

Abstract
HNO plays significant roles in many biological processes. Numerous heme proteins bind HNO, an important step for its biological functions. A systematic computational study was performed to provide the first detailed trends and origins of the effects of iron oxidation state, axial ligand, and protein environment on HNO binding. Results show that HNO binds much weaker with ferric porphyrins than corresponding ferrous systems, offering strong thermodynamic driving force for experimentally observed reductive nitrosylation. Axial ligand was found to influence HNO binding through its trans effect and charge donation effect. The protein environment significantly affects the HNO hydrogen bonding structures and properties. The predicted NMR and vibrational data are in excellent agreement with experiment. These novel and broad range of results shall facilitate studies of HNO binding in many heme proteins, models, and related metalloproteins.
Keywords: nitrogen oxides, bioinorganic chemistry, heme proteins, porphyrinoids, density functional calculations
Graphical Abstract
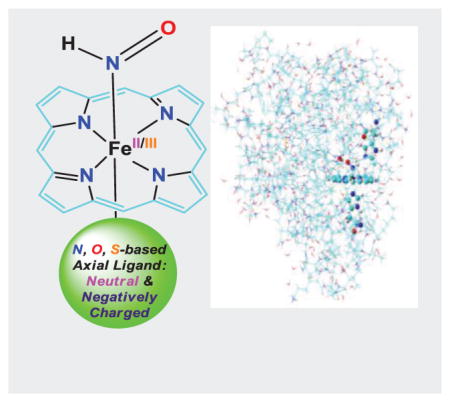
HNO binding is an important step for its biological functions. Our results for the first time directly show that 1) HNO preferably binds with ferrous porphyrins than corresponding ferric systems due to better metal to ligand back-donation; 2) Axial ligand can strongly influence HNO binding through its trans effect and charge donation effect; 3) The heme protein environment has a significant effect on HNO hydrogen bonding structures and properties in different proteins.
HNO plays significant roles in many biological processes, such as vascular relaxation, enzyme activity regulation, and neurological function regulation.[1] With a better vasodilative effect than NO and an increased contractility effect, HNO donors offer a promising new class of vasodilators and heart failure treatment.[2] It was also suggested as a potential treatment for reduction of neuron damage during stroke.[1a] Among its biological targets, heme proteins have been studied for several decades, such as nitrite and nitric oxide reductases involved in denitrification processes in plants, bacteria, and fungi,[1b, c] nitric oxide synthase, peroxidase, and cytochrome P450 nitric oxide reductase involving HNO as an intermediate in their catalytic cycles,[3] and metmyoglobin, methemoglobin, ferricytochrome c, oxymyoglobin, myoglobin, cytochrome P450, and horseradish peroxidase (HRP) used to scavenge HNO.[1a] Despite decades-long experimental work and recent computational work on geometric and electronic structures, affinities, spectroscopic properties of HNO bound protein complexes and models,[4] some critical questions remain to be answered. For instance, although it is well-known that ferrous nitrosyl compounds are more stable than corresponding ferric systems,[1a] such comparison of iron oxidation state on HNO binding has not been reported. Stable HNO binding was reported for several ferrous heme proteins,[5] but ferric hemes lead to reductive nitrosylation.[1a, 3c, 6] Despite this transient HNO binding in ferric hemes, the detailed comparisons of iron oxidation state on HNO binding may offer useful thermodynamic trend to help understand experimental results and what properties are most significantly affected. In addition, given the different axial ligands in numerous heme proteins, how such axial ligand affects HNO binding is yet to be uncovered. The effect from the protein environment beyond the proximal axial ligand and distal hydrogen bonding residue is also unknown. Therefore, a systematic computational study was performed here to address these questions.
As the first study to address such questions, our goal is to reveal HNO binding trends due to these effects and their origins, not accurate absolute values of binding energies. So, the HNO bound ferrous and ferric porphyrins with five axial ligands (see Scheme 1) were first studied using the previously reported DFT method for similar systems[4b, c] in full geometry optimization and frequency analysis (trends are the same when other methods and solvent effect were used, see Supporting Information for computational details and optimized 3D structures). The 5-methylimidazole (5-MeIm), NH2CH3, PhO−, and CH3S− ligands were used to model the axial His, Lys, Tyr, and Cys ligands in myoglobin, cytochrome c nitrite reductase, catalase, cytochrome P450 nitric oxide reductase, respectively, while CH3O− was used to compare with PhO− and CH3S−. Since there is a strong correlation between HNO binding electronic energies (ΔE’s) and Gibbs free energies (ΔG’s) with linear correlation coefficient R2=0.9885 (see Table S2 and Figure S1), the following discussion is based on ΔG’s.
Scheme 1.

Molecular structures of studied HNO complexes of ferrous (1–5) and ferric (6–10) porphyrins.
We first investigated the iron oxidation state effect. As shown in Table 1 and Figure 1, for all axial ligands studied here, HNO always preferably binds with ferrous porphyrins than corresponding ferric ones. It is interesting to note that the HNO binding ΔG’s between ferric and ferrous porphyrins are highly correlated with R2=0.9863, see Figure S2, suggesting a consistent iron oxidation state effect. This effect is the same as in the case of NO binding, [1a] which may not be surprising since the major driving force for HNO and NO binding in metalloporphyrins was recently found to be the same: metal back-donation to the anti-bonding π*NO orbital.[4c, d] However, this is the first time to directly show the iron oxidation state effect on HNO binding and the mostly influenced properties as described below. The weakened binding in ferric hemes are clearly demonstrated by their longer Fe-N(H)O distances (RFeN’s) in Table 1, compared to ferrous systems. Based on this major driving force, the higher oxidation state leads to decreased metal back-donation to HNO, and consequently less negative NO charges (QNO’s), shorter NO bond lengths (RNO’s), and larger NO vibrational frequencies (νNO’s), see Table 1. The strong interrelationships among RNO, QNO, and νNO are shown in Figure 2A and 2B with R2=0.9819 and 0.9881, respectively. Since this major electronic driving force involves mostly Fe to NO back-donation, we can see large geometric changes in RFeN of ~0.04 Å and RNO of ~0.03 Å (Table 1) vs. small changes in NH bond lengths (≤ 0.003 Å) and Fe-NO bond angles (≤ 2.3°) (Table S2) due to the iron oxidation state change.
Table 1.
Binding Energies, Geometric Parameters, Charges, and Vibrational Frequencies of FeII/III(Por)(HNO)(L) [a]
| L | ΔG (kcal/mol) | RFeN (Å) | RNO (Å) | QNO (e) | ΔQL (e) | υNO (cm−1) | |
|---|---|---|---|---|---|---|---|
| FeII | 5-MeIm | −28.44 | 1.805 | 1.246 | −0.226 | 0.093 | 1398 |
| NH2CH3 | −32.40 | 1.804 | 1.246 | −0.232 | 0.095 | 1397 | |
| CH3O− | −21.20 | 1.852 | 1.268 | −0.383 | 0.108 | 1302 | |
| PhO− | −21.24 | 1.823 | 1.261 | −0.333 | 0.075 | 1345 | |
| CH3S− | −23.84 | 1.862 | 1.264 | −0.365 | 0.138 | 1311 | |
| FeIII | 5-MeIm | −17.71 | 1.848 | 1.228 | −0.124 | 0.094 | 1452 |
| NH2CH3 | −22.17 | 1.845 | 1.228 | −0.121 | 0.103 | 1459 | |
| CH3O− | −11.57 | 1.893 | 1.238 | −0.208 | 0.189 | 1419 | |
| PhO− | −11.36 | 1.842 | 1.238 | −0.191 | 0.241 | 1424 | |
| CH3S− | −14.86 | 1.904 | 1.235 | −0.194 | 0.293 | 1424 |
L = axial ligand.
Figure 1.

Binding Gibbs free energies of HNO complexes 1–10. Red and blue data points are for ferric and ferrous complexes, respectively.
Figure 2.

Plots of νNO vs. RNO (A) and νNO vs. QNO (B) for complexes 1–10.
The relatively weak HNO binding in ferric porphyrins offers the first theoretical insight to support a facile thermodynamic driving force for reductive nitrosylation to generate the stable NO bound ferrous porphyrins when HNO meets with ferric heme proteins and models, as observed experimentally with rate constants on the orders of 104~107 M−1 s−1.[1a, 3c, 6] The varied reaction rates show that different proteins modulate the HNO interactions. Since HNO binding is the first and rate-limiting step in such interactions,[1a] it is important to study more details of protein effect in this process.
We next investigated the protein axial ligand effect on HNO binding, since this proximal residue can vary in different heme proteins, and is directly attached to Fe and trans to HNO, which may play a significant role in regulating HNO binding. Indeed, as shown in Figure 1, there is a clear difference between neutral and negatively charged ligands regarding their effects on HNO binding, as highlighted by green and orange ovals respectively. The average HNO binding ΔG’s for negatively charged ligands in both ferrous and ferric porphyrins are ca. 7–8 kcal/mol higher than those for neutral ligands, indicating less stable binding. This is probably a result of the stronger trans effect of negatively charged ligands compared to neutral ligands, as evidenced by their relatively longer Fe-N(H)O distances with an average of ~0.04 Å, see Table 1. It is interesting to note that experimental HNO consumption rates of ferric heme proteins with neutral His axial ligands (e.g. Mb, HRP) are higher than that of catalase with a negatively charged Tyr ligand.[3c] Since HNO binding is the first and rate-limiting step in such reactions,[1a] these experimental results suggest a favourable HNO binding with the neutral protein axial ligand, consistent with the trend revealed here.
Interestingly, axial ligand also has another effect: charge donation to metal center, resulting in positive ligand charge change upon HNO binding (ΔQL), see Table 1. These data show that a negatively charged ligand usually donates more charge than a neutral ligand. This enhances metal to HNO back-donation, resulting in a lengthened NO bond, increased NO charge, and smaller NO vibrational frequency as seen from Table 1. This effect is secondary compared to the above-mentioned trans effect of axial ligands with different formal charges on HNO binding. But it helps understand the mild difference for ligands with the same formal charge: a mildly improved HNO binding for NH2CH3 vs. 5-MeIm among neutral ligands, and for the sulfur ligand vs. the oxygen ligand among negatively charged ligands, due to their slightly stronger charge donation, see ΔQL data in Table 1.
Besides the proximal axial ligand, the distal hydrogen bonding residue was previously found to play an important role on HNO binding.[4b] However, the effects from other active site residues and the whole protein environment on HNO binding have not been reported. Because stable HNO binding was only reported for several ferrous heme proteins with spectroscopic characterizations,[5] we next used monomeric ferrous globins, myoglobin (Mb) and leghemoglobin (lgHb), with experimental 1H and 15N NMR spectroscopic data that are sensitive to differentiate various HNO binding modes,[4b] as the first step to examine these effects.
For Mb, we first studied large active site models with nearby residues that might influence HNO binding (see Supporting Information for details and optimized coordinates). Because both H and O of HNO can participate in hydrogen bonding (HB) and distal His can be either Nδ or Nε protonated, two mono HB modes (A – HNO···His, B – ONH···His) and two dual HB modes (C – H2O···HNO···His, D – OH2···ONH···His) were investigated. The use of water as the second HB partner in dual HB modes was based on our recent small active site model study: [4b] 1) only dual HB modes can reproduce experimental spectroscopic data, 2) the dual HB mode for HNO is actually common in previously reported HNO interactions,[4a] and 3) water is viable in proteins and no other nearby residue in Mb can be the second HB partner. Since Mb and lgHb have same axial ligand and distal His residue, it is important to study the large active site to learn the origin of their experimental NMR shift differences.[5b] As seen from Table 2, mode D (see Figure 3A) consistently yields the best predictions of experimental 1H and 15N NMR chemical shifts for the major MbHNO isomer shown here,[5b] and its slightly larger NMR shifts than those of C is also consistent with the experimental results that this major isomer has slightly larger shifts than a minor isomer discussed previously.[4b] The predicted νNO value also agrees well with experiment.[5c] For lgHb, again mode D gives the best predictions of experimental NMR data and the slightly downfield 1H and 15N NMR shifts in lgHbHNO vs. MbHNO.[5b]
Table 2.
Geometric Parameters and Spectroscopic Properties of HNO bound heme proteins
| [a] | HB Mode | RHis (Å) | Rwater (Å) | δH (ppm) | δN (ppm) | υNO (cm−1) | |
|---|---|---|---|---|---|---|---|
| Mb | Expt [b] | 14.93 | 661 | 1385 | |||
| Small [c] | A | 2.176 | / | 14.93 | 605 | 1377 | |
| B | 2.078 | / | 15.55 | 659 | 1402 | ||
| C | 2.145 | 2.031 | 15.04 | 647 | 1380 | ||
| D | 2.069 | 2.095 | 15.13 | 658 | 1384 | ||
| Large | A | 2.118 | / | 14.86 | 604 | 1374 | |
| B | 2.068 | / | 15.84 | 661 | 1396 | ||
| C | 2.095 | 2.118 | 15.02 | 644 | 1377 | ||
| D | 2.014 | 2.104 | 15.05 | 663 | 1374 | ||
| Whole | A | 2.160 | / | 14.66 | 600 | 1380 | |
| B | 2.055 | / | 15.45 | 657 | 1397 | ||
| C | 2.107 | 2.033 | 15.04 | 642 | 1383 | ||
| D | 1.999 | 1.960 | 15.30 | 662 | 1374 | ||
| LgHb | Expt [d] | 15.00 | 665 | ||||
| Large | A | 2.126 | / | 14.85 | 605 | 1374 | |
| B | 2.049 | / | 15.65 | 662 | 1392 | ||
| C | 2.085 | 2.044 | 14.84 | 649 | 1379 | ||
| D | 2.031 | 2.037 | 15.48 | 666 | 1376 |
Figure 3.

HB mode D structures of (A) large active site of MbHNO; (B) large active site of lgHbHNO; (C) MbHNO whole protein. Atom color scheme: C- cyan, N- blue, O- red, H – grey, Fe - black.
Above results show that the large active site models are helpful to study experimental spectroscopic data in Mb and lgHb. We then used such structures to reveal most significant differences in their HNO binding geometries. As shown in Table S3, the Fe-N(H)O distance, NO and NH bond lengths, and the Fe-N-O bond angle differences in each mode of these two proteins are all negligible (<0.005 Å and <1°). In contrast, HB geometries have much larger differences and the largest changes occur with HNO’s HB with water. As seen from Table 2, from MbHNO to lgHbHNO, the average HNO···water distance (Rwater) decreases as significant as ~0.07 Å, in contrast with ~0.01 Å difference in the average absolute HNO···His distances (RHis’s). This is probably due to the relatively smaller flexibility of distal His vs. water. These data also indicate a more compact active site in lgHbHNO. In fact, this feature is also reflected by the closest distances of nearby residues to HNO’s O atom involved in water HB: 3.03/5.21 Å for Leu65/Val105 in lgHbHNO vs. 4.43/5.69 Å for Leu29/Val68 in MbHNO. In addition, as shown in Figure 3A and 3B, the water’s H atom not involved in HB is pushed away from Leu65 in lgHbHNO compared to the toward Leu29 conformation in MbHNO, due to ~1.4 Å closer Leu in lgHbHNO. These results show that the nearby residues in the large active site models induce the structural differences between these two heme proteins, with changes mostly on HB geometries.
To compare with large active site models, the whole protein models for MbHNO were also investigated (see Supporting Information for computational details with one structure shown in Figure 3C). As shown in Table S3, again the optimized RFeN, RNO, RNH, and <FeNO data are basically not affected with only <0.01 Å and <1° differences. The significant geometric changes are also associated with HNO’s HB distances, with the water side HB again more affected than the distal His side.
In summary, this work provides the first detailed trends and origins of effects of iron oxidation state, axial ligand, and protein environment on HNO binding in heme proteins: 1) ferric hemes have much weaker binding than ferrous systems due to reduced metal back-donation to HNO, offering the first theoretical insight into the strong thermodynamic driving force for experimentally observed reductive nitrosylation to form stable NO bound ferrous porphyrins; 2) axial ligand influences HNO binding through its trans effect and charge donation effect, providing the first information how its formal charge and structural feature affects HNO binding; 3) the protein environment effect of other active site residues and whole protein was also revealed for the first time, with significant changes on HNO’s HB structures and properties, and is responsible for experimental spectral differences in different heme proteins. These novel and broad range of results shall facilitate many researches of HNO binding in heme proteins, models, and related metalloproteins.
Supplementary Material
Acknowledgments
This work was supported by an NIH grant GM085774 to YZ. YZ also thanks Liu Yang for her preliminary study of this project.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Dr. Rahul L. Khade, Department of Biomedical Engineering, Chemistry and Biological Sciences, Stevens Institute of Technology, 1 Castle Point on Hudson Hoboken, NJ 07030 (USA)
Yuwei Yang, Department of Biomedical Engineering, Chemistry and Biological Sciences, Stevens Institute of Technology, 1 Castle Point on Hudson Hoboken, NJ 07030 (USA).
Yelu Shi, Department of Biomedical Engineering, Chemistry and Biological Sciences, Stevens Institute of Technology, 1 Castle Point on Hudson Hoboken, NJ 07030 (USA).
Prof. Dr. Yong Zhang, Email: yong.zhang@stevens.edu, Department of Biomedical Engineering, Chemistry and Biological Sciences, Stevens Institute of Technology, 1 Castle Point on Hudson Hoboken, NJ 07030 (USA)
References
- 1.a) Miranda KM. Coord Chem Rev. 2005;249:433–455. [Google Scholar]; b) Averill BA. Chem Rev. 1996;96:2951–2964. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]; c) Farmer PJ, Sulc F. J Inorg Biochem. 2005;99:166–184. doi: 10.1016/j.jinorgbio.2004.11.005. [DOI] [PubMed] [Google Scholar]; d) Roncaroli F, Videla M, Slep LD, Olabe JA. Coord Chem Rev. 2007;251:1903–1930. [Google Scholar]; e) Flores-Santana W, Switzer C, Ridnour LA, Basudhar D, Mancardi D, Donzelli S, Thomas DD, Miranda KM, Fukuto JM, Wink DA. Arch Pharm Res. 2009;32:1139–1153. doi: 10.1007/s12272-009-1805-x. [DOI] [PubMed] [Google Scholar]; f) Doctorovich F, Bikiel D, Pellerino J, Suarez SA, Larsen A, Marti MA. Coord Chem Rev. 2011;255:2764–2784. [Google Scholar]; g) Doctorovich F, Bikiel DE, Pellegrino J, Suárez SA, Martí MA. Acc Chem Res. 2014;47:2907–2916. doi: 10.1021/ar500153c. [DOI] [PubMed] [Google Scholar]
- 2.Feelisch M. Proc Natl Acad Sci - U S A. 2003;100:4978–4980. doi: 10.1073/pnas.1031571100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Rusche KM, Spiering MM, Marletta MA. Biochemistry. 1998;37:15503–15512. doi: 10.1021/bi9813936. [DOI] [PubMed] [Google Scholar]; b) Huang JM, Sommers EM, Kim-Shapiro DB, King SB. J Am Chem Soc. 2002;124:3473–3480. doi: 10.1021/ja012271v. [DOI] [PubMed] [Google Scholar]; c) Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, Wink DA. Proc Natl Acad Sci U S A. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lehnert N, Praneeth VKK, Paulat F. J Comput Chem. 2006;27:1338–1351. doi: 10.1002/jcc.20400. [DOI] [PubMed] [Google Scholar]
- 4.a) Zhang Y. J Inorg Biochem. 2013;118:191–200. doi: 10.1016/j.jinorgbio.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang L, Ling Y, Zhang Y. J Am Chem Soc. 2011;133:13814–13817. doi: 10.1021/ja204072j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ling Y, Mills C, Weber R, Yang L, Zhang Y. J Am Chem Soc. 2010;132:1583–1591. doi: 10.1021/ja907342s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang L, Fang WH, Zhang Y. Chem Comm. 2012;48:3842–3844. doi: 10.1039/c2cc31016c. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Linder DP, Rodgers KR. Inorg Chem. 2005;44:8259–8264. doi: 10.1021/ic0504745. [DOI] [PubMed] [Google Scholar]; f) Goodrich LE, Lehnert N. J Inorg Biochem. 2013;118:179–186. doi: 10.1016/j.jinorgbio.2012.07.024. [DOI] [PubMed] [Google Scholar]; g) Goodrich LE, Roy S, Alp EE, Zhao J, Hu MY, Lehnert N. Inorg Chem. 2013;52:7766–7780. doi: 10.1021/ic400977h. [DOI] [PubMed] [Google Scholar]; h) Speelman AL, Lehnert N. Acc Chem Res. 2014;47:1106–1116. doi: 10.1021/ar400256u. [DOI] [PubMed] [Google Scholar]; i) Einsle O, Messerschmidt A, Huber R, Kroneck PMH, Neese F. J Am Chem Soc. 2002;124:11737–11745. doi: 10.1021/ja0206487. [DOI] [PubMed] [Google Scholar]; j) Silaghi-Dumitrescu R. Eur J Inorg Chem. 2003:1048–1052. [Google Scholar]
- 5.a) Kumar MR, Fukuto JM, Miranda KM, Farmer PJ. Inorg Chem. 2010;49:6283–6292. doi: 10.1021/ic902319d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kumar MR, Pervitsky D, Chen L, Poulos T, Kundu S, Hargrove MS, Rivera EJ, Diaz A, Colon JL, Farmer PJ. Biochemistry. 2009;48:5018–5025. doi: 10.1021/bi900122r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Immoos CE, Sulc F, Farmer PJ, Czarnecki K, Bocian DF, Levina A, Aitken JB, Armstrong RS, Lay PA. J Am Chem Soc. 2005;127:814–815. doi: 10.1021/ja0433727. [DOI] [PubMed] [Google Scholar]; d) Sulc F, Fleischer E, Farmer PJ, Ma DJ, La Mar GN. J Biol Inorg Chem. 2003;8:348–352. doi: 10.1007/s00775-002-0422-7. [DOI] [PubMed] [Google Scholar]; e) Lin R, Farmer PJ. J Am Chem Soc. 2000;122:2393–2394. [Google Scholar]
- 6.a) Bari SE, Marti MA, Amorebieta VT, Estrin DA, Doctorovich F. J Am Chem Soc. 2003;125:15272–15273. doi: 10.1021/ja036370f. [DOI] [PubMed] [Google Scholar]; b) Marti MA, Bari SE, Estrin DA, Doctorovich F. J Am Chem Soc. 2005;127:4680–4684. doi: 10.1021/ja044632n. [DOI] [PubMed] [Google Scholar]; c) Álvarez L, Suarez SA, Bikiel DE, Reboucas JS, Batini-Haberle I, Martí MA, Doctorovich F. Inorg Chem. 2014;53:7351–7360. doi: 10.1021/ic5007082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.