

Abstract
Background
Li-Fraumeni syndrome (LFS) is an autosomal dominant cancer predisposition syndrome characterized by very high lifetime cancer risk and early age at diagnosis of a wide cancer spectrum. Precise estimates for first and subsequent cancers risk are lacking.
Methods
The NCI’s LFS Study includes families meeting diagnostic criteria for LFS or Li-Fraumeni-like syndrome, and individuals with a germline TP53 mutation, choroid plexus carcinoma, adrenocortical carcinoma, or ≥3 cancers. We estimated cumulative risk and annual hazards for first and second cancer among TP53 mutation carriers (TP53+) using MATLAB.
Results
We evaluated 286 TP53+ individuals from 107 families. Cumulative cancer incidence was 50% by age 31 among TP53+ females and 46 among males, and nearly 100% by age 70 for both. Cancer risk was highest after age 20 for females, mostly due to breast cancer, while among males risk was higher in childhood and later adulthood. Among females, the cumulative incidence by age 70 for breast cancer, soft tissue sarcoma (STS), brain cancer, and osteosarcoma were 54%, 15%, 6%, and 5%, respectively. Among males, the incidence was 22%, 19%, and 11% for STS, brain cancer, and osteosarcoma. 49% of those with one cancer developed at least another cancer after a median of 10 years. Average age-specific risk of developing a second cancer was comparable to that of developing a first cancer.
Conclusions
Cumulative cancer risk in TP53+ individuals was high and varied by gender, age, and cancer type. Additional work, including prospective risk estimates, is needed to better inform personalized risk management.
Keywords: Li-Fraumeni syndrome, TP53, cumulative cancer risk, second cancer, second cancer risk
Introduction
Li-Fraumeni syndrome (LFS, OMIM#151623) is an autosomal dominant cancer predisposition syndrome.1 Clinical diagnostic criteria for the “classic” LFS kindred include a person with sarcoma diagnosed before age 45, with a first-degree relative with any cancer before age 45 and another first- or second-degree relative with a sarcoma at any age or another cancer before age 45.2 The less stringent Li-Fraumeni-like (LFL) criteria expand the proband’s cancer type to include childhood cancers, brain cancers, and adrenal cortical carcinoma (ACC), and change the relatives’ age at diagnosis to <60 years.3, 4
Germline mutations in TP53, the underlying molecular basis of LFS,5, 6 are identified in ~70% of families meeting the classic LFS diagnostic criteria7, 8 and ~40% of families meeting the LFL diagnostic criteria.3 The frequency of de novo mutations in TP53 is estimated to be between 7% and 20%.9 Guidelines for TP53 mutation testing have also been developed.10–15
LFS is characterized by early cancer diagnosis and high lifetime cancer risk, with osteosarcoma, soft-tissue sarcomas (STS), early-onset breast cancer, brain tumors, leukemia, and ACC being the core cancers.2, 16, 17 As more families with TP53 mutations are identified, the LFS cancer spectrum has expanded to include melanoma, lung, gastrointestinal tract, thyroid, ovarian, and other cancers.15, 17–19 Cumulative cancer risk associated with LFS has been estimated to be ~50% by age 40 and up to 90% by age 60,20 with females having higher risk than males.21–23 The risk of developing STS and brain cancer has been observed to be greatest in childhood, while risk of osteosarcoma was highest during adolescence, and female breast cancer risk increased significantly around age 20 and continued into older adulthood.15, 23 However, with more broadly defined diagnostic criteria, more inclusive testing criteria, and the introduction of cancer gene panel testing,24–26 less penetrant LFS kindreds are being identified, which will likely lead to changes in cancer risk estimates.
Individuals with LFS have substantial risk of multiple primary cancers;15, 17, 23, 27 however, risk estimates for subsequent cancer(s) after a first diagnosis are limited. Previous studies suggested that risk of second cancer increased with younger age at first cancer diagnosis23, 27 and some second malignancies were related to previous radiation therapy.28, 29 It is unclear whether the type of first cancer influences second cancer risk that is not associated with therapeutic radiation.
To resolve some of these uncertainties, we examined the cumulative first and second cancer risk among TP53 mutation carriers from families enrolled in the National Cancer Institute (NCI) LFS study.
Methods
Study Participants
The NCI LFS study (NCT01443468, http://lfs.cancer.gov), a long-term prospective cohort study, opened to accrual in August 2011. Eligibility criteria include meeting the diagnostic criteria for classic LFS or Birch’s LFL,3 or having a pathogenic germline TP53 mutation or a first- or second-degree relative with a mutation, or a personal history of choroid plexus carcinoma, ACC, or ≥3 primary cancers.
Written informed consent, including permission to use family information, was obtained from all participants. Parents provided written informed consent for children younger than 18. An assent was signed by children aged 13 to 18. This protocol was approved by the NCI Institutional Review Board.
Family history information
A detailed family history questionnaire (FHQ) was completed by the proband, or another family member with knowledge of the family information. The FHQ provides names, birth date, vital status, date/age of death (if deceased), and cancer history (type and year/age of diagnosis) for all first-, second-, and third degree relatives and any extended family members with available information. Information contributed by other family members was also recorded.
Cancer diagnosis confirmation
We attempted to confirm all cancer diagnoses through evaluation of pathology reports, surgical operative notes, consultation reports, clinic notes, and/or death certificates. Permission to obtain medical records was obtained from proxies of deceased family members.
Mutation status
Copies of TP53 testing reports were obtained for participants tested prior to enrollment. Genetic testing was performed after enrollment for participants not previously tested. Only individuals tested or inferred positive for a TP53 mutation (TP53+) were included in this report.
Statistical analysis
Cumulative cancer risks, overall survival, and cancer-free survival were estimated separately for females and males. For overall survival estimates, individuals were censored at age at last follow-up. For cancer-free survival, individuals were censored at age at death or last follow-up (only one participant died before cancer diagnosis or last follow-up). Several methods were used to handle participants with multiple cancers diagnosed synchronously: in Tables 1 and 2 and Figure 2 and S1, each cancer was counted separately; in all other analyses, the diagnoses were considered as a single event. Competing risks30 for first cancer diagnosis were estimated for breast cancer (females), prostate cancer (males), osteosarcoma, STS, brain cancer, ACC, lung cancer, leukemia, colorectal cancer, and others; competing risks of second cancer or death after first diagnosis were also estimated. Kaplan-Meier and product-limit curves31 and annual hazard curves32, 33 were estimated for each of the major cancer types, and for second cancers. If multiple primaries of the same cancer type were diagnosed at different times, the age at the first diagnosis was used in the cumulative risk estimates. In several analyses we aimed to compare males and females more directly by removing sex-specific cancers from consideration; for these analyses, individuals were included, but breast and prostate cancer diagnoses were ignored as events. We also constructed landmark plots for participants in age ranges 0–17, 18–29, 30–44, and 45+. The landmark plots present survival curves beginning at the start of each age interval. P-values for differences in survival curves were calculated using score tests from corresponding Cox proportional hazards models. Analyses were carried out using MATLAB (MATLAB: The Language of Technical Computing. Version R2014B. Natick, MA: The MathWorks, Inc.; 2014) and R34 version 3.2.3 with the survival package.35
Table 1.
Number and type of first cancer by age group at diagnosis
| 0–17 | 18–29 | 30–44 | 45+ | Total (Females/Males) | |
|---|---|---|---|---|---|
| ACC | 5 | 0 | 0 | 0 | 5 (3/2) |
| Brain | 10 | 8 | 2 | 3 | 23 (9/14) |
| Breast | 0 | 24 | 42 | 6 | 76 (76/0) |
| Colorectal | 0 | 2 | 3 | 4 | 9 (5/4) |
| Leukemia | 3 | 2 | 0 | 0 | 5 (4/1) |
| Lung | 0 | 0 | 0 | 4 | 4 (2/2) |
| OS | 11 | 6 | 1 | 0 | 18 (9/9) |
| Prostate | 0 | 0 | 0 | 2 | 2 (–/2) |
| STS | 12 | 10 | 9 | 8 | 41 (25/16) |
| Other | 2 | 7 | 6 | 11 | 28 (14/14) |
| Total | 43 | 59 | 63 | 38 | 211 (147/64) |
| Persons with 1st cancer diagnosis* | 41 | 57 | 59 | 33 | 193 (132/61) |
| Persons at risk | 286 | 207 | 128 | 42 | 286 (186/100) |
| Person-years | 4390 | 2053 | 1236 | 383 | 8062 (5114/2948) |
Some subjects had >1 cancer diagnosed synchronously.
ACC: adrenal cortical carcinoma; OS: osteosarcoma, STS: soft-tissue sarcoma
Other: Non-melanoma skin cancer (9), melanoma (3), non-Hodgkin’s lymphoma (3), kidney cancer (3), ovarian cancer (2), germ-cell tumor (2), liver cancer (1), neuroblastoma (1), thyroid cancer (1), tongue squamous cell cancer (1), gastric cancer (1), and carcinoid (1).
Table 2.
Number and type of second cancer diagnoses
| Females | Males | Total | |
|---|---|---|---|
| ACC | 1 | 0 | 1 |
| Brain | 3 | 6 | 9 |
| Breast | 42 | 0 | 42 |
| Colorectal | 0 | 2 | 2 |
| Leukemia | 1 | 1 | 2 |
| Lung | 9 | 0 | 9 |
| OS | 3 | 0 | 3 |
| Prostate | - | 4 | 4 |
| STS | 13 | 3 | 16 |
| Other | 9 | 13 | 22 |
| Total number of cancer diagnoses | 81 | 29 | 110 |
| Number of persons with 2nd cancer diagnosis | 69 | 26 | 95 |
| Persons at risk | 130* | 61 | 191 |
| Person-years | 851 | 544 | 1395 |
2 females with missing date of last follow-up were not included in the population at risk for second cancer
ACC: adrenal cortical carcinoma; OS: osteosarcoma, STS: soft-tissue sarcoma
Other: Non-melanoma skin cancer (7), melanoma (6), kidney cancer (2), non-Hodgkin’s lymphoma (1), ovarian cancer (1), thyroid (1), esophageal cancer (1), bladder cancer (1), Paget’s disease (1), and pancreatic cancer (1).
Figure 2.

Cumulative incidence of first cancer diagnosis among TP53+ females (A) and TP53+ males (B)
Results
A total of 107 families with germline TP53 mutations were included in this report. At enrollment, 46 (43%) families met LFS criteria, 41 (38%) met LFL criteria, 9 (8%) were individuals with ≥3 primary cancers and no significant family history, and 11 (10%) who tested positive for a TP53 mutation, without meeting any of the current diagnostic criteria or testing guidelines. 296 of the 1269 bloodline family members were TP53+ (263 tested negative for the familial mutation and 710 have not been tested), of whom ten were excluded for missing date of birth. For the 286 TP53+ individuals, 189 (63.8%) were females and 107 (36.2%) were males, with a median age at death or last follow-up of 35 (range 0–91 years).
Cancer Diagnosis Confirmation
A total of 403 cancer diagnoses were reported among the mutation carriers, of which 211 were the first primary cancers. 53% of the first primary cancers were confirmed by medical records (pathology reports, surgical operative notes, consultation reports, clinic notes, and/or death certificates). 15% pf the cancer diagnoses were self-reported and 32% were reported by family members. The rates of confirmation were 59% for second cancer diagnosis and 60% for subsequent cancer diagnoses (Supplemental Table1).
First cancer diagnosis and cumulative risk
Among the 286 TP53+ individuals, 193 had been diagnosed with at least one cancer. The number and type of first cancer by age-group at diagnosis are shown in Table 1. Among those younger than 18, STS was the most common diagnosis, followed by osteosarcoma and brain cancer. Among those between 18 and 44, breast cancer was the most common diagnosis, while that in the 45+ age group was STS. All five first cancer diagnoses of ACC occurred before age 18.
Cumulative cancer incidence was 50% by age 31 (95% CI 29–35) among TP53+ females and age 46 (95% CI 39–51) among TP53+ males, and approached 100% by age 70 for both groups. TP53+ males had higher risk of a first cancer diagnosis before age 25 and after age 50. In contrast, the risk of a first cancer was highest from age 20 to 50 for TP53+ females (Figure 1), with breast cancer being the most common first cancer diagnosis (Figure 2A). Among males, STS, osteosarcoma, and brain cancer were more commonly the first cancer diagnosed (Figure 2B).
Figure 1.
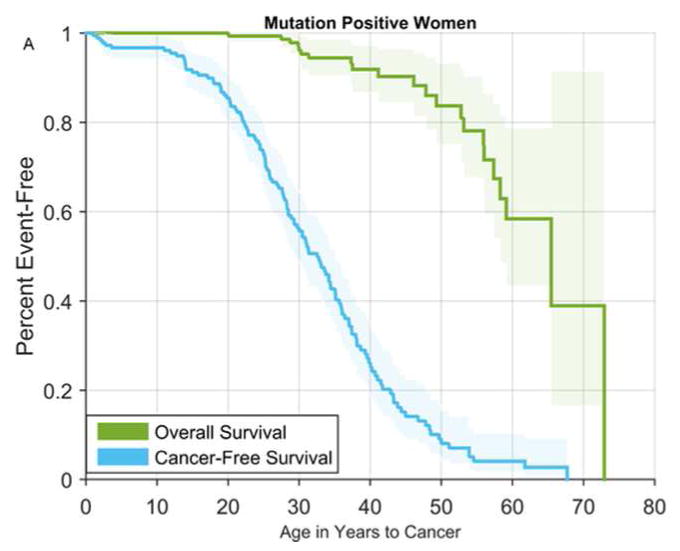

Overall and cancer-free survival among TP53+ females (A) and TP53+ males (B)
Specific cancer types diagnosed at any time
A total of 403 cancers were diagnosed among the 193 cancer-affected TP53+ individuals, with 95 (49%) having >1 cancer. Amongst the 403 cancer diagnoses, breast cancer was the most common, followed by STS, brain cancer, and osteosarcoma (Supplemental Figure S1). The cancer-specific estimate for cancer-free survival up to age 70 was ~15% for breast cancer for females; ~30% for females and ~60% for males for STS, ~80% for females and ~70% for males for brain cancer, and ~90% for both females and males for osteosarcoma (Figure 3).
Figure 3.

Overall estimated product-limit and hazard curves for brain cancer, STS, osteosarcoma, and breast cancer (Note: Hazard curve for breast cancer is on a different scale). STS: soft-tissue sarcoma; OS: osteosarcoma
The annual hazard varied by age, and was different by cancer type and gender. For female breast cancer, the annual hazard started to increase in the late teens and peaked at about age 40. The annual hazard for STS in males and females fluctuated throughout life, but increased significantly after age 40, especially for males. The variation in annual hazard followed a different pattern for brain cancer, with higher hazard in infancy and after age 50 for males and peaked in the second decade of life for females. The hazard for osteosarcoma was higher before age 10 for both genders (Figure 3).
Second primary cancers
Among the 193 mutation carriers with >1 cancer diagnosis, two did not have follow-up information after the first diagnosis and were excluded in the population at risk for second cancer. Ninety-five (50%) of the 191 individuals included developed a second cancer. Table 2 shows the number and type of the second diagnoses (See Supplemental Tables S2 for the number of cancers in each age-group at second cancer diagnosis by age-group at first cancer diagnosis, and Supplemental Table S3 and S4 for the number of second cancers by type of first cancer diagnosis). Overall, breast cancer was the most common second cancer diagnosis, followed by STS and brain and lung cancer (Table 2). All nine lung cancers were diagnosed in females, five of whom had a previous history of breast cancer. Cumulative second cancer risk was ~50% 10 years after the first diagnosis for both males and females, and was higher for females after 10 years (p=0.005); however, when breast and prostate cancers as either first or second malignancy were excluded, the cumulative risk was similar for both genders (p=0.9) (Figure 4A and 4D). Similarly, when all cancers were considered, age at diagnosis was younger and the annual hazard of developing a second cancer between 6 and 55 years after the first cancer diagnosis was higher for females, but this difference was no longer present when breast and prostate cancers were excluded (Figure 4B–C, E–F). There was no significant difference in the annual hazard of second cancer by type of first cancer diagnosis (data not shown). Furthermore, compared with males, females had higher risk of second cancer risk, but lower risk of death. The low risk of death among females was observed for all age-at-first-diagnosis groups (Supplemental Figure S2).
Figure 4.

Estimated Kaplan-Meier curve of time from first cancer to second cancer, product-limit for age at second cancer, and hazard curves for age at second cancer. All cancers included (A–C) and with breast and prostate cancer excluded (D–F). BC: breast cancer; PC: prostate cancer
The interval between the first and second cancer diagnosis varied depending on the age at first diagnosis and by gender. Among females, with all cancers included, the median time to second cancer diagnosis was 15 years for those with the first diagnosis <18 years and 10 years for the other age groups (p=0.4). When breast cancer was excluded, the median time to second cancer was longer for the younger age groups (p=0.004). Among males, those with the first cancer diagnosis in the <18 and 18–29 age groups had a longer median time to second cancer compared with the older age groups (p=0.004, Figure 5).
Figure 5.

Kaplan-Meier Curves of time from first cancer to second cancer, stratified by age at first cancer diagnosis; A: Females, B: Females, with breast cancer excluded, C: Males
Annual hazards of developing first and second cancer
The annual hazard for developing the first cancer was different for males and females, even after excluding breast and prostate cancer diagnoses. For females, the hazard increased throughout life, while for males, it was higher before age 10, remained low between the ages of 10 and 30, and then increased from age 30 to 60 (Supplemental Figure S3). Among both females and males, there were no differences in the annual hazard of having a first or second cancer for all cancers, and with breast and prostate cancer excluded.
When examining by specific cancer type, the annual hazard for STS was similar whether it was diagnosed as a first or second diagnosis (p=0.3), whereas the hazard for breast cancer was higher as a second versus a first cancer (p=0.01). The numbers precluded a formal analysis for other cancer types (Supplemental Figure S4).
The landmark plots (Supplemental Figure S5) show the percent cancer-free (first and second diagnosis) over age-groups 0–17, 18–29, 30–44, and 45+. In most cases, second cancer risk (conditional on participants already having a first cancer diagnosis) is somewhat higher than first cancer risk, particularly between age 30 and 44. These landmark plots agree with the hazard curves in Figure S3, but provide an alternate visual summary of hazard within specific time intervals.
Discussions
Our report provides one of the most detailed assessments of first and second cancer risks in LFS to date. We used competing risks methodology to examine cancer-specific cumulative incidence. In addition, we estimated age-specific hazard rates of first and second cancers. These graphs quantify cancer risks by age among persons with LFS who are alive and first or second cancer-free, which can be applied clinically in individualized risk discussions. Our estimates of second cancer risk are particularly relevant, given the currently limited data available.
Although the “classic” LFS-related cancers remained the more frequently diagnosed cancers among TP53+ individuals, the cancer spectrum in our study was quite broad. Similar to previous studies,15–19 we observed high cumulative cancer risk, with females having higher risk than males, mainly due to early-onset breast cancer. Unlike some previously published data,23 we did not observe a persistent difference in cumulative cancer risk between females and males after excluding breast and prostate cancer. This could be due to differences in the study populations. We also confirmed the recent observation that, among children, brain cancer, osteosarcoma, STS, and ACC were the most frequent diagnoses; while breast cancer and STS were the more common diagnoses among adults.15 Our estimates of the cancer-specific cumulative risk showed that they differed by gender. In addition, the annual hazards for the LFS core cancers differed from one another and varied by age. These observations provide a better understanding of the risk level overall and for specific cancers at a given age.
Among TP53+ females, breast cancer is by far the most common malignancy. Our findings showed that breast cancer risk increased significantly after the second decade, thus supporting the recommendation to begin breast cancer screening at age 20.14 Moreover, the cumulative breast cancer incidence was approximately 85% by age 60, a risk level comparable to that seen in females with germline mutations in BRCA1 and BRCA2. This information might help in the discussion regarding consideration of risk-reducing mastectomy as a risk management option.14 In addition to breast cancer risk, we also estimated cumulative risks and annual hazard for other cancer types, and showed that there were variations in risk based on gender and age. However, due to the relatively small number for the specific cancer cases and the retrospective nature of this study, additional research, with risk estimates based on prospective follow-up, is needed before personalized recommendations for targeted screening can be made.
The risk of subsequent malignancies after the first cancer diagnosis was ~50% and occurred from 0 to 49 years after the first diagnosis. The annual hazard for a second cancer was similar to that of the first cancer; thus, having had a cancer did not substantially alter the risk of developing cancer. However, we were not able to determine whether any of the second cancer diagnoses were related to treatment received for the first diagnosis. We did not observe any pattern of second cancer risk based on the first cancer type. These findings confirm that it is prudent to consider cancer screening for survivors with good prognosis from the previous diagnosis.
We also provided detailed percent cancer-free, both overall and by various age groups. Although these plots present estimates based on retrospectively collected data, this information may be useful in estimating future cancer risk for an individual based on their current age and cancer history. For example, a 25 year-old woman with no cancer history would have a different 5-year risk estimate than a 40-year old man with a previous cancer diagnosis.
This study has several strengths. Our cohort represents a large set of families with information on all family members systematically collected. We also attempted to confirm all reported cancer diagnoses with medical records and/or death certificates. In addition, the number of cancer diagnoses in this cohort was large enough to permit an exploration of cancer-specific risk for the more common LFS cancers as well as risk for subsequent cancer diagnosis.
Our study may be limited by referral/selection bias since the families enrolled might have been more readily identified due to an excess of cancer diagnoses among family members. While this bias is inevitable, there were several TP53+ families in our cohort that did not meet the classic LFS or LFL diagnostic criteria. Notably, several TP53+ participants were identified based on cancer gene panel testing, with no family history suggestive of LFS. Thus, our cohort might be likely to represent the LFS population seen in the clinic setting and encompass the true spectrum of cancer penetrance. Another limitation was that we only included in this analysis family members known to be TP53+. The mutation-unknown family members were not tested either because they were from older generations or died from a cancer-related cause before the mutation was identified in the family, or were alive, but chose not to be tested. Similarly, the overall survival estimates might be inflated because those with longer survival were more likely to be available for testing. Additional analyses of cancer penetrance using all family members and taking into account family structure will be important. Furthermore, we conducted a sensitivity analysis to examine the effect of family clustering on our analyses and found it to be negligible under a Cox Proportional Hazards model with frailty (data not shown). Finally, our findings are based on data collected retrospectively and some diagnoses reported by relatives could not be confirmed. Likewise, we only have limited data on previous treatments, and thus not able to ascertain whether any of the subsequent cancer diagnoses were treatment-related. With longer follow-up of the cohort, we will be able to prospectively estimate cancer risk and penetrance, taking into account treatment received and other potential risk modifiers.
Using data collected from this large cohort, we examined the risk of first and subsequent cancer, as well as risk of selected specific cancer types. These results will contribute to more accurate cancer risk estimates for individuals with LFS and help strengthen cancer risk management guidelines.
Supplementary Material
Figure S1: Distribution of all cancer diagnoses among TP53 mutation carriers. ACC: Adrenal cortical carcinoma. Other: Non-melanoma skin cancer (29), kidney cancer (7), pancreatic cancer (5), thyroid cancer (5), non-Hodgkin’s lymphoma (5), esophageal cancer (3), ovarian cancer (3), germ-cell tumor (2), liver cancer (1), neuroblastoma (1), tongue squamous cell cancer (1), gastric cancer (1), Paget’s disease (1), bladder cancer (1), thymoma (1), parotid tumor (1), neuroendocrine tumor (1), carcinoid (1), and cancer of unknown primary (1).
Figure S2: Competing risk for second cancer and death overall (A) and by age groups for females (B) and males (C)
Figure S3: Annual hazard of developing first and second cancer. A: Females, all cancers included; B: Females, breast cancer excluded; C: Males, all cancers included; D: Males, breast and prostate cancer excluded
Figure S4: Annual hazard curves for age at breast cancer (A) and STS (B) as a first cancer and second cancer. STS: soft-tissue sarcoma
Figure S5: Landmark product-limit plots for 1st and 2nd cancer for age groups 0–17, 18–29, 30–44, 45+; Panel A: Females; Panel B: Males
Acknowledgments
Funding: This work was supported by funding from the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health.
Footnotes
The authors report no conflict of interest
Author Contributions:
Phuong L. Mai: conception and design; data acquisition, analysis and interpretation; drafting of manuscript; final approval
Ana F. Best: analysis and interpretation of data; reviewing and editing of manuscript; final approval
June A. Peters: conception and design; data acquisition; reviewing and editing of manuscript; final approval
Rosamma DeCastro: conception and design; data acquisition; reviewing and editing of manuscript; final approval
Payal P. Khincha: conception and design; data acquisition; reviewing and editing of manuscript; final approval
Jennifer T. Loud: conception and design; data acquisition; reviewing and editing of manuscript; final approval
Renée C. Bremer: conception and design; data acquisition; reviewing and editing of manuscript; final approval
Philip S. Rosenberg: conception and design; data analysis and interpretation; reviewing and editing of manuscript; final approval
Sharon A. Savage: conception and design; data acquisition, analysis and interpretation; reviewing and editing of manuscript; final approval
References
- 1.Li FP, Fraumeni JFJ. Soft-tissue sarcomas, breast cancer, and other neoplasms: A familial syndrome? Ann Intern Med. 1969;71:747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 2.Li FP, Fraumeni JF, Jr, Mulvihill JJ, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362. [PubMed] [Google Scholar]
- 3.Birch JM, Hartley AL, Tricker KJ, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994;54:1298–1304. [PubMed] [Google Scholar]
- 4.Eeles RA. Germline mutations in the TP53 gene. Cancer Surv. 1995;25:101–124. [PubMed] [Google Scholar]
- 5.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990;348:747–749. doi: 10.1038/348747a0. [DOI] [PubMed] [Google Scholar]
- 7.Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 2003;21:313–320. doi: 10.1002/humu.10185. [DOI] [PubMed] [Google Scholar]
- 8.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez KD, Buzin CH, Noltner KA, et al. High frequency of de novo mutations in Li–Fraumeni syndrome. J Med Genet. 2009;46:689–693. doi: 10.1136/jmg.2008.058958. [DOI] [PubMed] [Google Scholar]
- 10.Chompret A, Abel A, Stoppa-Lyonnet D, et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet. 2001;38:43–47. doi: 10.1136/jmg.38.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tinat J, Bougeard G, Baert-Desurmont S, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol. 2009;27:e108–109. doi: 10.1200/JCO.2009.22.7967. author reply e110. [DOI] [PubMed] [Google Scholar]
- 12.Bougeard G, Sesboue R, Baert-Desurmont S, et al. Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet. 2008;45:535–538. doi: 10.1136/jmg.2008.057570. [DOI] [PubMed] [Google Scholar]
- 13.McCuaig JM, Armel SR, Novokmet A, et al. Routine TP53 testing for breast cancer under age 30: ready for prime time? Fam Cancer. 2012;11:607–613. doi: 10.1007/s10689-012-9557-z. [DOI] [PubMed] [Google Scholar]
- 14.National Comprehensive Cancer Network (NCCN) Genetic/familial high-risk assessment: breast and ovarian. doi: 10.6004/jnccn.2016.0018. Version 2.2015. Available from: http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. [DOI] [PubMed]
- 15.Bougeard G, Renaux-Petel M, Flaman J-M, et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol. 2015;33:2345–2352. doi: 10.1200/JCO.2014.59.5728. [DOI] [PubMed] [Google Scholar]
- 16.Olivier M, Goldgar DE, Sodha N, et al. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643–6650. [PubMed] [Google Scholar]
- 17.Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- 18.Nichols KE, Malkin D, Garber JE, Fraumeni JF, Li FP. Germline p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol Biomarkers Prev. 2001;10:83–87. [PubMed] [Google Scholar]
- 19.Ruijs MW, Verhoef S, Rookus MA, et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47:421–428. doi: 10.1136/jmg.2009.073429. [DOI] [PubMed] [Google Scholar]
- 20.Lustbader ED, Williams WR, Bondy ML, Strom S, Strong LC. Segregation analysis of cancer in families of childhood soft-tissue-sarcoma patients. Am J Hum Genet. 1992;51:344–356. [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C-C, Shete S, Amos CI, Strong LC. Joint effects of germ-line p53 mutation and sex on cancer risk in Li-Fraumeni Syndrome. Cancer Res. 2006;66:8287–8292. doi: 10.1158/0008-5472.CAN-05-4247. [DOI] [PubMed] [Google Scholar]
- 22.Fang S, Krahe R, Bachinski LL, Zhang B, Amos CI, Strong LC. Sex-specific effect of the TP53 PIN3 polymorphism on cancer risk in a cohort study of TP53 germline mutation carriers. Hum Genet. 2011;130:789–794. doi: 10.1007/s00439-011-1039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwang SJ, Lozano G, Amos CI, Strong LC. Germline p53 mutations in a cohort with childhood sarcoma: sex differences in cancer risk. Am J Hum Genet. 2003;72:975–983. doi: 10.1086/374567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Domchek SM, Bradbury A, Garber JE, Offit K, Robson ME. Multiplex genetic testing for cancer susceptibility: Out on the high wire without a net? J Clin Oncol. 2013;31:1267–1270. doi: 10.1200/JCO.2012.46.9403. [DOI] [PubMed] [Google Scholar]
- 25.Doherty J, Bonadies DC, Matloff ET. Testing for hereditary breast cancer: Panel or targeted testing? Experience from a clinical cancer genetics practice. J Genet Couns. 2015;24:683–687. doi: 10.1007/s10897-014-9796-2. [DOI] [PubMed] [Google Scholar]
- 26.Hall MJ, Forman AD, Pilarski R, Wiesner G, Giri VN. Gene panel testing for inherited cancer risk. J Natl Compr Canc Netw. 2014;12:1339–1346. doi: 10.6004/jnccn.2014.0128. [DOI] [PubMed] [Google Scholar]
- 27.Hisada M, Garber JE, Fung CY, Fraumeni JF, Jr, Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90:606–611. doi: 10.1093/jnci/90.8.606. [DOI] [PubMed] [Google Scholar]
- 28.Evans DG, Birch JM, Ramsden RT, Sharif S, Baser ME. Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. J Med Genet. 2006;43:289–294. doi: 10.1136/jmg.2005.036319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salmon A, Amikam D, Sodha N, et al. Rapid development of post-radiotherapy sarcoma and breast cancer in a patient with a novel germline ‘de-novo’ TP53 mutation. Clin Oncol. 2007;19:490–493. doi: 10.1016/j.clon.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Gaynor JJ, Feuer EJ, Tan CC, et al. On the use of cause-specific failure and conditional failure probabilities: Examples from clinical oncology data. J Am Stat Associat. 1993;88:400–409. [Google Scholar]
- 31.Wang M-C, Jewell NP, Tsai W-Y. Asymptotic properties of the product limit estimate under random truncation. Ann Stat. 1986;14:1597–1605. [Google Scholar]
- 32.Rosenberg PS. Hazard function estimation using B-splines. Biometrics. 1995;51:874–887. [PubMed] [Google Scholar]
- 33.Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003;101:822–826. doi: 10.1182/blood-2002-05-1498. [DOI] [PubMed] [Google Scholar]
- 34.R Core Team. R: A language and environment for statistical computing. 2015 Available from: https://www.R-project.org/
- 35.Therneau T. A Package for Survival Analysis in S. 2015 Version 2.38. Available from http://CRAN.R-project.org/package=survival.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Distribution of all cancer diagnoses among TP53 mutation carriers. ACC: Adrenal cortical carcinoma. Other: Non-melanoma skin cancer (29), kidney cancer (7), pancreatic cancer (5), thyroid cancer (5), non-Hodgkin’s lymphoma (5), esophageal cancer (3), ovarian cancer (3), germ-cell tumor (2), liver cancer (1), neuroblastoma (1), tongue squamous cell cancer (1), gastric cancer (1), Paget’s disease (1), bladder cancer (1), thymoma (1), parotid tumor (1), neuroendocrine tumor (1), carcinoid (1), and cancer of unknown primary (1).
Figure S2: Competing risk for second cancer and death overall (A) and by age groups for females (B) and males (C)
Figure S3: Annual hazard of developing first and second cancer. A: Females, all cancers included; B: Females, breast cancer excluded; C: Males, all cancers included; D: Males, breast and prostate cancer excluded
Figure S4: Annual hazard curves for age at breast cancer (A) and STS (B) as a first cancer and second cancer. STS: soft-tissue sarcoma
Figure S5: Landmark product-limit plots for 1st and 2nd cancer for age groups 0–17, 18–29, 30–44, 45+; Panel A: Females; Panel B: Males