

Abstract
Incorporation of the OCF3 group into organic molecules, especially aromatic and heteroaromatic compounds, is recognized as one of the major challenges in synthetic organic chemistry. Although many attempts have been made to develop efficient trifluoromethoxylation strategies, most of the current approaches still require use of highly toxic, thermally unstable reagents, or impractical reaction conditions. Herein, we highlight a recent contribution from our group towards the synthesis of (hetero)aryltrifluoromethyl ethers. Our protocol is scalable, operationally simple, and allows an easy access to a wide range of synthetically useful ortho-OCF3 aniline derivatives, as well as functionalized trifluoromethoxylated pyridines and pyrimidines under mild reaction conditions.
Keywords: amides, anilines, rearrangement, fluorine, trifluoromethoxylation, trifluoromethylation
Graphical Abstract
The intrinsic properties of the fluorine atom (small size, high electronegativity, high strength and polarity of C–F bond) have a great influence on physical and chemical properties of the parent molecules, with profound consequences on their biological activities. For example, incorporation of fluorine atom(s) into potential drug candidates often significantly improves their metabolic stability, lipo-philicity, and bioavailability.1 Since fluorine furnishes molecules with unique properties that cannot be attained using any other element, fluorine substitution has become a common strategy in the discovery and development of new drugs, agrochemicals, and specialized materials.
Among the fluorine-containing functional groups, the trifluoromethoxy (OCF3) group is becoming increasingly important. The growing interest in OCF3-bearing compounds, in particular (hetero)aryl trifluoromethyl ethers, results from the unique characteristics of the OCF3 group. The OCF3 group is one of the most lipophilic substituents, as indicated by its Hansch–Leo parameter [πx (SCF3) = +1.44, πx (SF5) = +1.23, πx (OCF3) = +1.04, πx (CF3) = +0.88, πx (OCH3) = −0.02].2 In addition to an increased lipophilicity, (hetero)aryl trifluoromethyl ethers also exhibit unique conformational properties. Due to the strong electron-withdrawing effect of fluorine, the lone-pair electrons on oxygen are drawn towards the trifluoromethyl group and are weakly interacting with the arene π-electron system. This nO → σ*C–F hyperconjugative interaction and the steric bulk of the CF3 group cause the O–CF3 bond to lie in the plane orthogonal to the arene ring (Figure 1).3 This unique orientation renders the OCF3 an electron-withdrawing group (χ = 3.7),4 and a strong para-directing substituent (the repulsion between the lone-pair electrons of fluorine atoms and the arene π electrons increases the electron density on the para position).5 The greater conformational flexibility of the OCF3 in comparison with a methoxy group can be beneficial to obtain additional binding affinity of biologically active aryl trifluoromethyl ethers.6 With these favorable druglike properties, a number of pharmaceuticals and agrochemicals containing the OCF3 group show enhanced efficacy often coupled with diminished side effects.7
Figure 1.

The nO → σ*C–F hyperconjugative interaction and the preferred conformation of trifluoromethoxyarene in which the O–CF3 bond lies in a plane orthogonal to the arene plane
Due to the intriguing properties of the OCF3 group, aryl trifluoromethyl ethers have found applications as structural elements of pharmaceuticals,7c,i,8 crop-protecting agents,7d,9 electro-optical materials,10 and soluble organic semiconductors.11 Nonetheless, the OCF3 group is probably still one of the least understood fluorine substituents and its facile introduction into organic compounds, in particular (hetero)aromatic ones is recognized as a major challenge in synthetic chemistry.12 The difficulty in the synthesis of (hetero)aryl trifluoromethyl ethers results from the lack of efficient and mild methods for the formation of both the carbon–OCF3 and the oxygen–CF3 bonds [the trifluoromethoxy group cannot be formed, analogously to the methoxy group, by simple treatment of trifluoroiodomethane with phenoxides, due to (i) electronic repulsion between three fluorine atoms and an incoming nucleophile; (ii) formation of energetically disfavored CF3 carbocation transition-state structure (TS); and (iii) the inversion of the electrostatic partial charges in CF3I because of the strong electron-with-drawing ability of the trifluoromethyl moiety].13 Consequently, only a few preparative methods for synthesis of tri-fluoromethoxylated (hetero)arenes have been developed over the past decades.
The first synthesis of aryl trifluoromethyl ethers was reported by Yagupolskii in 1955.14 The protocol involved chlorination of the methoxy group of electron-deficient anisoles with chlorine gas and phosphorus pentachloride at around 200 °C to afford aryl trichloromethyl ethers, which can be subsequently converted into aryl trifluoromethyl ethers by nucleophilic substitution of chlorine with fluorine using anhydrous HF or SbF3 in the presence of a catalytic amount of SbCl5 (Scheme 1, a).14,15 The Yagupolskii’s method was later improved by Yarovenko and Vasileva, who discovered that aryl trichloromethyl ethers can be readily accessed from aryl chlorothionoformates by chlorination (Scheme 1, b).15a This approach is also applicable to the synthesis of trifluoromethoxypyridines with chlorine atoms at the position α to the ring nitrogen, as recently demonstrated by Leroux and coworkers.16
Scheme 1.

Synthesis of aryl trifluoromethyl ethers via intermediate trichloromethoxyarenes
In 1964 Sheppard reported an alternative method towards structurally simple aryltrifluoromethyl ethers based on deoxyfluorination of phenol fluoroformates (Scheme 2, a).17 The intermediate fluoroformates can be prepared from phenols upon treatment with fluorophosgene and their conversion into the final aryl trifluoromethyl ethers is achieved with SbF3 or SF4/HF at 160 °C. About 30 years later, Hiyama developed a new protocol for the synthesis of aryl trifluoromethyl ethers via an oxidative desulfurization–fluorination strategy (Scheme 2, b).18 Exposure of aryl dithio-carbonates (xanthogenates) to a large excess of hydrogen fluoride–pyridine and 1,3-dibromo-5,5-dimethylhydantoin (DBH) or N-bromosuccinimide (NBS) afforded the desired trifluoromethoxylated arenes.
Scheme 2.

Synthesis of aryl trifluoromethyl ethers via (a) fluoroformates and (b) dithiocarbonates
Aryl trifluoromethyl ethers have also been prepared directly from unprotected phenols via electrophilic O-trifluoromethylation with electrophilic CF3 reagents. In 1996, Umemoto and coworkers reported that phenol derivatives can undergo O-trifluoromethylation with in situ generated O-(trifluoromethyl)dibenzofuranium salts in the presence of alkyl amine bases at −100 °C to −90 °C (Scheme 3, a).19 Togni et al. investigated trifluoromethylation of phenols with hypervalent iodine reagents: reaction of 2,4,6-trimethylphenol with 1-trifluoromethyl-1,2-benziodoxol-3-(1H)-one (Togni reagent II) resulted in the formation of the desired aryl trifluoromethyl ether in modest 15% yield due to the competing trifluoromethylation at the softer aromatic carbon centers (Scheme 3, b).20
Scheme 3.

O-Trifluoromethylation of phenols using electrophilic CF3 sources
The synthesis of aryl trifluoromethyl ethers via carbon–OCF3 bond formation has not been widely explored due to the thermal instability and poor nucleophilicity of the trifluoromethoxide.21 Nonetheless, in 2008, Kolomeitsev reported the synthesis of phenyl and naphthyl trifluoromethyl ethers via addition of trifluoromethoxide to in situ generated benzyne and α-naphthyne, respectively (Scheme 4, a).21a In 2011, Ritter and co-workers developed a new strategy for the synthesis of aryl trifluoromethyl ethers based on silver-mediated cross–coupling reaction of functionalized aryl stannanes and aryl boronic acids with in situ generated tris(dimethylamino)sulfonium trifluoromethoxide (Scheme 4, b).22 This approach can be employed towards synthesis of complex molecules, the most impressive examples being trifluoromethoxylated estrone and OCF3-bearing morphine derivative.
Scheme 4.

Synthesis of aryl trifluoromethyl ethers using trifluoromethoxide
In 2012, a direct trifluoromethoxylation of benzene using toxic gaseous trifluoromethyl perfluorite in the presence of a radical promoter perfluoromethyl vinyl ether have been reported, but the substrates are limited to simple arenes and, in most cases, the yields are low due to the competing arene fluorination reaction (Scheme 5).23
Scheme 5.

Radical trifluoromethoxylation reaction of arenes (2012)
Most recently, Qing and co-workers reported a direct synthesis of aryl trifluoromethyl ethers from unprotected phenols (Scheme 6).24 This silver-mediated O-trifluoromethylation reaction was achieved using TMSCF3 as the CF3 source, and Selectfluor® as well as N-fluorobenzenesulfonimide (NFSI) as exogenous oxidants. The major drawback of Qing’s protocol is the requirement of a large excess of the reagents, which renders the reaction extremely expensive and limits its practicality on large scale synthesis.
Scheme 6.

Synthesis of aryl trifluoromethyl ethers via silver-mediated oxidative trifluoromethylation
As shown above, classical approaches towards aryl trifluoromethyl ethers involve prefunctionalization at oxygen prior to the introduction of fluorine (Scheme 1 and Scheme 2). Many of these conventional routes entail use of highly toxic or corrosive reagents, including strong Lewis acids, and thus have a low functional-group tolerance and a narrow substrate scope. In addition, they require specialized equipment for handling some of the regents. The major limitation of the more modern approaches, in turn, is the need to utilize impractical reaction conditions due to the thermal instability of trifluoromethylating and trifluoromethoxylating reagents. The lack of easy-to-handle OCF3 radical precursors for radical trifluoromethoxylation is also a problem. As a result, many of OCF3-containing building blocks, especially OCF3-bearing (hetero)arenes, are prohibitively expensive, and the OCF3 group is not yet considered as a ‘classical’ group in all molecular screenings in medicinal chemistry and materials science. Clearly, there is a real need for preparative methods for the synthesis of OCF3-substituted aromatic and heteroaromatic building blocks that avoid use of highly toxic and thermally labile reagents. Transformations that can be conducted at room temperature, easily scaled up, and have a broad substrate scope are highly desired.
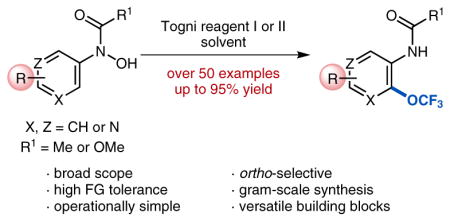
Recently, our group has successfully developed a novel protocol for trifluoromethoxylation of arenes and heteroarenes.25 Our approach relies on trifluoromethylation of protected N-(hetero)aryl-N-hydroxylamines followed by an intramolecular rearrangement of the intermediate N-(hetero)aryl-N-(trifluoromethoxy)amines (Scheme 7). This unique synthetic method provides an easy access towards ortho-trifluoromethoxylated anilines, as well as trifluoromethoxylated 3-aminopyridines and pyrimidines. Our procedure, which utilizes bench-stable reagents, is operationally simple, fully amenable to scale-up, and displays high levels of functional-group tolerance.
Scheme 7.

(Hetero)aryl trifluoromethoxylation via intramolecular OCF3-migration
Our initial investigation focused on development of a reliable reaction protocol for O-trifluoromethylation of protected N-aryl-N-hydroxylamines. Upon screening of different electrophilic CF3 sources, we obtained the O-trifluoromethylation products in high yields when protected N-aryl-N-hydroxylamines were treated with 1.2 equivalents of Togni reagent II in the presence of 10 mol% Cs2CO3 in CHCl3 (0.1 M) at room temperature (Scheme 8). We were pleased to find that our O-trifluoromethylation protocol tolerates a wide range of functional groups (nitrile, ketone, ester, amide, halogens, CF3 group, ether, α-amino acid ester, quinoline, and indole) and different nitrogen protecting groups (acetyl, benzoyl, and methoxycarbonyl).
Scheme 8.

Selected examples of arene substrates undergoing the O-trifluoromethylation–OCF3-migration reaction. a 50 °C. b 120 °C. c 140 °C. d Less than 5% para product was detected.
Having found the optimal conditions for the synthesis of protected N-aryl-N-(trifluoromethoxy)amines, we directed our attention to examining the OCF3-migration reaction. We observed that upon heating (50–140 °C) in MeNO2 (MeCN worked equally well when heating up to 80 °C was required), a wide range of N-aryl-N-trifluoromethoxyamine derivatives underwent OCF3-group migration to afford the trifluoromethoxylated aniline derivatives in good to excellent yields. The electron-poor arene substrates required higher reaction temperatures (either 120 °C or 140 °C) in order to achieve full conversion. In general, the rearrangement was highly ortho-selective, however, low levels of regiocontrol were obtained when two nonidentical ortho positions were present.
We were delighted to find that our trifluoromethylation–rearrangement strategy is also applicable to heteroaromatic substrates (Scheme 9). This time, we used 3,3-di-methyl-1-(trifluoromethyl)-1,2-benziodoxole (Togni reagent I) as the trifluoromethylating reagent since it gave us superior yields when N-protected hydroxylamines derived from 3- or 5-nitropyrimidines were used as starting materials. We also simplified the synthetic protocol and performed the two steps (i.e., O-trifluoromethylation and OCF3 migration) in one pot, without isolation of the intermediate N-heteroaryl-N-(trifluoromethoxy)amines. The trifluoromethoxylated pyridine and pyrimidine derivatives were formed in good to excellent yields. Our reaction protocol was highly functional-group tolerant and applicable to a wide range of highly substituted substrates including complex molecules such as estrone and Tadalafil derivatives. Notably, electron-rich pyridine rings (with electron-donating substituent α to pyridine nitrogen), both the trifluoromethylation and the rearrangement reaction took place at or below room temperature. In case of less electron-rich substrates, heating was required to promote the rearrangement step.
Scheme 9.

Selected examples of heteroarene substrates undergoing the O-trifluoromethylation–OCF3-migration reaction. a At 23 °C then 50 °C in CH2Cl2. b 4 °C in CH2Cl2 (0.01 M). c Following the O-trifluoromethylation reaction at 23 °C, the reaction mixture was concentrated, the residue was dissolved in MeNO2, and the resulting mixture was heated. d 120 °C. e 80 °C. f 60 °C. g c 0.01 M. h Atropisomeric ratio.
A proposed mechanism of our trifluoromethoxylation reaction is depicted in Scheme 10. Deprotonation of N-protected (hetero)aryl hydroxylamine is followed by a single-electron transfer (SET) to Togni reagents, which generates N-hydroxyl radical I, trifluoromethyl radical, and alkoxide. Recombination of N-hydroxyl radical I with trifluoromethyl radical affords the O-trifluoromethylated hydroxylamine.26 Our preliminary mechanistic studies suggested that the rearrangement reaction proceeds via thermally induced heterolytic cleavage of the N–O bond, followed by recombination of trifluoromethoxide with a nitrenium ion and tautomerization of intermediate II to generate the desired product.27
Scheme 10.

Proposed reaction mechanism: radical O-trifluoromethylation followed by OCF3-migration via an ion-pair formation
Prior to our report, the synthesis of OCF3-bearing heterocycles under mild conditions had remained elusive, and trifluoromethoxylated heteroarenes were extremely rare.16,24,28 Our trifluoromethylation–rearrangement protocol is thus a major breakthrough in the field. We believe that our synthetic strategy would enable an easy access to trifluoromethoxylated organic molecules that otherwise would be difficult or impossible to make. We envision that our method would be employed in the synthesis of complex bioactive molecules and hence accelerate drug discovery and development process. Nevertheless, our approach is not without limitations – it is not a direct arene trifluoromethoxylation strategy, but a four-step reaction sequence enabling the conversion of nitro(hetero)arenes into trifluoromethoxylated (hetero)arenes. The products obtained through our strategy always contain an amino group ortho to the OCF3 substituent. Although such an amino group could be used as a versatile synthetic handle for further functionalization reactions, its removal would be necessary in cases where the NH2 group was not desirable. Our trifluoromethylation–rearrangement strategy also requires use of Togni reagents, which are among the most expensive CF3 sources. Clearly, challenge still remains in development of more straightforward and economical trifluoromethoxylation protocols.
We anticipate that future efforts in the field would be directed at development of more stable and versatile trifluoromethoxylating reagents, either for a direct, late-stage trifluoromethoxylation of complex (hetero)arenes with OCF3 radical or potential transition-metal-catalyzed coupling reactions with prefunctionalized (hetero)arene substrates. In addition, new methods for direct O-trifluoromethylation of unprotected phenols with the aid of transition-metal catalysts would be sought after.
Acknowledgments
We acknowledge generous start-up funds from Stony Brook University in support of this work. K.N.L. is grateful for the Chemistry Graduate Research Fellowships provided by the Department of Chemistry.
Biography

Katarzyna N. Lee (left) was born in Warsaw, Poland. In 2011, she completed her MSc with honors in chemistry from the University College London. She continued her studies at Harvard University and received her MA degree in chemistry in 2013. She is currently pursuing her PhD degree at the Stony Brook University under the supervision of Prof. Ngai. Her research in the Ngai group has focused on development of novel trifluoromethoxylation methods.
Johnny W. Lee (middle) was born in New York, United States. In 2015, he obtained his BSc degree in chemistry from Stony Brook University, where he conducted undergraduate research in the laboratory of Prof. Ngai. In the same year, he began his PhD degree at Stony Brook University under the supervision of Prof. Ngai. At Stony Brook he is focused on development of novel fluorination strategies, with a specific interest in trifluoromethoxylation reactions.
Ming-Yu Ngai (right) was born in Fuching, China and graduated with BSc degree from the University of Hong Kong in 2003. After he received his PhD degree with honors in chemistry from the University of Texas at Austin under the guidance of Prof. Michael J. Krische in 2008, he worked with Prof. Barry M. Trost at Stanford University as the Croucher postdoctoral fellow (2009–2011) and with Prof. Tobias Ritter at Harvard University as a postdoctoral associate (2011–2013). In 2013, he was appointed as an Assistant Professor in the Department of Chemistry at Stony Brook University. His research focuses on creating new functional molecules with a specific interest in fluorinated compounds and exploring their properties as well as applications in the fields of chemistry, biology, and materials science.
References
- 1.(a) Thayer AM. Chem Eng News. 2006;84(23):15. [Google Scholar]; (b) Hagmann WK. J Med Chem. 2008;51:4359. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]; (c) O’Hagan D. Chem Soc Rev. 2008;37:308. [Google Scholar]; (d) Ojima I. Fluorine in Medicinal Chemistry and Chemical Biology. Wiley-Blackwell; Chichester: 2009. [Google Scholar]; (e) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (f) Liang T, Neumann CN, Ritter T. Angew Chem Int Ed. 2013;52:8214. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]; (g) Barnes-Seeman D, Beck J, Springer C. Curr Top Med Chem. 2014;14:855. doi: 10.2174/1568026614666140202204242. [DOI] [PubMed] [Google Scholar]; (h) Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 2.Hansch C, Leo A. Substituent Constants for Correlation Analysis in Chemistry and Biology. Part VII 339 Wiley; New York: 1979. [Google Scholar]
- 3.Federsel D, Herrmann A, Christen D, Sander S, Willner H, Oberhammer H. J Mol Struct. 2001;567:127. [Google Scholar]
- 4.Mcclinton MA, Mcclinton DA. Tetrahedron. 1992;48:6555. [Google Scholar]
- 5.Castagnetti E, Schlosser M. Chem Eur J. 2002;8:799. doi: 10.1002/1521-3765(20020215)8:4<799::aid-chem799>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 6.Muller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 7.(a) Oreilly P, Kobayashi S, Yamane S, Phillips WG, Raymond P, Castanho B. Brighton Crop Protection Conference: Pests and Diseases. 1992;1–3:427. [Google Scholar]; (b) Bensimon G, Lacomblez L, Meininger V, Bouche P, Delwaide C, Couratier P, Blin O, Viader F, Peyrostpaul H, David J, Maloteaux JM, Hugon J, Laterre EC, Rascol A, Clanet M, Vallat JM, Dumas A, Serratrice G, Lechevallier B, Peuch AJ, Nguyen T, Shu C, Bastien P, Papillon C, Durrleman S, Louvel E, Guillet P, Ledoux L, Orvoenfrija E, Dib M. New Engl J Med. 1994;330:585. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]; (c) Delorenzi FG. Pulm Pharmacol. 1994;7:129. [Google Scholar]; (d) Santel HJ, Bowden BA, Sorensen VM, Mueller KH. 1999 Brighton Conference: Weeds. 1999;1–3:23. [Google Scholar]; (e) Leroux F, Jeschke P, Schlosser M. Chem Rev. 2005;105:827. doi: 10.1021/cr040075b. [DOI] [PubMed] [Google Scholar]; (f) Jeschke P, Baston E, Leroux FR. Mini-Rev Med Chem. 2007;7:1027. doi: 10.2174/138955707782110150. [DOI] [PubMed] [Google Scholar]; (g) Beria I, Valsasina B, Brasca MG, Ceccarelli W, Colombo M, Cribioli S, Fachin G, Ferguson RD, Fiorentini F, Gianellini LM, Giorgini ML, Moll JK, Posteri H, Pezzetta D, Roletto F, Sola F, Tesei D, Caruso M. Bioorg Med Chem Lett. 2010;20:6489. doi: 10.1016/j.bmcl.2010.09.060. [DOI] [PubMed] [Google Scholar]; (h) Beria I, Bossi RT, Brasca MG, Caruso M, Ceccarelli W, Fachin G, Fasolini M, Forte B, Fiorentini F, Pesenti E, Pezzetta D, Posteri H, Scolaro A, Depaolini SR, Valsasina B. Bioorg Med Chem Lett. 2011;21:2969. doi: 10.1016/j.bmcl.2011.03.054. [DOI] [PubMed] [Google Scholar]; (i) Landelle G, Panossian A, Leroux FR. Curr Top Med Chem. 2014;14:941. doi: 10.2174/1568026614666140202210016. [DOI] [PubMed] [Google Scholar]
- 8.(a) Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. Nature (London, UK) 2000;405:962. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]; (b) Valsasina B, Beria I, Alli C, Alzani R, Avanzi N, Ballinari D, Cappella P, Caruso M, Casolaro A, Ciavolella A, Cucchi U, De Ponti A, Felder E, Fiorentini F, Galvani A, Gianellini LM, Giorgini ML, Isacchi A, Lansen J, Pesenti E, Rizzi S, Rocchetti M, Sola F, Moll J. Mol Cancer Ther. 2012;11:1006. doi: 10.1158/1535-7163.MCT-11-0765. [DOI] [PubMed] [Google Scholar]
- 9.(a) Howard J, Wall R. Bull Entomol Res. 1995;85:71. [Google Scholar]; (b) Amir OG, Peveling R. J Appl Entomol. 2004;128:242. [Google Scholar]; (c) Chen Y, Zhang AF, Wang WX, Zhang Y, Gao TC. Ann Appl Biol. 2012;161:247. [Google Scholar]; (d) Sagheer M, Yasir M, Mansoor-ul-Hasan, Ashfaq M. Pak J Agr Sci. 2012;49:173. [Google Scholar]
- 10.Kirsch P, Bremer M. Angew Chem Int Ed. 2000;39:4217. doi: 10.1002/1521-3773(20001201)39:23<4216::AID-ANIE4216>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 11.Mamada M, Shima H, Yoneda Y, Shimano T, Yamada N, Kakita K, Machida T, Tanaka Y, Aotsuka S, Kumaki D, Tokito S. Chem Mater. 2015;27:141. [Google Scholar]
- 12.Leroux FR, Manteau B, Vors JP, Pazenok S. Beilstein J Org Chem. 2008;4(13) doi: 10.3762/bjoc.4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirk KL. J Fluorine Chem. 2006;127:1013. [Google Scholar]
- 14.Yagupolskii LM. Dokl Akad Nauk SSSR. 1955;105:100. [Google Scholar]
- 15.(a) Yarovenko NN, Vasileva AS. Zh Obshch Khim. 1958;28:2502. [Google Scholar]; (b) Yagupolskii LM, Troitskaya VI. J Gen Chem USSR. 1961;31:845. [Google Scholar]; (c) Yagupolskii LM, Orda VV. Zh Obshch Khim. 1964;34:1979. [Google Scholar]
- 16.Manteau B, Genix P, Brelot L, Vors JP, Pazenok S, Giornal F, Leuenberger C, Leroux FR. Eur J Org Chem. 2010:6043. [Google Scholar]
- 17.Sheppard WA. J Org Chem. 1964;29:1. [Google Scholar]
- 18.(a) Kuroboshi M, Suzuki K, Hiyama T. Tetrahedron Lett. 1992;33:4173. [Google Scholar]; (b) Kanie K, Tanaka Y, Suzuki K, Kuroboshi M, Hiyama T. Bull Chem Soc Jpn. 2000;73:471. [Google Scholar]; (c) Kuroboshi M, Kanie K, Hiyama T. Adv Synth Catal. 2001;343:235. [Google Scholar]
- 19.Umemoto T, Adachi K, Ishihara S. J Org Chem. 2007;72:6905. doi: 10.1021/jo070896r. [DOI] [PubMed] [Google Scholar]
- 20.Stanek K, Koller R, Togni A. J Org Chem. 2008;73:7678. doi: 10.1021/jo8014825. [DOI] [PubMed] [Google Scholar]
- 21.(a) Kolomeitsev AA, Vorobyev M, Gillandt H. Tetrahedron Lett. 2008;49:449. [Google Scholar]; (b) Zhang CP, Vicic DA. Organometallics. 2012;31:7812. [Google Scholar]
- 22.Huang C, Liang T, Harada S, Lee E, Ritter T. J Am Chem Soc. 2011;133:13308. doi: 10.1021/ja204861a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venturini F, Navarrini W, Famulari A, Sansotera M, Dardani P, Tortelli V. J Fluorine Chem. 2012;140:43. [Google Scholar]
- 24.Liu JB, Chen C, Chu L, Chen ZH, Xu XH, Qing FL. Angew Chem Int Ed. 2015;54:11839. doi: 10.1002/anie.201506329. [DOI] [PubMed] [Google Scholar]
- 25.(a) Hojczyk KN, Feng P, Zhan C, Ngai MY. Angew Chem Int Ed. 2014;53:14559. doi: 10.1002/anie.201409375. [DOI] [PubMed] [Google Scholar]; (b) Feng P, Lee KN, Lee JW, Zhan C, Ngai M-Y. Chem Sci. 2015 doi: 10.1039/C5SC02983J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matoušek V, Pietrasiak E, Sigrist L, Czarniecki B, Togni A. Eur J Org Chem. 2014:3087. [Google Scholar]
- 27.Tabolin AA, Ioffe SL. Chem Rev. 2014:5426. doi: 10.1021/cr400196x. [DOI] [PubMed] [Google Scholar]
- 28.(a) Barton DHR, Hesse RH, Jackman GP, Pechet MM. J Chem Soc, Perkin Trans 1. 1977:2604. [Google Scholar]; (b) Chambers RD, Grievson B, Drakesmith FG, Powell RL. J Fluorine Chem. 1985;29:323. [Google Scholar]; (c) Fuss A, Koch V. Synthesis. 1990:604. [Google Scholar]; (d) Morimoto K, Makino K, Sakata G. J Fluorine Chem. 1992;59:417. [Google Scholar]; (e) Guiadeen D, Kothandaraman S, Yang LH, Mills SG, MacCoss M. Tetrahedron Lett. 2008;49:6368. [Google Scholar]