkers, who investigated the effects of pancratistatin on normal and cancer cells, providing evidence supporting selective toxicity of this isocarbostyril to cancer cells.22–24 In contrast, such chemotherapeutic drugs as paclitaxel and etoposide show toxicity to both cell types. The authors speculate that pancratistatin may take advantage of the differences between mitochondria in cancerous and non-cancerous cells to selectively induce apoptosis in the former cell types. These experiments bode well for the clinical development of this anticancer agent.19,20,25,26
Because of the structural complexity of the natural product, several research groups have synthesized and evaluated for anticancer activity numerous simplified pancratistatin analogues. Unfortunately, so far such efforts have not resulted in compounds with medicinal potential comparable to that of the natural product itself. All three rings, A, B and C (Fig. 1) have been targeted for modifications. Removal of the oxygen substituents on ring A leads to significant reduction of potency. 7-Deoxypancratistatin is about 10-fold less potent,27 while analogue 1 (Fig. 2) with a single alkoxy group, prepared by Hudlicky and coworkers,28 is 100-fold less cytotoxic than pancratistatin. The interesting bioisosteric replacement of the methylenedioxybenzene ring with the indole moiety as in compound 2, also prepared by Hudlicky and coworkers, leads to essentially complete loss of in vitro anticancer activity.29,30
Fig 2.

Numerous analogues have been prepared to evaluate the criticality of the trans B–C ring junction stereochemistry and the integrity of ring B itself for the anticancer activity of pancratistatin. Compound 3 lacking the C10a–C10b bond31 and 4, a lactone analogue of narciclasine synthesized by Chapleur and his coworkers,32 did not show any significant anticancer activity (Fig. 3). C10b-(S)-epimers of 7-deoxypancratistatin33 and pancratistatin34 (5 and 7) as well as the analogue possessing the C10b-(R)-hydroxyl (6)35 exhibited significantly reduced potencies or were completely inactive. From the above data it can be concluded that the R-stereochemistry of the C10a–C10b bond of pancratistatin is crucial for activity.
Fig 3.

Recently, Kornienko and coworkers36 synthesized ring B opened analogues 8a–e, 9a–e and 10a–e, which possess this essential C10a–C10b bond with the correct stereochemistry (Fig. 4). Unfortunately, none of the analogues showed any significant anticancer activity.
Fig 4.

Finally, synthetic work aimed at simplification of ring C by various investigators produced several deoxygenated analogues (Fig. 5). Compounds 11, 12 and 13 were prepared by McNulty and coworkers,37,38 while diol 14 was reported by Hudlicky and coworkers.39 11 and 12 were found inactive, 13 is on average 2–3 orders of magnitude less potent than pancratistatin in the NCI 60 cell line screen, while 14 only showed low micromolar growth-inhibitory potency against the murine P388 cell line but was completely inactive against a panel of human cancer cell lines.
Fig 5.

The lack of promising activity associated with the above-mentioned structurally simplified analogues underscores the importance of all elements of complexity present in the structure of pancratistatin. This makes the development of a practical chemical synthesis of the natural isocarbostyril an important research direction, which has been pursued by several dozen groups worldwide. Several excellent reviews discussing the synthetic work in the area of Amaryllidaceae anticancer constituents have appeared.2,3,4,40,41,42 The purpose of this review is a detailed description and comparative evaluation of the nine total syntheses of pancratistatin reported to date.
While pancratistatin is not a large molecule, it has a number of elements of complexity. Perhaps, the combination of these two factors is what makes this target a formidable challenge. The small overall structure harbors six stereogenic centers, all of which are located on ring C. The pentasubstituted aromatic ring A requires a regiocontrolled introduction of the aromatic substituents, such that the groups installed first dictate the positioning of the subsequent aromatic derivatization. Furthermore, the lactam ring B is highly strained, because the trans fusion with ring C distorts the planarity created by the four atoms in the sp2-hybridization state (C10a, C6a, C6 and C5). While with larger molecules different individual segments can be worked on independently, the high density of structural complexity in pancratistatin requires a thoroughly designed synthetic plan, in which each step must be scrutinized in the context of the entire molecule rather than its small portion.
Nine strategies have resulted in completed synthetic pathways to pancratistatin. The description of each synthesis will be split into two sections. The first one, entitled “synthetic sequence and tactical nuances,” will detail the entire pathway along with various problems encountered by investigators and their corresponding solutions. The second section, entitled “evaluation of the strategy,” will summarize and evaluate how the structural complexity issues were resolved. Of these, we will focus on (a) choice of the starting material, (b) strategy for achieving the asymmetric synthesis if only (+)-enantiomer of the natural product was prepared, (c) installation of stereocenters in the stereo-congested ring C, (d) strategy for closure of ring B, containing the strained lactam moiety, and (d) regioselective introduction of the substituents in the pentasubstituted aromatic ring A.
I. FIRST SYNTHESIS OF (±)-PANCRATISTATIN BY DANISHEFSKY’S GROUP (1989)
1. Synthetic Sequence and Tactical Nuances
Danishefsky and Lee accomplished the first total synthesis of racemic pancratistatin in 1989, starting from readily available pyrogallol (15, Scheme 1).43 The major features of the synthetic design involve the Diels-Alder reaction to construct ring C and the Overman rearrangement to introduce the nitrogen with the correct stereochemistry in lactam ring B.
Scheme 1.

Reaction of 15 with triethylorthoformate gave orthoester 16, whose carbamoylation was achieved with diethylcarbamoyl chloride (Scheme 1). Subsequent methanolysis of 17 and installation of the methylenedioxy moiety by treatment of the intermediate diol 18 with methylene bromide in the presence of potassium carbonate and copper (II) oxide afforded selectively protected trioxybenzene 19. The carbonyl group of the carbamate functionality then served as a handle for directed lithiation of the adjacent free ortho-position in 19 with sec-butyl lithium to generate an intermediate aryl lithium species, which underwent the Snieckus rearrangement44 to yield tetra-substituted aromatic compound 20. The phenolic hydroxyl was protected using TBS group and second metalation with sec-butyl lithium followed by addition of DMF gave penta-substituted aromatic aldehyde 22 in 70% yield. Thus, the selective protection of 15 and the sequential lithiation strategy allowed for the introduction of all substituents in the aromatic ring with complete regiocontrol.
To prepare the required diene 24 for the Diels-Alder cycloaddition, aldehyde 22 was treated with allylmagnesium bromide and the resulting alcohol group was converted to the mesylate ester (Scheme 2). Elimination of the mesylate to install the double bond was achieved by treatment with DBU to form the desired diene 24. The Diels-Alder reaction of diene 24 with β-nitrovinyl sulfone 25, a known acetylenic dienophile equivalent,45,46 proceeded in 96% yield to give cycloadduct 26. Radical elimination of the nitro and the benzenesulfonyl groups was accomplished by treatment of 26 with AIBN/Bu3SnH. In preparation for iodolactonization the TBS ether was removed using Bu4NF to give phenol 28. Removal of the silyl group was necessary, since halolactonization under various conditions was not successful due to the increased repulsive forces between the large OTBS group and the hypothetical intermediate diethyl imminium ion as shown in 32 (Fig. 6).
Scheme 2.

Fig 6.

Still, iodolactonization of 28 was not successful. Here it was reasoned that the amidic carbonyl in 28 was not nucleophilic enough to accomplish the ring opening of the iodonium ion. Stannylation of the phenolic hydroxyl increased the nucleophilicity of the carbonyl oxygen47 and the lactonization proceeded in 67% yield in the presence of I2 in THF to form 29. This transformation allowed the investigators to introduce the cis-C10b–C1 relationship. The phenolic OH was now protected as a benzyl ether to give 30 in 85% yield. Efforts towards elimination of HI to install the C2–C3 double bond by treating with a base resulted in the formation of the fully aromatic ring C in 34 (Fig. 7).
Fig 7.

To solve the aromatization problem, the double bond was dihydroxylated using catalytic OsO4 and NMO to give 31 as a single stereoisomer in 90% yield. This transformation led to trouble-free elimination of HI when 31 was treated with DBU to give 35 (Scheme 3).
Scheme 3.

Before introducing the C2–C3 cis functionality, the investigators prepared for the regiospecific reductive elimination of the C4a and C4 heteroatoms by converting the cis-C4a–C4 diol into trans-bromoacetate by treatment of 35 with 2-acetoxy-iso-butyryl bromide.48,49 This reaction resulted in formation of 37 in 63% yield along with 25% yield of the allylic isomer 36. Dihydroxylation of 37 with OsO4/NMO proceeded selectively from the less hindered α face due to the presence of the C1- and C4-substituents blocking the β face to give 38.
To introduce the C4a-α-amino moiety, the researchers relied on the Overman rearrangement50 that could be accomplished by the reductive removal of the trans bromoacetate, conversion of the allylic hydroxyl to imidate followed by suprafacial [3,3]-sigmatropic rearrangement to give the desired compound 41 (Scheme 4).
Scheme 4.

However, the reaction of 39 with trichloroacetonitrile led to the formation of the cyclic orthoamide as a mixture of two diastereomers 42 and 43 which did not undergo the Overman rearrangement (Scheme 5).
Scheme 5.
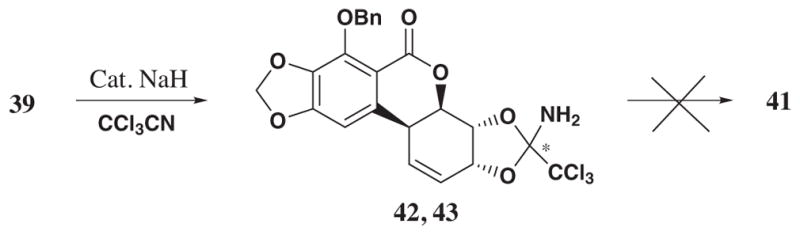
To overcome the formation of the orthoamide, the C2 hydroxyl had to be blocked. This was achieved by the reaction of 38 with bis(di-n-butyltin)oxide and further treatment of the resultant stannylene with p-methoxybenzyl bromide to give 44 in 84% yield.
The C2 hydroxyl group was now protected as benzyl ether and the PMB protection was removed by the treatment of 45 with DDQ to give 46 (Scheme 6). Upon treatment with Zn dust acetoxy bromide 46 formed the desired C2-protected allylic alcohol 47. Reaction of 47 with NaH and trichloroacetonitrile gave 48 in 74% yield and, as expected, 48 under thermal conditions underwent suprafacial [3,3]-sigmatropic reaction to give 49 in 56% yield. Dihydroxylation of 49 to install the C3–C4 hydroxyl functional groups proceeded from the β face due to the presence of the C2 and C4a α-functionality. This concluded the installation of all six stereocenters in ring C. Under basic conditions the lactone ring was opened to form the intermediate amino acid 51 that in the presence of DCC cyclized to form the trans fused lactam 52 in 82% yield. Deprotection of the benzyl ether gave racemic pancratistatin with an overall yield of 0.13% over 27 steps.
Scheme 6.

2. Evaluation of the Strategy
a) Starting Material
The synthetic effort started with inexpensive achiral pyrogallol (15, Scheme 7), which incorporates three oxygen-bearing centers found in ring A of pancratistatin.
Scheme 7.

b) Asymmetric Strategy
Racemic target natural product was synthesized.
c) Installation of Stereocenters
Ring C was formed by the Diels–Alder reaction and iodolactonization of the diene 27 allowed the introduction of the C10b-C1 cis geometry in pancratistatin. Various problems encountered at this stage were resolved by deprotection of the bulky silyl group and stannylating the phenolic hydroxyl to improve the nucleophilicity necessary to open the iodonium ion ring to form the lactone 29. The key step resulting in installation of the C4a-nitrogen functionality involved the [3,3]-sigmatropic rearrangement of imidate 48. This suprafacial transformation gave the trans C10b-C4a relationship. Finally β-orientation of the C4a and C2 substituents in 37 guided the dihydroxylation of the double bond from the less hindered α-face to introduce the C3–C4 cis hydroxyl groups.
d) Lactam Formation
The C6 carbonyl of pancratistatin was introduced early in the synthesis was engaged as a lactone with the C1 hydroxyl. Hydrolysis of the lactone after installing the C4a amino group with the right stereochemistry allowed for the formation of the trans lactam in the presence of the coupling reagent DCC.
e) Regioselective Introduction of the Substituents on the Aromatic Ring
The starting material pyrogallol has three hydroxyl groups, two of which were selectively protected as methylene-dioxy moiety and the third one was carbamoylated. Snieckus rearrangement of 19 installed the C6 carbonyl and another metalation reaction of 21 led to the introduction of the aldehyde group that was transformed into C10a–C10b bond later in the synthesis.
II. CHEMOENZYMATIC SYNTHESIS OF (+)-PANCRATISTATIN BY HUDLICKY’S GROUP (1995)
1. Synthetic Sequence and Tactical Nuances
Hudlicky’s group successfully employed a trans aziridine ring opening process with a higher order cuprate reagent to achieve the first asymmetric synthesis of (+)-pancratistatin.51,52
The synthesis commenced (Scheme 8) from bromobenzene or chlorobenzene and their oxidation to enantiomerically pure diols 49a,b, by the whole cell fermentation with Pseudomonas putida 39/D1.53 These were protected as their respective acetonides to form 50a,b. To reach the desired aziridines 55a,b, two independent paths were pursued. One involved treatment of 50a,b with NBS followed by aqueous work-up, which gave bromohydrins 51. The latter were reacted with sodium azide to afford hydroxy azides 52, possibly via the intermediacy of the β-epoxides. Subsequent mesylation and azide reduction was accompanied by aziridine formation to give 54. These were tosylated to give tosyl aziridines 55a,b. A much shorter route involved the direct treatment of 50a,b with Yamada’s iodonium ylide54 to give α-aziridines 55a,b in 54% and 20% yields respectively. Bromide 55a, which was obtained in significantly higher yield than the corresponding chloride 55b, was further dehalogenated to furnish vinylaziridine 56 in good yield.
Scheme 8.

The aromatic ring A of pancratistatin was synthesized using piperonic acid (57, Scheme 9) as a starting material. Treatment of 57 with thionyl chloride followed by diethyl or dimethyl amines gave amides 58, 59. The phenolic OH at C7 of pancratistatin was installed by o-lithiation of the amides using sec-BuLi/TMEDA and quenching with trimethylborate followed by oxidation with hydrogen peroxide. The resulting phenols 60 were protected as ethers 61, 62 and 63. Second lithiation at the other ortho position of the amide directing groups and transmetalation using CuCN gave the higher order cuprates 64a–c. The researchers investigated the nucleophilic ring opening of vinylaziridines under various conditions55,56,57 and found that lithium dimethyl-cyanocuprate gave syn-1,4-addition product, while lithium diphenylcyanocuprate resulted in formation of anti-1,2-addition product. With these encouraging results obtained with higher order cuprates, the reactions of 64a–c with aziridine 56 were performed. As expected the anti-1,2-addition products 65, 66 and 67 were obtained in 49%, 75% and 72% yields, and this accomplished the construction of the carbon skeleton of pancratistatin.
Scheme 9.

Various routes were explored to achieve the cyclization to form ring B. Transamidation using sec-BuLi at −15 °C utilized earlier by Heathcock and coworkers58 for the synthesis of pancratistatin models failed to induce cyclization in 65, possibly due to the allylic and benzylic nature of the C10b hydrogen, and therefore, its enhanced acidity. Efforts to convert the benzamide group into a more versatile group that could facilitate cyclization failed, since the benzamide was resistant to various hydrolysis and reduction conditions, possibly due to the presence of the bulky ortho substituents.
After extensive experimentation the investigators focused on TBS-protected benzamide 66 to complete the synthesis of pancratistatin. Removal of the silyl protection using TBAF (Scheme 10) and reduction of the benzamide to the corresponding aldehyde using sodium bis(2-methoxyethoxy)aluminum hydride (SMEAH) gave aldehyde 68. Protection of the phenolic hydroxyl as benzyl ether gave 69, which existed as a mixture with hemiaminal 70. Oxidation of this mixture with Jones reagent gave 71. Various attempts to remove the tosyl-protecting group under a number of reductive conditions led to disappointing results reforming hemiacetal 70. It was reasoned that the lactam ring in 71 is highly strained forcing the sp2 carbonyl to form a tetrahedral intermediate, and therefore, removal of the tosyl group at an earlier stage was imperative.
Scheme 10.

At this stage it was also observed that compounds 65 and 66 existed as α and β atropisomers. Boc protection of 66 formed a mixture of atropisomers 72α and 72β in 68% yield (Scheme 11). The α isomer in which the carbonyl oxygen participates in hydrogen bonding with the sulfonamide NH showed much reduced reactivity due to steric congestion as can be seen in Fig. 8.
Scheme 11.

Fig 8.

Treatment of the mixture of 72 with Na/anthracene led to the formation of desilylated β-atropoisomer 73 and α-isomer 74 in 82% combined yield. α-Isomer 74 was isolated and desilylated using TBAF to give 73 as a single compound.
Reduction of benzamide 73 using SMEAH/morpholine then gave aldehyde 75 (Scheme 12). The latter was protected as benzyl ether and oxidized with sodium chlorite to give acid 77. Subsequent methylation using diazomethane afforded metyl ester 78 in quantitative yield. The presence of the bulky Boc protecting group, however, disfavored cyclization of the acid or the ester under various conditions. The removal of the Boc protecting group to form the primary amine using trifluoroacetic acid led to the desired cyclization, but also resulted in migration of the olefin to the conjugated C10b-C1 position. To avoid further complications, the acetonide group was cleaved and epoxidation of diol 79 using t-BuOOH/VO(acac)2 formed β-epoxide 80 as a single compound. Finally, refluxing 80 in water in the presence of catalytic sodium benzoate resulted in a number of desired transformations. These conditions led to the cleavage of the Boc group, cyclization to form the lactam as well as transdiaxial opening of the epoxide at C2 position to give pancratistatin in 51% yield for this transformation. Overall, the target natural product was reached in 14 steps and 2 % yield starting from bromobenzene.
Scheme 12.

2. Evaluation of the Strategy
a) Starting Material
The synthesis began with the microbial oxidation of readily available halobenzenes to give compounds 49, which have two hydroxyl groups found in pancratistatin already present. In addition, the two double bonds differ in reactivity patterns making selective transformations possible.
b) Asymmetric Strategy
Toluene dioxygenase enzyme-mediated enantiospecific dihydroxylation of halobenzenes formed the basis for the introduction of asymmetry in the molecule. Easily accomplished in one step, enzymatic oxidation of aromatic groups efficiently introduced C3–C4 cis hydroxyl groups, which control the stereochemical outcome of the crucial aziridination reaction.
c) Installation of Stereocenters
C3–C4 stereocenters are already present in diol 49. Protecting the cis diol with isopropylidene moiety imparted steric blocking of the β-face of the diol forcing aziridination from the less hindered α-face. Regiocontrolled SN2 opening of the aziridine led to the inversion at C10b, allowing the construction of C10a–C10b bond in the right configuration. Allylic alcohol-directed epoxidation gave β-epoxide 80, whose transdiaxial opening by H2O resulted in the incorporation of C1–C2 hydroxyls with the required stereochemistry.
d) Lactam Formation
Though the authors envisioned the lactamization process at a much earlier stage of the synthesis, the failure to achieve the desired ring closure by trans-amidation and the facility of formation of the sp3 hybridized hemiacetal 70 made the researchers redesign their strategy. In addition, the attempted cyclization by attack of the primary amine onto the methyl ester, led to the migration of the olefin to form the more stable conjugated system. However, epoxidation of the olefin and opening of the epoxide with H2O/cat.BzONa facilitated lactamization without the accompanying olefin migration, and along with the concomitant epoxide opening provided an excellent solution to the earlier setbacks in the synthesis.
e) Regioselective Introduction of the Substituents on the Aromatic Ring
The arylcuprate required for the opening of the aziridine was synthesized from piperonic acid. Two sequential ortho-lithiation reactions of amides 58 and 59 accomplished the regioselective installation of the C7 hydroxy group of pancratistatin and gave rise to the formation of the necessary copper-carbon bond.
III. DESYMMETRIZATION ROUTE TO (+)-PANCRATISTATIN BY TROST’S GROUP (1995)
1. Synthetic Sequence and Tactical Nuances
Trost and coworkers described an asymmetric synthesis of (+)-pancratistatin59 utilizing a palladium-catalyzed desymmetrization methodology developed by this group earlier.60 The starting material of the synthesis was benzoquinone, which was converted to key conduritol derivative 89 following a known five-step procedure in 69% overall yield (Scheme 14).61
Scheme 14.

Diol 89 was treated with n-BuLi and methylchloroformate to form symmetric dicarbonate 90 (Scheme 15), which was subjected to a desymmetrization process in the presence of the palladium complex derived from the chiral ligand A (Fig. 9) and TMSN3 as a source of azide nucleophile.
Scheme 15.

Fig 9.

Azide 91 was obtained in >95% ee and 82% yield. The earlier mechanistic studies of Trost and coworkers revealed that ionization is the enantiodetermining step in the catalytic cycle of this highly selective process.60 In addition, these investigators previously proposed a mnemonic rule62 to predict the favored pathway for the enantioselective ionization, and the stereochemical outcome of the transformation 90 → 91 is fully consistent with these studies. Thus, the absolute sense of chirality for the amide ligand A can be reduced to representation B, highlighting the positioning of the large (L) and small (S) substituents on the bis-phosphine skeleton (Fig. 9). The energy difference of the two diastereomeric transition states, one leading to the departure of the pro-S carbonate (path “a” in Figure 9) and the other that of the pro-R carbonate (path “b”), is controlled by the steric interaction of the substituents L and S with the rest of the cyclohexene ring, as the palladium complex turns counter-clockwise (a) or clockwise (b) respectively. The steric clash of the substituent L with the acetonide moiety virtually prevents the type “b” rotation and, consequently, the ionization of the pro-R carbonate.
With the key intermediate 91 in hand, regio- and diastereocontrolled SN2′ introduction of the aryl moiety was achieved by reaction of the requisite Grignard reagent with 91 in the presence of CuCN. Dihydroxylation of the resulting olefin 92 from the sterically more accessible α-face gave diol 93, whose protection as bis-triethylsilyl ether proceeded quantitatively. The next crucial step in the synthesis was an intramolecular cyclization of the isocyanate, derived from the azido group in 94, to form the lactam ring of pancratistatin. However, all efforts to induce such cyclization failed, leading to mixtures of products that appeared to have lost the acetonide protection. The investigators suspected that the ether oxygen of acetonide is more nucleophilic compared to the aromatic ring. Therefore, an alternate route involving the conversion of the π-nucleophilic aromatic ring to a more powerful σ-nucleophilic species was undertaken. Thus, bromination of 94 ortho63 to the methoxy group using NBS gave 95 in 75% yield. Isocyanate 96 was prepared from 95 using trimethyphosphine-mediated azide reduction and treatment with phosgene, and without purification 96 was reacted with t-BuLi. The lithium-bromine exchange was faster than the nucleophilic addition of t-BuLi to isocyanate and the lithiated species immediately underwent intramolecular cyclization to form the desired lactam 97. To achieve the right stereochemistry at the C1 position of pancratistatin, the silyl groups were removed and the resultant diol 98 was converted to cyclic sulfate 99. It was hoped that the SN2 opening with an oxygen-based nucleophile would occur at C1 position to avoid the steric interaction with the isopropylidene group. Indeed, nucleophilic ring opening of 99 using cesium benzoate occurred as predicted to give a benzoate, whose subsequent treatment with dilute sulfuric acid resulted in simultaneous cleavage of the alkyl sulfate and acetonide moieties to give 100 in 85% overall yield. Finally, methanolysis of the benzoate ester and cleavage of the methyl ether with lithium iodide yielded pancratistatin in 85% yield over the last two steps. The synthesis proceeded in 19 steps and 8% overall yield from benzoquinone.
2. Evaluation of the Strategy
a) Starting Material
Although the synthesis began with a complex conduritol derivative 89, containing four stereocenters, the meso-symmetry of the latter allowed for its concise five-step preparation from benzophenone using a protocol reported previously by Rutledge and coworkers.61
b) Asymmetric Strategy
The critical stereochemical design element of the pathway involved a palladium-catalyzed desymmetrization of conduritol 89. The advantage of this process over a conventional kinetic resolution approach is undoubtedly the possibility of conversion of the entire meso starting material to just one enantiomer of azide 91. The investigators reported the enantiomeric excess value of > 95% for this transformation. Together with the high yield of 83% this approach rendered the asymmetric strategy an extremely efficient approach for the synthesis of pancratistatin in single enantiomeric form.
c) Installation of Stereocenters
In azide 91 the C3, C4 and C4a stereocenters are already in place. The regio- and diastereocontrolled allylic substitution of the C2-carbonate with an aryl-copper reagent resulted in formation of the C10b stereocenter with the correct configuration in 92. The β-orientation of the C3 and C10b substituents allowed for facile installation of the two oxygen-bearing stereocenters at C1 and C2 from the α-face of the double bond. Finally, steric shielding of the C2 carbon by the acetonide moiety resulted in efficient stereochemistry inversion at C1 exclusively using ring opening of cyclic sulfate in 99 with benzoate ion.
d) Lactam Formation
The direct attack by the π-nucleophilc aromatic ring onto the isocyanate group failed, possibly due to the presence of a nearby oxygen at C4 and the subsequent deprotection of the acetonide moiety. However, conversion of the π-nucleophilic aromatic ring to a σ-nucleophilic one provided a nice solution to this problem, although it led to the necessity of prior bromination of the aromatic ring ortho to the methoxy group.
e) Regioselective Introduction of the Substituents on the Aromatic Ring
The aromatic bromide necessary for the preparation of the cuprate had been described in the literature before,64,65 and therefore, azide 92 already had four out of five substituents correctly positioned on the aromatic ring. The problem of regioselectivity of the introduction the fifth substituent was solved by highly selective bromination at position C6a. A strong para-directing aptitude of the methylene-dioxy group, compared to the alkoxy one, had been previously observed by other investigators66–69 and this phenomenon is most likely due to a better overlap of the lone pair of electrons on oxygen atoms of the methylenedioxy group with the p-orbitals of the benzene ring.
IV. FORMAL SYNTHESIS OF (+)-PANCRATISTATIN BY HASELTINE’S GROUP (1997)
1. Synthetic Sequence and Tactical Nuances
In 1997 Haseltine and coworkers reported a total synthesis of (+)-pancratistatin,70 which led to an intermediate in the Danishefsky’s pathway. Thus, the synthetic sequence constitutes a formal synthesis of (+)-pancratistatin. Ring C of pancratistatin was constructed using the retro-Diels-Alder reaction of acetonide 105, following the strategy developed by Knapp and coworkers (Scheme 17).71 In contrast to the thermally induced retro Diels-Ader reaction (Scheme 14), this original procedure utilized the “Evans Accelerating Effect” of the alkoxide brought about by treatment of alcohol 105 with potassium hydride. Desymmetrization using P30 lipase with isopropenyl acetate, silylation of the free hydroxyl and deacetylation afforded 107 in 93% yield over three steps.
Scheme 17.

The aromatic portion of pancratistatin was prepared from piperonol 108 (Scheme 18). The original plan involved the introduction of the C7 phenolic hydroxyl at an early stage by o-lithiation of the ethoxyethyl-protected piperonol and treatment with BH3•THF to generate an intermediate arylborohydride, which was then oxidized with H2O2. The hydroxyl group in 110 was protected as benzyl ether and the ethoxyethyl protection was removed to give 112.
Scheme 18.

Trichloroacetimidate methodology was subsequently used to join the piperonol unit 112 with the conduritol derivative 107 to give 114, which was desilylated with TBAF. Intramolecular electrophilic aromatic substitution was envisioned for the construction of the C10b-C10a bond. Such process was brought about by conversion of the allylic alcohol to the corresponding triflate ester using triflic anhydride and 2,6-di-tert-butylpyridine. The desired cyclized product 116 was obtained only in low 8% yield, together with 50% of the rearrangement product 117. The formation of 117 was explained by the SN2′ attack on the olefin at position C6a of the aromatic ring (ipso to the tether) with the subsequent 1,2-shift of the alkoxymethyl group and deprotonation to reestablish the aromatic system. Furthermore, resonance activation of the C6a position by the presence of the benzyloxy group at C7 was thought to be responsible for this undesired process.
The investigators reasoned that to avoid the resonance activation of the C6a position, the C7 oxygen functionality would have to be installed later in the synthesis. Because a halogen at C7 could be converted into a hydroxyl at a later stage, halogenation of metalated 121 and 122 was carried out to give 123 and 124. Upon treatment with HBr 123, 124 and a non-halogenated precursor gave piperonyl bromides 125–127 (Scheme 19). Williamson ether formation of 125–127 with conduritol 107 proceeded in much better yields compared to the earlier imidate strategy to furnish coupled products 128–130. Cyclization induced by treatment with Tf2O gave the desired pentacyclic compounds along with the rearrangement products 132 and 134. Importantly, compound 130, lacking any substituent at the C7 aromatic position, cleanly gave the desired cyclization product 135 in 73% yield without any accompanying rearrangement. To introduce the C7 hydroxyl in 135 the researchers followed the same strategy that led to 110 from 109. Previous work by Xu and co-workers72 showed that isochromane structures such as 135 could be regioselectively coupled with alcohols in the presence of DDQ. This process is facilitated by the presence of alkoxy substituents in the para position. Thus, coupling of methoxyethanol with 135 in the presence of DDQ gave rise to diastereomerically pure acetal 136. Lithiation guided by the alkoxy appendage was selective for the C7 position and the intermediate aryl lithium species was oxygenated in the usual manner. The hydroxyl group was then protected as its benzyl ether and the alkoxy linkage was cleaved to give 138 in 71% yield.
Scheme 19.

Since 138 is very similar to Danishefsky’s compound 4743 now the focus was to achieve the conversion of 138 to 47 (Scheme 20). Oxidation of lactol 138 to lactone 139 was followed by acetonide hydrolysis. Protection of the allylic hydroxyl as a methoxyethoxymethyl ether allowed for the benzyl protection of the second hydroxyl. Finally, removal of the MEM group afforded 47 in overall 25% yield from 138. Formal synthesis of pancratistatin was achieved.
Scheme 20.

2. Evaluation of the Strategy
a) Starting material
The synthesis utilized acetonide 105 that was made in four steps from benzoquinone using the procedure developed by Knapp and coworkers.71 The retro Diels-Alder of 105 afforded protected conduritol 89 in 65–75% yield, which is the same starting material utilized by Trost and coworkers in their synthesis of (+)-pancratistatin.
b) Asymmetric Strategy
Enzymatic acetylation73 using P30 lipase and isopropenyl acetate led to desymmetrization of meso diol 89 to form enantiopure acetate 106 in high 94% yield.
c) Installation of Stereocenters
An intramolecular electrophilc aromatic SN2′ substitution process was used to introduce the aromatic group. Unlike Trost’s synthesis,62 in which the acetonide cis diol was equivalent to the C3–C4 hydroxyls in pancratistatin, in the Haseltine’s synthesis the conduritol unit was modified to obtain Danishefsky’s late stage intermediate 47 in which the acetonide oxygens correspond to the C2–C3 groups in pancratistatin. Syn-specific Overman rearrangement of the acetimidate, formed with the unprotected C3 hydroxyl group in 47, resulted in the introduction of the C4a nitrogen.
d) Lactam Formation
This would be similar to the Danishefsky’s synthesis, namely hydrolysis of the lactone to give the intermediate amino acid followed by cyclization in the presence of DCC to form the trans-lactam.
e) Regioselective Introduction of the Substituents on the Aromatic Ring
Aromatic ring made from piperonol already has the methylenedioxy moiety and the C6a bond. The investigators planned to introduce the C7 hydroxyl before the intramolecular coupling of the rings A and C portions. However, the failure to achieve the desired cyclization to form the C10a–C10b bond forced the C7 installation at a later stage in the synthesis. The direct o-lithiation of 135 was not successful, which prompted the researchers to introduce the ethoxymethoxy tether, which resolved the problem efficiently.
V. SYNTHESIS OF (+)-PANCRATISTATIN BY MAGNUS’ GROUP BASED ON β-AZIDONATION (1998)
1. Synthetic Sequence and Tactical Nuances
The synthesis of pancratistatin by Magnus and Sebhat74,75 is based on utilization of the β-azidonation reaction using hypervalent iodine and trimethylsilyl azide developed in this group earlier.76 The synthesis began with the bromination of o-vanillin 143 with Br2/AcOH to give 144, which was oxidized with H2O2/NaOH to form diol 145 (Scheme 22). Methylenation of diol 145 using BrCH2Cl in the presence of potassium carbonate yielded aryl bromide 146 in an overall yield of 65% from 144.
Scheme 22.

Lithiation of aryl bromide 146 followed by the addition to ketone 147 gave tertiary alcohol 148 in 85% yield. The latter was dehydrated using POCl3 and DBU in pyridine and the resulting double bond was hydrogenated. Removal of the acetal-protecting group by acid hydrolysis generated ketone 151 in good yield. Asymmetric lithium enolate formation was achieved by the treatment of 151 with lithium (+)-bis(α-methylbenzyl)amide in the presence of LiCl. The enolate was trapped with TIPSOTf to give silyl enol ether 152 in ≥ 85% enantiomeric excess. Earlier mechanistric studies by this group showed that silyl enol ethers in the presence of (PhIO)n/TMSN3 undergo hydride abstraction at the more stabilized β carbon to form positively charged intermediate 162, rather than the removal of the hydride to afford the non-stabilized intermediate 163 (Fig. 10).76
Fig 10.

The β-azidation process indeed proceeded selectively to produce cis and trans diastereomers 153 in a 1:3.5 ratio (Scheme 22). The mixture of inseparable diastereomers was reduced using LiAlH4 and the resulting amino group was protected as a carbamate using methyl chloroformate in pyridine to afford a separable mixture of epimers, from which pure trans isomer 154 was isolated in 56% yield (Scheme 23). Unfortunately, trialkylsilyl enol ether 154 in the presence of mCPBA preferentially underwent axial epoxidation and after a series of intermediate reactions gave 155, which had the wrong stereochemistry at C4. This was confirmed by reducing the keto group in 156 using NaBH4 to give two isomers α:β in 72 and 28 % yields. The major diastereomer upon the Bischler-Napieralski cyclization formed three products 158, 159 and 160. X-ray crystal structure of 159 unambiguously established the incorrect stereochemistry at both C3 and C4 positions.
Scheme 23.

To obtain the correct configuration at C4, the axial –OCOAr group in 156 was epimerized to the more stable equatorial position by treatment of 156 with t-BuOK/HMPA to give 164 in 91% yield (Scheme 24).
Scheme 24.

Further functionalization of the C ring of pancratistatin was continued by formation of TMS enol ether 165, which was converted to α:β-unsaturated ketone 167 by treatment with PhSeOCOCF3 and oxidation of the resultant selenide 166 with H2O2 in pyridine to afford 167 in 85% yield from 164 (Scheme 24). Epoxidation of 167 under mild alkaline conditions and L-selectride reduction of the carbonyl formed 169 as a single diastereomer. Acetylation of 169 and regioselective opening of the epoxide ring by the C1 axial attack of sodium benzoate gave an intermediate alcohol, which was protected as acetate to give 171. Modified Bishler-Napieralski conditions using Tf2O/DMAP77 induced lactam formation to give 173 along with its regioisomer 172 (7:1) in 60 % total yield as an inseparable mixture. Removal of the phenolic methyl ether with BBr3 allowed for the separation of the regioisomers and gave exclusively 174, since 172 did not react. Lastly, deprotection of the acetate esters with NaOMe in MeOH afforded (+)-pancratistatin in an overall yield of 1.2% over the 22 step synthesis.
2. Evaluation of the Strategy
a) Starting Material
The synthesis started with the known arylbromide 146, possessing three oxygen substituents found in the target molecule. 146 was synthesized in three steps from inexpensive o-vanillin.
b) Asymmetric Strategy
Enantioselective enolate formation with lithium (+)-bis(α-methylbenzyl) amide using ketone 151 and subsequent trapping with TIPSCl gave silyl enol ether 152 in more than 85% enantiomeric excess. The β-azidonation process affording azides 153 secured the required absolute stereochemistry.
c) Installation of Stereocenters
After obtaining the required C10b stereochemistry using asymmetric deprotonation with a chiral lithium base, the researchers performed epoxidation of silyl enol ether 153, which proceeded in the axial manner to give the unwanted configuration of the C4 stereocenter. Equilibration with a strong base brought about epimerization at C4, which can be explained on the basis of the preference for the bulky ester moiety to occupy the equatorial position as opposed to the axial one in 156. Epoxidation of C1, C2 double bond from the α-face to give 168, followed by selective reduction of the C3 keto group from the α-face and transdiaxial SN2 ring opening of the epoxide with benzoate ion at C1 led to the installation of all stereocenters in ring C of pancratistatin.
d) Lactam Formation
Regioselective Bischler-Napieralski process resulted in ring B closure by affording a 7:1 inseparable mixture of C6a and C10 cyclization products. However, only the desired C6a compound underwent the deprotection of the phenolic methyl ether with BBr3, in a process involving a possible assistance of the neighboring amidic carbonyl.
e) Regioselective Introduction of the Substituents on the Aromatic Ring
Arylbromide 146, synthesized using a literature procedure possessed three oxygens in place and the bromine substituent at the pancratistatin position C10a leading to facile creation of the C10a-C10b bond by lithium-halogen exchange and the addition of the resulting aryllithium to ketone 151.
VI. ARYL ENAMIDE PHOTOCYCLIZATION APPROACH TO (+)-PANCRATISTATIN BY RIGBY’S GROUP (2000)
1. Synthetic Sequence and Tactical Nuances
In 2000, Rigby and coworkers reported a new synthetic route using a hydrogen bond controlled aryl enamide photocyclization approach78 to synthesize both (+)-pancratistain and its natural congener (+)-narciclasine from a common intermediate.79
A key intermediate isocyanate 182 was synthesized using a previously published procedure80 from acid 181 starting from commercially available ester 175. Upon treatment with NBS and Bu3SnH, 175 formed diene 176 in 75% yield (Scheme 26). Oxidation to generate diepoxide 177 followed by treatment with sodium methoxide resulted in racemic syn-epoxy alcohol 178 in 42% yield. Enzymatic resolution of the esterified alcohol 178 using cholesterol esterase gave enantiomerically pure syn-epoxy alcohol 179 with > 93% enantiomeric excess and 40% yield. Silyl protection of the free hydroxyl and saponification gave syn-epoxy acid 181. Finally, the Curtius rearrangement of an acyl azide derived from carboxylic acid 181 gave isocyanate 182.
Scheme 26.

The aromatic portion of pancratistatin was prepared in three steps from 2,3-dihydroxybenzaldehyde (183, Scheme 27). Protection of the diol using CH2Br2 and potassium carbonate followed by oxidation of the aldehyde group under Bayer-Villiger conditions led to phenol 185 in 63% yield. Bromination in the presence of silver trifluoroacetate was selective ortho to the hydroxyl group, which was then protected as ethoxyethyl ether for subsequent lithiation.
Scheme 27.

Lithiated arylbromide 187 was then reacted with isocyanate 182 to give enamide 188 in 52% yield (Scheme 28). Protection of the NH of the amide group as PMB ether and removal of the ethoxyethyl protection formed 189, the desired precursor for the key photocyclization reaction. Removal of the ethoxyethyl group was necessary to allow for H-bonding between the amide carbonyl and the phenolic hydroxyl. The investigators reasoned that this would restrict rotation about the aryl-amide bond and increase the desired rotamer population of the secondary amide. In the event, irradiation of 189 at 254 nm in benzene brought about the desired cyclization giving the trans fused B-C ring junction in a modest yield of 30%. While the bulky TBS ether at C1 provided the necessary stereocontrol at the C10b position, the trans B-C ring fusion in the product 190 is most likely the result of stereospecific requirements of the two concerted processes involved (Scheme 29). 189 should undergo conrotatory 6π electrocyclic ring closure due to photoexcitation to give intermediate 198. This is followed by the suprafacial [1,5]-hydrogen shift, which restores the aromatic system and results in the observed trans fusion.
Scheme 28.

Scheme 29.

With A and B rings in place, the focus was turned to functionalizing the C ring. Inversion of the alcohol stereochemistry at C1 required removal of the silyl group and protection of the phenolic hydroxyl to give 192 in 83% yield over two steps (Scheme 28). Alcohol 192 failed to undergo inversion under the Mitsunobu conditions, and therefore, the investigators opted for oxidation of the hydroxyl followed by the ketone reduction from the α-face to give the desired stereochemistry at C1 in 193. The procedure involving oxidation using the mild Dess-Martin reagent with the immediate in situ reduction with NaBH4 at −20 °C was critical for the success of this transformation to avoid the epimerization at C10b, which would form the less strained cis fused ring.80 The C1 hydroxyl was protected as benzyl ether and allylic alcohol 195 was made in 84% yield by opening the epoxide with in situ generated phenyl selenide followed by elimination of the selenoxide. Stereoselective cis-dihydroxylation of the olefin to install the C3-C4 diol and deprotection of benzyl and N-p-methoxybenzyl groups by hydrogenolysis gave 197 in 78% yield over two steps. Similar to the synthesis by Trost’s group, removal of the C7 methyl group was accomplished by refluxing 197 with LiCl in DMF to give pancratistatin in 78% yield. Overall Rigby and coworkers completed the synthesis of pancratistatin in 23 steps with an overall yield of 0.35%.
2. Evaluation of the Strategy
a) Starting Material
The synthetic design involved the use of enantiomerically pure vinyl isocyanate 182, which was a key intermediate in Berchtold’s synthesis of (−)-chorismic acid.81–83 182 was prepared from rather inexpensive commercially available ester 175.
b) Asymmetric Strategy
Enzymatic resolution of racemic epoxy ester 178 with cholesterol esterase gave enantiomerically pure material. An important drawback of the strategy is the fact that kinetic resolution was performed on an intermediate obtained in five steps, thus resulting in significant loss of the material.84
c) Installation of Stereocenters
The key vinyl isocyanate already has three stereocenters in place. Although the stereochemistry at C1 was opposite at this stage in the synthesis, it was critical for the installation of C10b and C4a chiral centers through the aryl enamide cyclization. Inversion of C1 stereochemistry was achieved by oxidation and immediate in situ reduction of the carbonyl to avoid epimerization of the trans lactam. Opening of the epoxide with phenyl selenide and elimination of the intermediate selenoxide formed allylic alcohol 195 with the desired C2 geometry. Final functionalization of the C ring was achieved by simple dihydroxylation using OsO4 to install the C3-C4 hydroxyl groups at the β-face of 195. Nitrogen functionality at C4a was introduced by Curtius rearrangement of the acid 181 to form isocyanate 182.
d) Lactam Formation
The C1 silyloxy group-guided conrotatory electrocyclic ring closure of the enamide, and this accomplished the lactam formation. Steric shielding of the bottom face in enamide 189 by the bulky OTBS favored cyclization to give the correct trans diastereomer.
e) Regioselective Introduction of the Substituents on the Aromatic Ring
Diol of 2,3-dihydroxybenzaldehyde 183 was protected using dibromomethane and potassium carbonate to form the C9-C10 methylenedioxy ring of pancratistatin. The aldehyde functionality was converted to phenolic hydroxyl, which controlled the regioselective bromination of 185 ortho to the OH group and para to one of the oxygens of the methylenedioxy moiety. Halogen metal transfer with n-BuLi allowed coupling of 187 with 182 to form the C6-C6a bond. Lastly, photocyclization formed the crucial C10a-C10b bond.
VII. RELAY SYNTHESIS OF (+)-PANCRATISTATIN FROM ITS NATURAL CONGENER (+)-NARCICLASINE BY PETTIT’S GROUP (2001)
1. Synthetic Sequence and Tactical Nuances
Pettit and coworkers developed a new route to pancratistatin85 using its more naturally abundant congener narciclasine as the starting material. The synthesis commenced with the protection of the C3,C4-vicinal diol as an acetonide in 97% yield (Scheme 31). The C2 and C7 hydroxyl groups were acetylated with acetic anhydride in pyridine to give 200 along with minor quantities of monoacetylated compound 201. Epoxidation of 200 using mCPBA formed the desired α-diastereomer 202 in 55% yield. Hydrogenolysis of the epoxide in the presence of 10% Pd/C followed by reaction with methanolic potassium carbonate formed four products 203-206, which were separated by column chromatography and characterized.
Scheme 31.

Diol 204 was the only compound out of the mixture with the correct stereochemistry of the aromatic group at C10b. It was obtained with an overall yield of 15% starting from narciclasine. However, the configuration at the C1 position required inversion. To this end 204 was treated with thionyl chloride to form cyclic sulfite 207, which was further oxidized to cyclic sulfate 208 in 47% yield using catalytic RuCl3·3H2O and 3.5 equivalents of NaIO4. The nucleophilic attack of PhCO2Cs on the cyclic sulfate proceeded at the C1 carbon. This was followed by acid hydrolysis of the alkyl sulfate, which concomitantly cleaved the isopropylidene protecting group to give benzoate 209 in 74% yield. Removal of the benzoate ester group using K2CO3 in MeOH gave (+)-pancratistatin with an overall yield of 3.6 % in 10 steps starting from narciclasine.
2. Evaluation of the Strategy
a) Starting Material
Narciclasine is a close structural analogue of pancratistatin. The only difference is the presence in narciclasine of the C1-C10b double bond, and therefore, reduced structural complexity due to the absence of C1 and C10b stereocenters. Although narciclasine can be obtained more easily than pancratistatin using isolation from plant sources, its availability is still rather limited. One could envision the ideal relay synthesis of pancratistatin from narciclasine in one step by a regio- and stereoselective hydration of the C1-C10b double bond.
b) Asymmetric Strategy
No strategy was required.
c) Installation of Stereocenters
Narciclasine possesses four stereocenters, which are also found in pancratistatin. To install the remaining two stereocenters at C1 and C10b, Pettit and coworkers performed epoxidation that occurred from the α-face of the double bond giving 202 due to the steric shielding of the top face by the C3,C4-acetonide ring. Hydrogenolysis was not selective resulting in a mixture of compounds. Only one of the four products had the right configuration at C10b. Nucleophilic ring opening of the cyclic sulfate by cesium benzoate in 208 gave the desired stereochemistry at C1.
d) Lactam Formation
None was required.
e) Regioselective Introduction of the Substituents on the AromaticRing
None was required.
VIII. [3,3]-SIGMATROPIC REARRANGEMENT APPROACH TO (+)-PANCRATISTATIN BY KIM’S GROUP (2002)
1. Synthetic Sequence and Tactical Nuances
Kim and coworkers accomplished a synthesis of (±)-pancratistatin86,87 utilizing the Claisen rearrangement as a key step.
The synthesis began with bromide 214 prepared from methyl gallate using previously published methods (Scheme 33).88 Initially bromide 214 was converted to a phosphonium ylide and reacted with acrolein dimer 216 resulting in the formation of a minor product trans-olefin 217 along with the major cis-olefin 223 in 1:4.6 ratio in the presence of KOH and catalytic 18-crown-6 in 85% yield. Based on earlier studies by Büchi,89 the investigators proposed that the Claisen rearrangement of the Z-olefin 223 would proceed through a boat-like transition state to afford trans-aldehyde 224 with the correct C4a-C10b trans geometry (Fig. 11). However, the Z-olefin was reluctant to undergo rearrangement even at 250 °C while the E-olefin under similar conditions yielded the cis-disubstituted cyclohexane 218. The high-energy transition state involving unfavorable steric interaction between the aromatic group and the hydrogen atom on the dihydropyran ring was thought to be responsible for the lack of reactivity of the Z-olefin.
Scheme 33.

Fig 11.

Aldehyde 218 was therefore chosen for further functionalization of ring C. The reaction of Horner-Wadsworth-Emmons ylide 215 with aldehyde 216 gave E-olefin 217 exclusively (Scheme 33). To functionalize the C ring aldehyde 218 was oxidized to acid 219 in 90% yield using NaClO2. Iodolactionization of 219 using KI3 gave intermediate iodo compound 220, which after treatment with DBU formed unsaturated lactone 221. Reflux of 221 in methanol not only led to lactone ring opening to install C1-C10b cis relationship, but also resulted in epimerization at C4a to give ester 222 with three of the six stereocenters in pancratistatin assembled.
In an attempt to synthesize enantiopure (+)-pancratistatin, aryl bromide 146 was coupled with a chiral building block (R)-22590 and the resulting alcohol was esterified with 5-hexenoic acid 227 to yield (+)-228. The latter underwent [3,3]-sigmatropic rearrangement when treated with LDA and TBSCl forming a mixture of 230 and 231 in 77% yield (Scheme 34). Ring closing metathesis using Grubbs’ ruthenium catalyst afforded a mixture of cyclohexenes 219 and 232. Iodolactonization of the mixture gave (+)-221 along with unreacted 232 that could be separated at this stage. However, the investigators encountered problems with scaling up the synthesis of (+)-221, and therefore, the synthesis was completed using racemic ester 222.
Scheme 34.

Hydrolysis of the ester 222 using 1N LiOH furnished acid 233 in 99% yield (Scheme 35). Reaction of diphenyphosphoryl azide with 233 formed the intermediate isocyanate by way of the Curtius rearrangement, which gave carbamate 234 after treatment with NaOMe in 82% yield. Epoxidation of 235 under various conditions was not successful. The investigators opted for dihydroxylation, which proceeded from the less hindered α-face to afford 237 (Scheme 36).
Scheme 35.

Scheme 36.

Converting diol 237 to cyclic sulfate 238 and reacting the latter with DBU followed by acidic workup led to the installation of the C3-C4 double bond (Scheme 36). As was the case in the previous syntheses of pancratistatin, dihydroxylation occurred from β-face to afford 240 which has fully functionalized C and A rings. Similar to Magnus’s synthesis, acetyl protection of the hydroxyl groups followed by Bischler-Napieralski cyclization using Tf2O/DMAP accomplished the lactam formation and two regioisomers 243 and 242 were formed in 7:1 ratio. Removal of the phenolic methyl group allowed for facile separation of the two regioisomers. Final deprotection with sodium methoxide gave pancratistatin in 3.6% overall yield over 16 steps.
2. Evaluation of the Strategy
Starting Material
The synthesis began with the Horner-Wadsworth-Emmons process of phosphonate 215 made in five steps from methyl gallate 210 and acrolein dimer 216. Phosphonate 215 has all three oxygens installed in proper positions.
a) Asymmetric Strategy
The investigators initially started with the synthesis of racemic pancratistatin. After having worked out the Claisen rearrangement and iodolactonization processes they attempted an asymmetric synthesis using aryl bromide 146 and (R)-225. Enantiopure (R)-225 had been previously prepared by the same investigators utilizing enzymatic resolution of racemic γ-hydroxy vinylstannane 246 in > 99% ee (Scheme 38).90 However, the problems with purification of the mixture of 230 and 231 and scale-up of the synthesis forced the investigators to pursue the synthesis of (±)-pancratistatin.
Scheme 38.

b) Installation of Stereocenters
[3,3]-sigmatropic rearrangement of the E-olefin 217 installed the aromatic ring at C10b and the aldehyde group at C4a cis to each other. The C1 hydroxyl was efficiently introduced by iodolactonization of the acid 219 to form bicyclic lactone 221, which after methanolysis formed 222 with C1, C10b and C4a stereocenters installed in the desired configuration. After the disappointment with epoxidation of 235, the investigators found success by dihydroxylation and cyclic sulfate elimination reactions to introduce the C2 hydroxyl. Simple dihydroxylation of the 239 completed the functionalization of ring C of pancratistatin.
c) Lactam Formation
Curtius rearrangement of the azide formed by the reaction of acid 233 with DPPA gave the intermediate isocyanate, which upon treatment with sodium methoxide afforded carbamate 235. Lactamization was accomplished by the Bischler-Napieralski process at a much later stage in the synthesis similar to the precedent of Magnus. Lactam 243 was formed along with its regioisomer 242 in 7:1 selectivity.
d) Regioselective Introduction of the Substituents on the Aromatic Ring
Methyl gallate already has three oxygen groups as hydroxyl functionality. Two of them were protected as a methylenedioxy ring and the remaining alcohol was protected as methyl ether.
IX. SHORTEST SYNTHESIS OF (+)-PANCRATISTATIN BY LI’S GROUP (2006)
1. Synthetic Sequence and Tactical Nuances
The starting material for Li’s synthesis91 of pancratistatin was pinitol 249, which is expensive but has the right stereochemistry at five out of the six carbons present in the cyclitol portion of pancratistatin (Scheme 39). Compound 251 was prepared in two steps from pinitol by selective protection of the trans and cis hydroxy functionality using the bulky TIPDS and acetonide protecting groups. It is noteworthy that TIPDS is a selective protecting group for trans diols.94 The configuration of the only remaining hydroxyl was inverted using the Mitsunobu reaction with methyl sulfonate as nucleophile, followed by the SN2 substitution of the sulfonate with azide ion to install the C4a nitrogen center in pancratistatin, utilizing the procedure by Aher and Pore.93 The silyl group was then removed and the diol functionality in 253 was converted to the cyclic sulfate to give 254. The Staudinger reduction process for converting the azide to free amine generated the crucial intermediate 255 that would undergo a coupling reaction in a later part of the synthesis.
Scheme 39.

Ring A part of pancratistatin was introduced using the intermediate 256, which was synthesized according to the previously known method (Scheme 40).94 Removal of the PMB protection and phosgene mediated coupling with amine 255, yielded amide 259, in which the phenolic hydroxyl was protected as MOM ether to give 260 (Scheme 41). The crucial reaction of this synthesis involved the intramolecular ring opening of the cyclic sulfate at the C10b carbon. Nucleophilic ring opening using aryllithium and arylmagnesium species led to various side reactions due to the basic nature of these reagents. However, the low basicity and high nucleophilicity of the arylcerium reagents compared to the aryllithium or arylmagnesium ones makes it a softer nucleophile and ideal for intramolecular ring opening reactions.95 There had been no previous precedence of organocerium reagents being used for such reactions. Using organocerium methodology, compound 261 was obtained in 72% yield from 260. Removing all the protecting groups using BBr3 and methanol completed the synthesis of pancratistatin. This sequence represents the shortest synthesis of (+)-pancratistatin so far involving only 13 steps and proceeding in 9% overall yield from (+)-pinitol.
Scheme 40.

Scheme 41.

2. Evaluation of the Strategy
a) Starting Material
One of the synthetic challenges presented by pancratistatin is the presence of six stereocenters on ring C. Li’s synthesis of pancratistatin utilized (+)-pinitol as the starting material. Although relatively expensive, it has four stereocenters already present in pancratistatin.
b) Asymmetric Strategy
The investigators approached the synthesis starting with optically pure (+)-pinitol. Unlike the previous syntheses of pancratistatin (except Pettit’s synthesis), where the researchers started with achiral starting materials and introduced asymmetry by enzymatic or chemical desymmetrization, Li and his coworkers approached the synthesis of (+)-pancratistatin utilizing a chiral pool starting material (+)-pinitol.
c) Installation of Stereocenters
(+)-Pinitol has the correct stereochemistry at C1, C2, C3, C4 carbons and opposite geometry at C4a and C10b positions. Mitsunobu inversion of C4a followed by azide substitution successfully installed the C4a nitrogen present in pancratistatin. To attain inversion of C10b, the investigators functionalized the trans C1 and C4a diol as a cyclic sulfate. Nucleophilic ring opening using arylcerium allowed for the coupling of the aromatic and the cyclitol portions of pancratistatin with the inversion of configuration at C10b.
d) Lactam Formation
Phosgene mediated coupling of the amine 255 and the presumed intermediate acid chloride 258 afforded the crucial amide 259. It was envisioned that generation of the carbanion in 260 by halogen metal exchange would cause nucleophilic ring opening of the cyclic sulfate closing the B ring of pancratistatin. The investigators could not accomplish this step with traditional reagents such as ArLi and ArMg, which are strong bases and led to a complex mixture of products. The authors developed a new methodology using organocerium, a softer nucleophile, for the desired ring opening to obtain 261 in 72% yield.
e) Regioselective Introduction of the Substituents on the Aromatic Ring
Aromatic ring A was synthesized from 2,3-dihydroxybenzaldehyde following the procedure developed by Coleman and Gurrala (Scheme 42).94 Formation of the presumed acid chloride 258 occurred ortho to the phenolic hydroxyl and para to an oxygen of the methylenedioxy moiety.
Scheme 42.

X. CONCLUSION
The above discussion of the successful strategies, resulting in completed syntheses of the promising medicinal agent (+)-pancratistatin, illustrates the challenge of developing a practical scalable pathway suitable for preparation of multigram quantities of this natural product. Pancratistatin has been in preclinical development for over 20 years with two major hurdles towards its advancement to clinical trials, namely its poor aqueous solubility (53 μg/mL) and lack of availability from natural sources. The first problem has been addressed in a variety of ways including the addition of DMSO or complex formation with nicotinamide to enhance the aqueous solubility.96 In addition, the use of a 40% aqueous solution of hydroxypropyl-β-cyclodextrin (HPCD) solubilizes pancratistatin up to 1.2 mg/mL.97 The best solution was, however, offered by Pettit’s group with the preparation of phosphate prodrugs 266 and 267 (Fig. 12), which were designed to undergo non-specific dephosphorylation in biological systems with the release of pancratistatin.25,26 These prodrugs have already shown efficacy in experimental in vivo cancer systems and their clinical development is anticipated.20,21 Both 266 and 267, however, are prepared from the natural product itself and the availability of pancratistatin has become an even more pressing necessity.
Fig 12.

The nine successful synthetic pathways to pancratistatin are summarized in Table. Apart from a short 10-step relay synthesis of (+)-pancratistatin from its natural congener (+)-narciclasine, only two other synthetic sequences involve less than 15 steps. This number has been suggested by Hudlicky,98 and it serves as a good standard for evaluation of practicality of synthetic endeavors. The synthesis of Hudlicky, albeit short, is rather low yielding and very arduous. A number of important issues have to be resolved before the synthesis can be scaled-up. The shortest synthesis so far is the most recent report from Li’s group. It proceeds in 13 steps and good 9% overall yield. The main drawback is the expense of the starting material, (+)-pinitol. The practicality, however, should also be assessed on the basis of the number of chromatographic purifications. The investigators are encouraged to combine sequential steps, so that crude material is utilized as often as possible, as well as replace chromatography with recrystallization whenever possible. On the basis of this criterion, none of the reported syntheses stands out. The publications from Trost’s and Li’s groups, however, are in a communication format and full procedures have not been disclosed. Therefore, no evaluation can be done until full articles appear.
Table.
| Year | Author | # of Stepsa | Yielda % | Starting Material | Price per 10 grams | # of Column Purifications |
|---|---|---|---|---|---|---|
| 1989 | Danishefsky | 27 | 0.16 | pyrogallol | $3.71 | 27 |
| 1995 | Hudlicky | 14 | 2 | bromobenzene | $1 | 13 |
| 1995 | Trost | 20 | 8 | benzoquinone | $31 | NAb |
| 1997 | Haseltine | 24 | 0.97 | benzoquinone | $31 | 16 |
| 1998 | Magnus | 22 | 1.2 | o-vanillin | $6 | 17 |
| 2000 | Rigby | 23 | 0.35 | methylcyclohex-3-enecarboxylate | $18 | 17 |
| 2001 | Pettit | 10 | 3.6 | narciclasine | NAc | 3 |
| 2002 | Kim | 21 | 4 | methyl gallate | $12 | 15 |
| 2006 | Li | 13 | 9 | (+)-pinitol | $1,027 | NAb |
Number of steps and overall yield from commercially available starting material for the longest linear sequence. One-pot procedures are counted as one step. If the utilized starting material is not commercially available, but had been reported previously by other investigators, the number of steps and yields were taken from the cited sources.
Procedures are unavailable.
Isolated from plant sources.
Finally, the investigators are encouraged to continue optimizing their synthetic routes in search for practicality and scalability. The challenge of pancratistatin clearly requires multigenerational synthetic effort to facilitate its long-awaited advancement to human clinical trials.
Scheme 13.

Scheme 16.

Scheme 21.

Scheme 25.

Scheme 30.

Scheme 32.

Scheme 37.

Acknowledgments
US National Institutes of Health (CA-99957 and RR-16480) are gratefully acknowledged for financial support.
ABBREVIATIONS
- Ac
acetyl
- AIBN
azobisisobutyronitrile
- Bn
benzyl
- BOC
t-butoxycarbonyl
- CSA
camphorsulfonic acid
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCC
N,N′-dicyclohexylcarbodiimide
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DMP
Dess-Martin periodinane
- DPPA
diphenylphosphorylazide
- LDA
lithium diisopropylamine
- mCPBA
m-chloroperbenzoic acid
- MsCl
methanesulfonyl chloride
- NBS
N-bromosuccinimide
- NMO
N-methylmorpholine-N-oxide
- PMB
p-methoxybenzyl
- TBAF
tetra-n-butylammonium fluoride
- TBSCl
t-butyldimethylsilyl chloride
- Tf2O
triflic anhydride
- TIPDSCl2
1,1,3,3-tetraisopropyl-1,3-dichlorosiloxane
- TIPS
triisopropylsilyl
- TMEDA
tetramethylethylenediamine
- TMS
trimethylsilyl
- TsCl
p-toluenesulfonyl chloride
References
- 1.Hartwell JL. Lloydia. 1967;30:379. [Google Scholar]
- 2.Martin SF. In: The Alkaloids. Brossi AR, editor. Vol. 30. Academic Press; New York: 1987. p. 251. [Google Scholar]
- 3.Hoshino O. In: The Alkaloids. Cordell GA, editor. Vol. 51. Academic Press; London: 1998. p. 323. [Google Scholar]
- 4.Cook JW, Loudon JD. In: The Alkaloids. Holmes HL, editor. Vol. 2. Academic Press; New York: 1952. p. 331. [Google Scholar]
- 5.Okamoto T, Torii Y, Isogai Y. Chem Pharm Bull. 1968;16:1860. doi: 10.1248/cpb.16.1860. [DOI] [PubMed] [Google Scholar]
- 6.Ceriotti G. Nature. 1967;213:595. doi: 10.1038/213595a0. [DOI] [PubMed] [Google Scholar]
- 7.Carrasco L, Fresno M, Vazquez D. FEBS Lett. 1975;52:236. doi: 10.1016/0014-5793(75)80813-1. [DOI] [PubMed] [Google Scholar]
- 8.Jimenez A, Sanchez L, Vazquez D. FEBS Lett. 1975;55:53. doi: 10.1016/0014-5793(75)80955-0. [DOI] [PubMed] [Google Scholar]
- 9.Jimenez A, Santos A, Alonso G, Vazquez D. Biochim Biophys Acta. 1976;425:342. doi: 10.1016/0005-2787(76)90261-6. [DOI] [PubMed] [Google Scholar]
- 10.Mondon A, Krohn K. Chem Ber. 1975;108:445. [Google Scholar]
- 11.Pettit GR, Cragg GM, Singh SB, Duke JA, Doubek DL. J Nat Prod. 1990;53:176. doi: 10.1021/np50067a026. [DOI] [PubMed] [Google Scholar]
- 12.Pettit GR, Pettit GR, III, Bachaus RA, Boyd MR, Meerow AW. J Nat Prod. 1993;56:1682. doi: 10.1021/np50100a004. [DOI] [PubMed] [Google Scholar]
- 13.Pettit GR, Gaddamidi V, Cragg GM, Herald DL, Sagawa Y. J Chem Soc, Chem Commun. 1984:1693. [Google Scholar]
- 14.Pettit GR, Gaddamidi V, Herald DL, Singh SB, Cragg GM, Schimdt JM, Boettner FE, William M, Sagawa Y. J Nat Prod. 1986;49:995. doi: 10.1021/np50048a005. [DOI] [PubMed] [Google Scholar]
- 15.Ghosal S, Singh S, Kumar Y, Srivastava RS. Phytochemistry. 1989;28:611. [Google Scholar]
- 16.Pettit GR, Pettit GR, III, Groszek G, Backhaus RA, Doubek DL, Barr RJ. J Nat Prod. 1995;58:756. doi: 10.1021/np50119a017. [DOI] [PubMed] [Google Scholar]
- 17.Pettit GR, Meng Y, Herald DL, Knight JC, Day JF. J Nat Prod. 2005;68:1256. doi: 10.1021/np0402367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabrielsen B, Monath TP, Huggins JW, Kefauver DF, Pettit GR, Groszek G, Hollingshead M, Kirsi JJ, Shannon Wm, Shubert EM, Dare J, Ugarkar B, Ussery MA, Phelan MJ. J Nat Prod. 1992;55:1569. doi: 10.1021/np50089a003. [DOI] [PubMed] [Google Scholar]
- 19.Quarzane-Amara M, Franetich JF, Mazier D, Pettit GR, Meijer L, Doerig C, Desportes-Livage I. Antimicrob Agents Chemother. 2001;45:3409. doi: 10.1128/AAC.45.12.3409-3415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bibby MC, Holwell SE, Pettit GR. Anti-Vascular and Anti-Tumor Effects of the Novel Agent Pancratistatin Phosphate. Biological Basis for Antiangiogenic Therapy Conference; Milan, Italy. November 8–10, 1999. [Google Scholar]
- 21.Shnyder SD, Cooper PA, Millington NJ, Gill JH, Pettit GR, Bibby MC. Clin Cancer Res. 2005;11(24 Suppl):8971s. [Google Scholar]
- 22.Kekre N, Griffin C, McNulty J, Pandey S. Cancer Chemother Pharmacol. 2005;56:29. doi: 10.1007/s00280-004-0941-8. [DOI] [PubMed] [Google Scholar]
- 23.McLachlan A, Kekre N, McNulty J, Pandey S. Apoptosis. 2005;10:619. doi: 10.1007/s10495-005-1896-x. [DOI] [PubMed] [Google Scholar]
- 24.Pandey S, Kekre N, Naderi J, McNulty J. Artif Cells, Blood Substitues, Immobilization Biotechnol. 2005;33:279. doi: 10.1081/bio-200066621. [DOI] [PubMed] [Google Scholar]
- 25.Pettit GR, Freeman S, Simpson MJ, Thompson MA, Boyd MR, Williams MD, Pettit GR, III, Doubek DL. Anti-Cancer Drug Des. 1995;10:243. [PubMed] [Google Scholar]
- 26.Pettit GR, Orr B, Ducki S. Anti-Cancer Drug Des. 2000;15:389. [PubMed] [Google Scholar]
- 27.Hudlicky T, Moser M, Banfield SC, Rinner U, Chapuis J-C, Pettit GR. Can J Chem. 2006;84:1313. [Google Scholar]
- 28.Rinner U, Hillebrener HL, Adams DR, Hudlicky T, Pettit GR. Bioorg Med Chem Lett. 2004;14:2911. doi: 10.1016/j.bmcl.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 29.Rinner U, Hudlicky T, Gordon H, Pettit GR. Angew Chem Int Ed. 2004;43:5342. doi: 10.1002/anie.200460218. [DOI] [PubMed] [Google Scholar]
- 30.Hudlicky T, Rinner U, Finn KJ, Ghiviriga I. J Org Chem. 2004;70:3490. doi: 10.1021/jo040292c. [DOI] [PubMed] [Google Scholar]
- 31.Chrétien F, Ibn-Ahmed S, Masion A, Chapleur Y. Tetrahedron. 1993;49:7463. [Google Scholar]
- 32.Ibn-Ahmed S, Khaldi M, Chrétien F, Chapleur Y. J Org Chem. 2004;69:6722. doi: 10.1021/jo049153l. [DOI] [PubMed] [Google Scholar]
- 33.Hudlicky T, Rinner U, Gonzales D, Akgun H, Schilling S, Siengalewicz P, Martinot TA, Pettit GR. J Org Chem. 2002;67:8726. doi: 10.1021/jo020129m. [DOI] [PubMed] [Google Scholar]
- 34.Pettit GR, Melody N, Herald DL, Knight JC, Chapuis JC. J Nat Prod. 2007;70:417. doi: 10.1021/np068046e. [DOI] [PubMed] [Google Scholar]
- 35.Pettit GR, Melody N, Herald DL, Schmidt JM, Pettit RK, Chapuis J-C. Heterocycles. 2002;56:139. [Google Scholar]
- 36.Kireev AS, Nadein ON, Agustin V, Bush NE, Evidente A, Manpadi M, Ogasawara M, Rastogi SK, Rogelj S, Shors ST, Kornienko A. J Org Chem. 2006;71:5694. doi: 10.1021/jo0607562. [DOI] [PubMed] [Google Scholar]
- 37.McNulty J, Mao J, Gibe R, Mo R, Wolf S, Pettit GR, Herald DL, Boyd MR. Bioorg Med Chem Lett. 2001;11:169. doi: 10.1016/s0960-894x(00)00614-4. [DOI] [PubMed] [Google Scholar]
- 38.McNulty J, Larichev V, Pandey S. Bioorg Med Chem Lett. 2005;15:5315. doi: 10.1016/j.bmcl.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 39.Rinner U, Hillebrenner HL, Adams DR, Hudlicky T, Pettit GR. Bioorg Med Chem Lett. 2004;14:2911. doi: 10.1016/j.bmcl.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 40.Rinner U, Hudlicky T. Synlett. 2005:365. [Google Scholar]
- 41.Chapleur Y, Chrétien F, Ibn-Ahmed S, Khaldi M. Curr Org Syn. 2006;3:341. doi: 10.1021/jo049153l. [DOI] [PubMed] [Google Scholar]
- 42.Polt R. In: Organic Synthesis: Theory and Applications. Hudlicky T, editor. Vol. 3. JAI Press; Greenwich: 1997. p. 109. [Google Scholar]
- 43.Danishefsky S, Lee JY. J Am Chem Soc. 1989;111:4829. [Google Scholar]
- 44.Sibi MP, Snieckus V. J Org Chem. 1983;48:1935. [Google Scholar]
- 45.Ono N, Kamimura A, Kaji A. Tetrahedron Lett. 1986:1595. [Google Scholar]
- 46.Ono N, Kamimura A, Kaji A. J Org Chem. 1986;51:2139. [Google Scholar]
- 47.David S, Hanessian S. Tetrahedron. 1985;41:643. [Google Scholar]
- 48.Russell AF, Greenberg S, Moffatt JG. J Am Chem Soc. 1973;95:4025. doi: 10.1021/ja00793a032. [DOI] [PubMed] [Google Scholar]
- 49.Robins MJ, Hansske F, Low NH, Park JI. Tetrahedron Lett. 1984:367. [Google Scholar]
- 50.(a) Overman LE. J Am Chem Soc. 1974;96:597. [Google Scholar]; (b) Overman LE. Tetrahedron Lett. 1975:1149. [Google Scholar]; (c) Overman LE. J Am Chem Soc. 1976;94:2901. [Google Scholar]
- 51.Tian X, Hudlicky T, Konigsberger K. J Am Chem Soc. 1995;117:3643. [Google Scholar]
- 52.Hudlicky T, Tian X, Konigsberger K, Maurya R, Rouden J, Fan B. J Am Chem Soc. 1996;118:10752. [Google Scholar]
- 53.Gibson DT, Hensley M, Yosioka H, Mabry TJ. Biochemistry. 1970;9:1626. doi: 10.1021/bi00809a023. [DOI] [PubMed] [Google Scholar]
- 54.Yamada Y, Yamamoto T, Okawara M. Chem Lett. 1975:361. [Google Scholar]
- 55.Hudlicky T, Tian X, Konigsberger K, Rouden J. J Org Chem. 1994;59:4037. [Google Scholar]
- 56.Hudlicky T, Reed JW. In: In Comprehensive Synthesis: Selectivity, Strategy & Efficiency in Modern Organic Chemistry. Trost BM, editor. Vol. 5. Pergamon Press; Oxford: 1991. p. 899. [Google Scholar]
- 57.Hudlicky T, Sinai-Zingde G, Seoane G. Synth Commun. 1987;17:1155. [Google Scholar]
- 58.Lopes RSC, Lopes CC, Heathcock CH. Tetrahedron Lett. 1992;33:6775. [Google Scholar]
- 59.Trost BM, Pulley SR. J Am Chem Soc. 1995;117:10143. [Google Scholar]
- 60.Trost BM, Van Vranken DL, Bingel C. J Am Chem Soc. 1992;114:9327. [Google Scholar]
- 61.Cambie RC, Renner ND, Rutledge PS, Woodgate PD. Synth Commun. 1989;19:537. [Google Scholar]
- 62.Trost BM, Li L, Guile SD. J Am Chem Soc. 1992;114:8745. [Google Scholar]
- 63.Pearson DE, Wysong RD, Breder CV. J Org Chem. 1967;32:2358. [Google Scholar]
- 64.Comber MF, Sargent MV. Aust J Chem. 1985;38:1481. [Google Scholar]
- 65.Iinuma M, Tanaka T, Matsuura S. Yakugaku Zasshi. 1983;103:997. doi: 10.1248/yakushi1947.103.9_994. [DOI] [PubMed] [Google Scholar]
- 66.Hudson BJF, Robinson R. J Chem Soc. 1941:715. [Google Scholar]
- 67.Arnold RT, Bordwell F. J Am Chem Soc. 1942;64:2983. [Google Scholar]
- 68.Brownell WB, Weston AW. J Am Chem Soc. 1951;73:4971. [Google Scholar]
- 69.Benington F, Morin RD. J Org Chem. 1962;27:142. [Google Scholar]
- 70.Doyle TJ, Hendrix M, VanDerveer D, Javanmard S, Haseltine J. Tetrahedron. 1997;53:11153. [Google Scholar]
- 71.Knapp S, Ornaf RM, Rodriques KE. J Am Chem Soc. 1983;105:5494. [Google Scholar]
- 72.Xu YC, Lebeau E, Gillard JW, Attardo G. Tetrahedron Lett. 1993;34:3841. [Google Scholar]
- 73.Johnson RC, Plé PA, Adams JP. J Chem Soc, Chem Commun. 1991:1006. [Google Scholar]
- 74.Magnus P, Sebhat IK. J Am Chem Soc. 1998;120:5341. [Google Scholar]
- 75.Magnus P, Sebhat IK. Tetrahedron. 1998;54:15509. [Google Scholar]
- 76.Magnus P, Lacour J, Evans PA, Roe MB, Hulme C. J Am Chem Soc. 1996;118:3406. [Google Scholar]
- 77.Banwell MG, Cowden CJ, Gable RW. J Chem Soc Perkin Trans 1. 1994:3515. [Google Scholar]
- 78.Rigby JH, Gupta V. J Org Chem. 1995;60:7392. [Google Scholar]
- 79.Rigby JH, Maharoof USM, Mateo ME. J Am Chem Soc. 2000;122:6624. [Google Scholar]
- 80.Thompson C, Kallmerten J. J Org Chem. 1990;55:6076. [Google Scholar]
- 81.Mcgowen DA, Berchtold GA. J Org Chem. 1981;46:2381. [Google Scholar]
- 82.Hoare JH, Policastro PP, Berchtold GA. J Am Chem Soc. 1983;105:6264. [Google Scholar]
- 83.Pawlak JL, Berchtold GA. J Org Chem. 1987;52:1765. [Google Scholar]
- 84.Martin VS, Woodard SS, Katsuki T, Yamada Y, Ikeda M, Sharpless KB. J Am Chem Soc. 1981;103:6237. [Google Scholar]
- 85.Pettit GR, Melody N, Herald DL. J Org Chem. 2001;66:2583. doi: 10.1021/jo000710n. [DOI] [PubMed] [Google Scholar]
- 86.Ko H, Kim E, Kim D. Org Lett. 2002;4:1343. doi: 10.1021/ol0256419. [DOI] [PubMed] [Google Scholar]
- 87.Ko H, Kim E, Park JE, Kim D, Kim S. J Org Chem. 2004;69:112. doi: 10.1021/jo035371n. [DOI] [PubMed] [Google Scholar]
- 88.Lalami K, Dhal R, Brown E. Heterocycles. 1988;27:1131. [Google Scholar]
- 89.Büchi G, Powell JE., Jr J Am Chem Soc. 1970;92:3126. [Google Scholar]
- 90.Lee T, Kim S. Tetrahedron: Asymmetry. 2003;14:1951. doi: 10.1016/S0957-4166(03)00040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li M, Wu A, Zhou P. Tetrahedron Lett. 2006;47:3707. [Google Scholar]
- 92.Watanabe Y, Mitani M, Morita T, Ozaki S. J Chem Soc, Chem Commun. 1989:482. [Google Scholar]
- 93.Aher NG, Pore VS. Synlett. 2005:2155. [Google Scholar]
- 94.Coleman RS, Gurrala SR. Org Lett. 2005;7:1849. doi: 10.1021/ol050476t. [DOI] [PubMed] [Google Scholar]
- 95.Tillet JG. Chem Rev. 1976;76:747. [Google Scholar]
- 96.Beijnen JH, Flora KP, Halbert GW, Henrar REC, Slack JA. Br J Cancer. 1995;72:210. doi: 10.1038/bjc.1995.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Torres-Labandeira JJ, Davignon P, Pitha J. J Pharm Sci. 1991;80:384. doi: 10.1002/jps.2600800421. [DOI] [PubMed] [Google Scholar]
- 98.Hudlicky T. Chem Rev. 1996;96:3. doi: 10.1021/cr950012g. [DOI] [PubMed] [Google Scholar]