

Abstract
Maternal vascular adaptation to pregnancy is critically important in order to expand the capacity for blood flow through the uteroplacental unit to meet the needs of the developing fetus. Failure of the maternal vasculature to properly adapt can result in hypertensive disorders of pregnancy such as preeclampsia (PE). Herein, we review the endocrinology of maternal adaptation to pregnancy and contrast this with that of PE. Our focus is specifically on those hormones with directly impact endothelial cell function and dysfunction, as endothelial cell dysfunction is a hallmark of PE. A variety of growth factors and cytokines are present in normal vascular adaptation to pregnancy but have also been shown to be circulating at abnormal levels in PE pregnancies. Many of these factors promote endothelial dysfunction when present at abnormal levels by acutely inhibiting key Ca2+ signaling events and chronically promoting the breakdown of endothelial cell-cell contacts. Increasingly, our understanding of how the contributions of the placenta, immune cells, and the endothelium itself promote the endocrine milieu of PE is becoming more clear. We then describe in detail how the complex endocrine environment of PE effects endothelial cell function, why this has contributed to the difficulty in fully understanding and treating this disorder, and how a focus on signaling convergence points of many hormones may be a more successful treatment strategy.
1. Introduction
Preeclampsia (PE), a hypertensive disorder of pregnancy, has been described for centuries and the physiology studied intensively for decades. Still, effective therapies have proven elusive. One barrier to our understanding of this disease of pregnancy is our incomplete understanding of what constitutes a healthy pregnancy. Drastic anatomic and physiologic changes occur in the mother, including substantial redistribution of blood flow to meet the needs of the developing fetus. In basic terms, we can describe the gross changes which permit a healthy maternal vasculature in pregnancy, but our understanding is more limited in terms of how the body coordinates the necessary adaptive processes from the molecular level to the organ/organ system level. A growing body of evidence points to disruption of these incompletely understood processes (s) as a potential mediators of the symptoms of PE. More specifically, while many studies point to the vascular endothelium as a tissue that is profoundly changed (develops enhanced or ‘adapted’ function) by the environment of healthy pregnancies, it can also become dysfunctional in pregnancies which develop preeclampsia. In this review, we attempt to describe the critical endothelial changes necessary to provide a healthy pregnancy. We will begin with a narrow focus on the endocrine regulation of healthy pregnancy, so that we will then be able to highlight the endocrine disruption which has more recently been described in PE pregnancies. We will finish by reviewing mechanisms by which this altered endocrinology of PE may promote physiologic changes underlying the clinical symptoms most commonly manifest in PE pregnancies, and how that may be targeted for novel therapy.
2. Physiologic and Anatomic Adaptation of Vasculature to Pregnancy
2.1 Implantation and Early Vascular Remodeling
While it could certainly be argued that hormonal changes associated with the menstrual cycle prepare the uterus for any impending pregnancy, maternal recognition of pregnancy and corresponding early vascular adaptation only begins at embryo implantation into the decidua. This occurs shortly after the embryo descends into the uterus from oviduct. Soon after implantation, cytotrophoblast invasion into the endometrium initiates the early stages of placentation (Page et al. 1972). Syncitiotrophoblast migration then begins remodeling of decidual uterine spiral arteries in the first trimester, forming sinuses which will eventually become placental villi. In the second trimester, myometrial spiral arteries are remodeled from high-resistance, coiled vessels to dilated low-resistance vessels (Robertson et al. 1975). Transformation of the myometrial spiral arteries greatly increases and indeed slows blood flow to the intervillous space of the developing placenta, facilitating exchange of gas and nutrients with the growing fetal circulation while protecting the fetal vessels themselves.
2.2 Physiologic Adaptation to Pregnancy
Beyond the placenta, pregnancy also has profound effects on uterine vascular and, to a lesser degree, systemic vascular physiology. With the hormonal changes of pregnancy comes an increase in maternal blood volume and increased cardiac output, which is normally matched or even slightly exceeded by a drop in vascular resistance. Maternal blood volume begins to rise early in pregnancy (6–8 weeks gestation), until reaching a maximum as much as 50% greater than during the non-pregnant state. Cardiac output increases with the rise in heart rate and stroke volume, totaling a 30–50% increase during pregnancy. The majority of the gestational change in cardiac output occurs early in pregnancy, typically in the first 8 weeks of gestation (Clark et al. 1989). The reason a gestational increase in blood volume and cardiac output does not increase blood pressure, is that there is a general decrease in vascular resistance, and while a drop in vascular resistance occurs throughout the body, there is a disproportionately large drop in vascular resistance in the uterine circulation. The outcome is a greater proportion of the cardiac output goes to the uteroplacental unit to meet the needs of the growing fetus. Estimates of the dramatic changes in uterine blood flow during pregnancy range from 30 to 50 fold increases compared to the non-pregnant state (reviewed in (Bird et al. 2003; Sladek et al. 1997)). The drop in vascular resistance is often primarily attributed to remodeling of the spiral arterioles mentioned above, but further drops in uterine vascular resistance are also achieved through mechanisms of uterine vascular remodeling and sustained vasodilation. While many researchers focus on the importance of changes in the smaller ‘resistance vessels’, it should also be noted in the face of a 30–50 fold increase in flow, this control point is only maintained because distributing vessels show a considerable increase in diameter and changes in both tone and increased vasodilatory capacity.
2.3 Anatomic Adaptation to Pregnancy
Angiogenesis is by definition the growth of new vessels from preexisting vessels. Most vasculature in healthy adult tissues is rather quiescent in this respect, with maintenance of existing vasculature being far more common than creation of new vessels. Uterine vessel growth throughout the menstrual cycle and during pregnancy are well-documented exceptions. Many suggest angiogenesis occurs as the result of an overall net abundance of pro-angiogenic signals over that of anti-angiogenic signals. In tumor angiogenesis, which dominates the literature, these signals are generally thought to consist of growth factors and cytokines. While these signals are also at the heart of angiogenesis in the gravid uterus, there are also those who believe that the hormones of pregnancy (namely, human chorionic gonadotropin, estradiol, and progesterone) can assist in tipping the balance towards angiogenesis (Fraser and Lunn 2000; Pepper 1997), in addition to the locally elevated growth factors and cytokines present at the site of implantation and placentation. Further physiological adaptation of the uterine vasculature includes outward hypertrophy and vessel lengthening (Osol and Mandala 2009; Palmer et al. 1992). Outward hypertrophy refers to increased vessel diameter, typically through vascular smooth muscle cell hypertrophy, which is likely accompanied by endothelial hyperplasia (endothelial hypertrophy is also possible, although it is yet to be studied in human pregnancy) to cover the increased surface area of the outwardly growing vessel. The maternal uterine arteries are also thought to elongate, at least as reported in animal studies (reviewed in (Osol and Mandala 2009)). The net result of outward hypertrophy (decreased resistance), vessel lengthening (increased resistance), and angiogenesis (decreased resistance) during pregnancy is an increase of overall vessel cross-sectional area, and therefore a drop in vascular resistance in the tissue. It is this local drop in vascular resistance that preferentially shunts blood to the uterus and thus to the placenta, ensuring adequate gas, nutrient, and waste exchange in a healthy pregnancy. In the ewe, where it has been studied most thoroughly, uterine artery angiogenesis primarily occurs in the early stages of pregnancy, as the structures created will be necessary to support the greater amount of uterine blood flow demanded by the fetus in late gestation (Bird et al. 2003; Magness and Zheng 1996). By late gestation (3rd trimester) the vessel architecture of the uterus is essentially developed and any further increases in blood flow are now dependent on a sustained vasodilation of those existing vessels. Clinically, it is the mid to late gestation period when most cases of PE present themselves to physicians, and PE subjects are known to show reduced capacity for vasodilation (Kenny et al. 2002; Krupp et al. 2013; Yamamoto et al. 2010).
2.4 Vasodilation in Pregnancy
Throughout pregnancy, but especially from mid gestation through parturition, adequate uteroplacental blood flow is highly dependent on vasodilation. It is clear that pregnancy adaptive increases in endothelial cell vasodilator production are necessary to maintain a healthy pregnancy. Indeed, to prevent hypertension, increased vasodilator production also occurs throughout the systemic circulation (Poston et al. 1995; Suzuki et al. 2002) and a failure of systemic vascular endothelial function has even been reported in hand vein endothelial cells of PE subjects (Mahdy et al. 1998). The vascular endothelium of the uterine arteries can increase their capacity to produce vasodilators to multiple agonists by increasing the expression of key mediators of vasodilator production. Concomitantly, the endothelium also reorganize the post receptor signaling in such a way that they can achieve greater and more sustained signaling responses to vasodilator-stimulating signals (i.e. hormones, shear stress, and mechanical stress) in order to activate in turn greater and more sustained vasodilator synthesis (Bird et al. 2003). Of particular note, a common activating mechanism for many vasodilators is elevation of cellular [Ca2+]i, and one important way [Ca2+]i responses can be amplified and sustained is through promoting intercellular signaling through gap junctions. Once more coordinated and synchronous signaling events are achieved, there is not only an increase in vasodilator output per cell but also a recruitment of more cells to respond to external stimuli. More specifically, periodic, transient Ca2+ burst events have been shown in endothelial cells in response to vasodilatory stimuli, and to be highly dependent on gap junction coupling through the connexin 43 (Cx43) isoform (Yi et al. 2010). If Cx43 function is blocked, sustained Ca2+ burst responses are lost, and increased vasodilator production due to pregnancy adaptation returns to non-pregnant levels. The following sections will now discuss how common vasodilators are regulated in pregnancy to maintain adequate uterine blood flow to the placenta.
2.4.1 Nitric Oxide
Nitric oxide (NO) production in the endothelium is catalyzed by the enzyme endothelial nitric oxide synthase (eNOS) to produce NO and L-citrulline from L-arginine, O2, and NADPH. The role of NO in regulating vascular tone is as a soluble gas which diffuses from the endothelium to the vascular smooth muscle, promoting cGMP production which ultimately results in smooth muscle relaxation. Phosphorylation events at multiple positions on eNOS, along with elevated [Ca2+]i levels largely determine endothelial NO output. Tran et al investigated the role of specific eNOS phosphorylation sites on Ca2+ sensitivity of the enzyme in vitro (Tran et al. 2009). While phosphorylation of eNOS itself was not sufficient to cause activation, the phosphorylations of S1179 and S617 in particular increased the sensitivity of eNOS to Ca2+ dependent activation. Both these phosphorylation events have been shown to occur in uterine artery endothelial cells during pregnancy in response to vasodilatory stimuli (Cale and Bird 2006). Studies have shown reduced uterine blood flow in pregnant animals after infusion with the eNOS inhibitor L-NAME (Sladek et al. 1997), linking endothelial NO production to pregnancy adapted vasodilation. Indeed, the role of NO in pregnancy adaptation has been reviewed extensively (Boeldt et al. 2011). Of note, while it is true that eNOS expression levels are increased during pregnancy in uterine artery endothelium (Magness et al. 1997), changes in Ca2+ signaling are the necessary trigger for NO production. Certainly increased expression raises the capacity of endothelium to make NO, and we have shown this contributes directly to increased NO production in pregnancy adapted uterine arteries (Yi et al. 2005). Nonetheless, it is equally important to note that in cell culture, endothelial cells from the uterine arteries of pregnant ewes lose the vast majority of pregnancy adapted eNOS expression difference from those cells derived from non-pregnant ewes and yet they still maintain pregnancy adapted elevations in eNOS activation when stimulated with Ca2+ mobilizing agonists (Sullivan et al. 2006). This appears to be because greater cell-cell coupling by Cx43 observed in pregnancy is retained in culture and Cx43 blockade both in freshly isolated vessels (Morschauser et al. 2014) and in cells in culture (Yi et al. 2010) removes Ca2+ bursting and enhanced NO output in parallel. Such observations reinforce quite clearly the critical importance of sustained Ca2+ responses to enhance eNOS activity in pregnancy and raises the question of whether a failure of Cx43 enhanced Ca2+ signaling could in turn contribute to endothelial dysfunction in PE pregnancy (Bird et al. 2013).
2.4.2 Prostacyclin
Prostacyclin (PGI2) is a potent vasodilator derived from endothelial arachidonic acid metabolism. In general the impact of prostacyclin on vascular tone can best be explained by the thromboxane (TX) A2/PGI2 ratio (reviewed in (Mehta and Griendling 2007). TXA2 acts as a vasoconstrictor on the vascular smooth muscle (VSM), while PGI2 functions as a vasodilator. Both TXA2 and PGI2 have a very short half-life, and as such only indirect measures can be made of stable metabolites in the circulation/media (TXB2 and 6-keto PGF2α, respectively). Because the effect of PGI2 is tightly tied to TXA2, metabolites, both must be measured and compared to draw conclusions for net effects on VSM tone. Unfortunately there are no techniques that allow monitoring of PG or TX class molecules in real time. While the system is more difficult to monitor, there is strong evidence that PGI2 plays an important role in pregnancy adapted vasodilator responses. Some groups have found elevated levels of PGI2 metabolites in pregnancy when compared to the non-pregnant state (Goodman et al. 1982; Lewis et al. 1980). There are many reports of upregulation of cyclooxygenase (COX) enzymes (predominantly COX-1 in endothelial cells) during pregnancy, which increase throughout gestation, so increasing the cellular capacity for PGI2 production (Habermehl et al. 2000; Janowiak et al. 1998; Magness et al. 2000). Lastly, PGI2 production is known to be the result of cPLA2 activation, and cPLA2 in turn also undergoes Ca2+ sensitive activation (Bird et al. 2000). Even though PGI2 cannot be studied directly in real time, as we can for NO, and certainly not on an individual cell basis, it is highly likely that the changes in Ca2+ signaling within cells and between cells which regulate NO production apply much in the same manner as PGI2.
2.4.3 EDHF
Endothelium derived hyperpolarizing factor (EDHF) has also been implicated as a third major player in endothelial cell-mediated vasodilator production in pregnancy. It has also become clear, however, that ‘EDHF’ is not a single factor, but a spectrum of responses that are otherwise neither NO or PGI2 mediated, but still result in smooth muscle relaxation. Currently, it seems that although EDHF and NO/PGI2 are most likely redundant pathways, a significant portion can be attributed to EDHF and it is fair to say in the absence of EDHF the maternal circulation would struggle to provide adequate blood flow to the fetus. Progress in the EDHF field is limited by this ongoing debate on the identity of the active compound(s), and tools to monitor it are comparatively crude. While not unanimous, a number of studies agree that pregnancy-specific upregulation of agonist induced vasodilation includes an EDHF component (Reviewed in (Morton and Davidge 2013)). As with NO and PGI2, Ca2+ signaling plays an important part in mediating at least some forms of EDHF production, further complicating the role of each respective vasodilator in pregnancy adaptation. Of note, 2-APB which effectively inhibits both [Ca2+]i burst signaling and corresponding NO production in uterine artery endothelium (Gifford et al. 2006; Yi et al. 2005) has also been argued to be an effective inhibitor of EDHF function (Griffith 2004). An important consideration in the role of EDHF in pregnancy adapted vasodilator production is that the relative contribution of NO, PGI2, or EDHF may be dependent on vascular bed or vessel size (Morton and Davidge 2013), and this is observed in both humans and in animal models. And while EDHF plays an important role in augmenting increased uterine blood flow, if not causing it in certain situations, it also seems likely changes in [Ca2+]i signaling may be equally important to NO, PGI2 and EDHF function given the action of 2-APB in each case. Consistent with this, it is also relevant that we reported recently that the endothelial cells of umbilical cords from PE women at late term show defects in sustained and coordinated endothelial Ca2+ bursting (Krupp et al. 2013) and while NO output was measured and a decrease observed, we did not measure PGI2 or EDHF.
3. Hormonal Control of Vascular Adaptation
3.1 Sex Steroids
Underlying the extensive remodeling of the uteroplacental vasculature during pregnancy are numerous endocrine changes. These endocrine changes include fluctuations in circulating sex steroids; which truly begin to change through the menstrual cycle in preparation for pregnancy. Estrogens and progesterone are known to have profound effects on the maternal vasculature, especially in the uterine arteries. As the circulating levels of these hormones change through pregnancy, so do their effects on maternal vascular function.
3.1.1 Estrogen
Estrogen in its various forms has a number of important effects on maternal vascular adaptations to pregnancy. In a rat model, de novo synthesis of estrogen during decidualization assists in angiogenesis at the implantation site (Das et al. 2009), which is essential for maintaining early pregnancy. As gestation continues and the critical regulator of vascular resistance switches from angiogenesis and vasodilation to almost exclusively vasodilation, so too do circulation estrogens. Estrogens are well known to promote vasodilator production, but estrogen alone is not effective at activation of Ca2+ signaling in UAEC (Chen et al. 2004). Rather the effects of estrogen on NO and PGI2 production may be more through genomic regulation downstream of estrogen receptor (ER)α and ERβ by directly promoting eNOS and COX-1 expression. Exposure of endothelial cells to estrogens increases eNOS and COX-1 espression and therefore raises vasodilatory capacity. Studies looking at estrogen effects in an ovine model on eNOS expression in high estrogen states such as the follicular phase of the menstrual cycle have clearly shown that estrogen promotes increased eNOS expression and this relates directly to increased NO output even without a change in Ca2+ bursting. (Yi et al. 2005). However, in late pregnancy much of the further increased NO production due to pregnancy adaptation occurs due to estrogen-independent changes in Ca2+ signaling. Estrogen may also promote vascular endothelial growth factor (VEGF) production by endothelial cells and so support vascular remodeling indirectly (Kazi and Koos 2007).
Beyond ERα and ERβ, others have sought to implicate the g-protein coupled estrogen receptor, GPR30 in maternal regulation of blood flow, but species and tissue-specific changes in GPR30 expression and action make for a controversial story. In the rat uterus, GPR30 has been implicated in estrogen-mediated reduction in vascular tone (Tropea et al. 2015) but some have suggested this may not be the case in humans (Corcoran et al. 2014). Others still have shown a role for GPR30 in regulation of vascular tone in human mammary arteries (Haas et al. 2009). It is critical that the switch from estrogens promoting angiogenesis to vasodilation occurs as gestation progresses because, as noted earlier, increases in uteroplacental blood flow become more reliant on vasodilation than angiogenesis. Since estrogen levels rise dramatically through mid-gestation (O’Leary et al. 1991; Smith et al. 2009), it would be counterproductive for increased estrogen to drive angiogenesis as the fetus/placenta drive vasodilation.
3.1.2 Progesterone
Progesterone levels rise through pregnancy, elevating rapidly as the gestation nears term before crashing to allow parturition (O’Leary et al. 1991; Smith et al. 2009). Early in pregnancy, it is likely progesterone plays a role in decidualization at the implantation site, as progesterone receptor is expressed in the endothelium of the decidua (Wang et al. 1992). Kristiansson and Wang (Kristiansson and Wang 2001) correlated higher progesterone levels in early pregnancy with lower blood pressures later in pregnancy, though they were quick to point out the lack of causative evidence. Progesterone is also able to promote vasodilator production through stimulation of eNOS and thus NO production (Simoncini et al. 2007) as well as increased expression of activity of COX-1, which regulates PGI2 production (Hermenegildo et al. 2005). This is not necessarily the case in all tissues, vascular beds, or models of pregnancy adaptation though. Others have shown rapid effects of progesterone on vascular tone through membrane-bound non-genomic progesterone receptors, which consisted of rapid vasodilation responses (Thomas and Pang 2013). In rats, progesterone blunted the pressor response to vasoconstrictors such as angiotensin II and norepinephrine (Nakamura et al. 1988; Novak and Kaufman 1991). It is important to note that in the progesterone dominated luteal phase in sheep, both eNOS expression and Ca2+ responses are at a minimum in uterine artery endothelium itself (Yi et al. 2005), suggesting elevated progesterone alone may not be a strong driver of vascular adaptation to pregnancy.
3.1.3 Androgens
The role of androgens in pregnancy adaptation is poorly understood. It is possible that androgens play no discernible role in healthy vascular adaptation to pregnancy. There are, however, a handful of papers that point to androgens as a negative regulator of blood flow in PE pregnancies, which will be addressed in a later section.
3.2 Cyclic Nucleotides
Both NO and PGI2 have been shown clearly to induce the production and secretion of both cGMP and cAMP in VSM (Reviewed in (Pelligrino and Wang 1998)). Cyclic nucleotides (cAMP and cGMP) are most well known as inhibitors of vascular tone in the VSM, but they can also play an important role in promoting adaptation of endothelium. Part of sustaining uterine blood flow through sustained vasodilation requires upregulation of functional Cx43 in the plasma membrane. Cyclic nucleotides are also known to drive Cx43 expression inside the cell as well as connexisome movement and placement into functional Gap junction plaques in the plasma membrane (Paulson et al. 2000). The observations of Magness and coworkers that Cx43 expression is increased in the uterine vascular endothelium along with eNOS in late gestation (Morschauser et al. 2014) combined with our own observation of increased Ca2+ bursting in the same UA Endo preparations (Yi et al. 2010) suggest healthy vascular function may become self-reinforcing through cyclic nucleotide feed forward loop that will further amplify and sustain vasodilator production. This is particularly true in the uterine circulation where both eNOS and Cx43 expression are also elevated in the uterine artery closest to the site of placentation (Morschauser et al. 2014), but cyclic nucleotides spilling from the uterus may also explain pregnancy adaptive effects in the systemic circulation where substantial increases in cyclic nucleotides are still detectable (Shaul, et al. 1992) and increased and more sustained Ca2+ signaling is also observed in maternal (Mahdy et al. 1998) and even fetal venous endothelium (Steinert et al. 2002).
3.3 Hormones of Inflammation
Increasingly, and certainly in the last twenty years, normal pregnancy is acknowledged to be a mild inflammatory state (Conrad and Benyo 1997; Tosun et al. 2010). Immune cells and their byproducts are known to interact with invading trophoblasts and endothelial cells, and in most instances this is a positive effect. In early pregnancy, immune cells play an important role in implantation and establishment of the placenta. The decidua is known to contain a large number of immune cells, including helper T cells, natural killer cells, dendritic cells, and macrophages (Ashkar et al. 2000; Hanna et al. 2006; Plaks et al. 2008; Shimada et al. 2006). These cells are known to produce many growth factors and cytokines including placental growth factor (PlGF), VEGF, tumor necrosis factor (TNF)α, interleukin (IL)-1β, IL-6, IL-8, all of which may be important for establishing placentation and the neovascularization which accompanies it (Abrahams et al. 2004; Dekel et al. 2010; Yang et al. 2003). Additionally, these immune cell-derived growth factors and cytokines are important for vascular remodeling to provide adequate blood supply to the fetus, especially in response to hypoxic oxygen gradients (Kharfi et al. 2003; Page 2002). VEGF in particular is very well understood as a driver of angiogenesis and endothelial cell proliferation, which are central to vascular remodeling in pregnancy. VEGF is also known as a somewhat weak agonist for vasodilator production (through its ability to weakly mobilize Ca2+), and this may be beneficial for late-pregnancy support of uterine blood flow. Later in pregnancy, the role of inflammatory hormones are less well defined, though circulating concentrations change throughout gestation (Gillespie et al. 2016). Of note, abnormally high levels may even be detrimental.
4. Preeclampsia
Preeclampsia is a disease of human pregnancy typically characterized by hypertension and proteinuria. This disease is characterized by an insufficient drop in uterine vascular resistance, primarily in late gestation. Most reports estimate the incidence of PE at 3–5% of pregnancies in the United States and up to 10% of pregnancies worldwide (Wallis et al. 2008). PE is associated with both maternal and fetal morbidity and mortality. Up to 15% of global maternal deaths can be attributed to PE. Maternal morbidities include renal failure, liver failure, stroke, and cardiac arrest (Matter F 2000). Fetal complications are mostly the result of intrauterine growth restriction, preterm birth, and can be severe enough for stillbirth (Jabeen et al. 2011). Early stages of the disease involve improper remodeling of the spiral arteries at the implantation site. Although the etiology of PE is still debated, many believe it is often the result of improper immune and hormonal responses which progresses over the period of gestation, such that there is incomplete remodeling of the entire uterine vasculature, as well as corresponding endothelial dysfunction. Endothelial dysfunction as indicated by a lack of enhanced vasodilation is detected early in pregnancy and later dysfunction can include a breakdown of the endothelial monolayer and loss of vascular integrity which is commonly reported in the clinic as proteinuria.
4.1 Diagnosis and Symptoms
Mild PE is defined as gestational blood pressure greater than 140/90 on two separate occasions at a minimum of 4 hours apart after 20 weeks gestation along with proteinuria (>1+ protein on dipstick or >300mg in 24hrs or protein/creatinine ratio ≥ 0.3); or in the absence of proteinuria, thrombocytopenia, renal insufficiency, impaired liver function, pulmonary edema, or cerebral or visual problems. Severe PE includes even higher blood pressure (160/110) as well as confounding factors such as edema and seizures (Sibai et al. 2003). Current treatment often consists of anti-hypertensives, bed rest, and ultimately caesarian section preterm delivery. Existing treatments, however, are for the symptoms, and not the cause of PE. Antihypertensives are aimed at relaxing the vascular smooth muscle and commonly include use of nifedipine, an L-channel blocker. (This is not a problem from an endothelial standpoint since most capacitative Ca2+ entry in endothelial cells is mediated through TRPC channels (Gifford et al. 2006)). Nonetheless, control of the condition is a challenge to clinicians, and the symptoms of PE typically remain until delivery of the placenta, so posing a continuing threat to mother and child.
4.2 Outcomes and Long-term Problems
Beyond the immediate threat, there are also potential negative outcomes, or increased risk at the least, associated with PE which may surface later in life for both the mother and fetus. There is an increased risk of hypertension (3 fold, (Canti et al. 2012; Drost et al. 2012) and cardiovascular disease (2 fold, (Ray et al. 2005) later in life, in women who were diagnosed with PE. While it may be argued PE indicated a predisposition to hypertension in the mother, other studies have also pointed to increased risk of hypertension and cardiovascular disease in children from PE pregnancies, especially among those who were delivered preterm (Irgens et al. 2001; Lawlor et al. 2012). Barker theorized that limited organ development and/or abnormal programming of organs and tissues occurs in such fetus and newborn infants gestated in the environment of a diseased pregnancy. Based on epidemiological studies from around the globe comparing birth records with adult onset disease he suggested that the uterus prepares the child for the world into which they will be born, and so these children would be maladapted for the ‘normal’ conditions outside the womb and would therefore be prone to disorders such as hypertension and heart disease later in life (Barker 1990). This theory has come to be commonly known as the Barker hypothesis, and is often cited as a critical reason why diseases affecting the gestational environment must continue to be investigated, even if modern obstetric practices can reduce immediate perinatal mortality.
4.3 Endothelial Dysfunction in Preeclampsia
One enduring aspect of PE that has been studied intensively for decades is endothelial dysfunction. Endothelial dysfunction contributes to all major symptoms of PE (hypertension, edema, proteinuria, improper platelet aggregation). The key physiologic functions of endothelial cells to control vascular function by sensing the blood composition as well as providing a physical barrier to the improper movement of water, ions, proteins, and cells from the blood into the vessel wall are likely now compromised in PE. In that case, and angiogenesis and vasodilation in response to stimuli such as decreased oxygen tension and mechanical stress due to shear forces are would no longer be sufficient. It appears this is a failure of endothelium to show normal pregnancy adaptation, given that PE subjects fail to develop insensitivity to vasoconstrictors, and both enhanced [Ca2+]i signaling and enhanced vasodilator production are reduced or lacking. The lower levels of circulating cyclic nucleotides also suggest feed forward enhancement of endothelial cell coupling and so Ca2+ bursting is lost, and certainly endothelial cells of PE subjects do show reduced Ca2+ bursting in parallel with reduced NO production (Krupp et al. 2013). The net result of the increased blood volume of pregnancy but insufficient drops in uterine and systemic vascular resistance contributes greatly to the hypertensive component of PE. Other vascular beds also see a lack of vasodilator production, but may be especially sensitive to other measures of endothelial dysfunction. In the kidney for example, glomerular endothelial cells often lose their barrier function integrity (beyond that of their typical fenestrations) and become pathologically permissive to protein movement into the urine (Wang et al. 2015). In extreme cases of endothelial monolayer barrier breakdown, at the blood-brain barrier, improper movement of proteins and ions can promote seizures (Hammer and Cipolla 2015). Though often not as severe, these same effects can manifest in other parts of the body and in the extremities can result in edema (Brown 1995). Often a secondary result of endothelial dysfunction in PE is hyper coagulation due to release of thrombotic factors from damaged endothelial cells and increased leukocyte traffic to the injured tissue. We will not focus on thrombosis in PE in this review and instead refer to recent reviews in the area (Jodkowska et al. 2015). Taken together, the indicators of endothelial dysfunction in PE often feed forward onto each other, exacerbating the condition.
4.4 Vasodilators in Preeclampsia
The central importance vasodilators play in pregnancy adaptation make them a likely suspect as a culprit in the endothelial pathologies of PE. In fact, all three of the major vasodilators outlined above as necessary components of normal pregnancy adaptations have been shown to be abnormally regulated in PE. It is important to note that while most studies in PE have focused on their role in uteroplacental tissues, many of the same deficiencies in vasodilator production are seen throughout the body.
Numerous studies have implicated decreased NO production or bioavailability in relation to PE pregnancies. While no clear trend in eNOS expression levels are observed, endothelial cells collected from PE pregnancies or exposed to maternal serum from PE pregnancies produce less NO than their counterparts from normal pregnancies (Hayman et al. 2000; Krupp et al. 2013). One study showed that even when eNOS expression is increased in endothelial cells exposed to PE serum, overall bioavailability of NO may still be decreased due to increased arginase expression and increased asymmetric dimethylarginine observed in early onset PE (Goulopoulou and Davidge 2015). Shear stress-induced NO-dependent vasorelaxation is also reduced in human myometrial arteries from PE patients (Kublickiene et al. 2000). Others have shown that agonist-stimulated NO production in reduced in PE-derived endothelial cells. Akar et al (Akar et al. 1994) showed that there is some increase in basal NO production in umbilical arteries from PE pregnancies, but a decrease in the greater agonist stimulated NO production. They reported no change in the umbilical vein, although this contrasts with other reports in the umbilical vein that clearly show decreased agonist-stimulated NO production in umbilical vein endothelial cells derived from PE pregnancies relates directly to deficiencies in Ca2+ signaling (Krupp et al. 2013; Steinert et al. 2002). Mahdy et al (Mahdy et al. 1998) showed similar results for Ca2+ in human hand vein endothelial cells, suggesting that impaired Ca2+ signaling (and NO production) is not constrained to the maternal uterine and umbilical vasculature alone, but indeed extends to the maternal systemic arterial and venous vasculature as well.
Another potent vasodilator, PGI2, has also been shown extensively to be reduced in PE pregnancies, and this apparently precedes the development of PE. PGI2 itself has a very short half-life, but plasma and urinary concentrations of PGI2 metabolites are decreased in PE pregnancies due to widespread reductions in PGI2 production in many vascular beds, such as uterine, subcutaneous, placental, and umbilical (Downing et al. 1980; Remuzzi et al. 1980). There are two predominant causes for decreased PGI2 production. Firstly, prostacyclin synthase, like eNOS is dependent on increased intracellular Ca2+, and as Steinert, Mahdy, and Krupp had have shown, Ca2+ signaling is impaired in endothelial cells from PE pregnancies (Krupp et al. 2013; Mahdy et al. 1998; Steinert et al. 2002). Secondly, increased oxidative stress associated with PE also inhibits prostacyclin synthase, thus reducing PGI2 production (Baker et al. 1996; Lorentzen et al. 1991). Much has also been written about TXA2/PGI2 ratio, which has been shown to be increased in PE due to both an increase in TXA2 production as well as PGI2 reduction. However, it appears that decreased PGI2 may be more important physiologically, as low dose aspirin administration to fight increased TXA2 levels while sparing PGI2 yields disappointing results in clinical trials (Mills et al. 1999; Sibai et al. 1993).
While the imprecisely defined nature of EDHF makes it difficult to clearly state its role in the vascular pathogenesis of PE, those that have undertaken this task have generally found EDHF-mediated vasorelaxation to be reduced in vessels from PE pregnancies. Luksha et al (Luksha et al. 2009; Luksha et al. 2008) have looked extensively at small subcutaneous and myometrial arteries from women with PE. They showed that myometrial arteries but not subcutaneous arteries have a reduced EDHF component in PE when compared to normal pregnancy. However, in the study on subcutaneous arteries, PE women could be broken down in to subgroups based on their relative contribution of EDHF to vasorelaxation. In subcutaneous arteries, those with small contributions from EDHF were highly dependent on myoendothelial gap junctions, whereas those more dependent on EDHF were more dependent on H2O2 and arachidonic acid metabolites. In the myometrial arteries, the myoendthelial gap junctions played a very large role, but H202 may compensate to some degree in PE pregnancies. Another study on myometrial arteries also noted that EDHF is an important component of pregnancy adaptation, but appears to be missing in PE (Kenny et al. 2002). Overall, the loss of EDHF-mediated vasorelaxation in PE may be more profound in smaller vessels, where the relative contribution of NO and PGI2 is typically reduced in favor of EDHF in normal pregnancy. Nonetheless the common theme of EDHF function depending on Gap junction function and the loss of EDHF function in PE is consistent with the observations regarding PE associated loss of NO and PGI2 vasodilators.
5. Hormones of Preeclampsia-related Endothelial Dysfunction
While it is clear the failure to achieve proper endothelial adaptation is associated with PE, the question is why? The root cause of PE-related endothelial dysfunction has long been elusive, though many theories have been postulated. At this time, research in this field may best be summarized as a case of “which came first, the chicken or the egg?”. Nonetheless, once insufficient blood flow to the uterus is established as the norm, secondary events can contribute to maintaining a lack of endothelial adaptation otherwise so critical for healthy pregnancy. It is those adverse events which we now focus upon.
5.1 Sex Steroids and PE
The role of estrogens and progesterone, despite their importance in pregnancy adaptation to at least control vasodilatory capacity in the uterine vascular endothelium, is unclear. This is partly due to complications in measuring free vs conjugated estrogens. Limited studies have shown a decrease in conjugated estrone and estriol, but not estradiol in peripheral serum from PE women (Rosing and Carlstrom 1984). Others have shown decreased estradiol in urine, but serum levels of estrogens were unclear (reviewed in (Rosing and Carlstrom 1984). Troisi et al (Troisi et al. 2003a; Troisi et al. 2003b) investigated both maternal and cord blood serum from normal and PE pregnancies and showed no change in estrone, estradiol, or estriol levels. The literature in progesterone levels in PE is also sparse. Rosing et al (Rosing and Carlstrom 1984) found no change in unconjugated progesterone in PE pregnancies when compared to normal pregnancies.
There appears to be a more convincing case that androgens may play a role in the vascular pathology of PE. Androgens have been implicated in the promotion of hypertension, possibly through sensitization to pressors as well as decreased PGI2 production (Acromite et al. 1999), so it is therefore unsurprising that there may be a link between androgens and PE. Both androstenedione and testosterone in the unconjugated form were shown to be elevated in PE vs normal pregnancy (Troisi et al. 2003b), but were unchanged in cord blood serum. Others have also shown increased androgens in PE (Acromite et al. 1999; Salamalekis et al. 2006; Sharifzadeh et al. 2012), and has been linked to dysregulation of p450 aromatase (Sathishkumar et al. 2012; Steier et al. 2002). Nonetheless, it is not clear how much this is a cause of PE and how much a consequence.
5.2 Cyclic Nucleotides
Circulating levels of cyclic nucleotides are also a matter of conflicting results between multiple studies. Most studies looked at levels in maternal plasma and some have found increased cGMP (Sandrim et al. 2011; Schneider et al. 1996), while others see no significant change from normal pregnancy levels in PE pregnancy (Dusse et al. 2013; Schiessl et al. 2006). One study saw decreased cAMP levels in maternal plasma in PE pregnancies (Yamamoto et al. 2010). The relative lack of information on cyclic nucleotides in PE makes it difficult to draw any firm conclusions on what role they may or may not play in the vascular pathogenesis of PE. However, measurement of cyclic nucleotides suffers from differences in methods of detection, which is not such an issue in normal pregnancy adaptation where very large changes in circulating cyclic nucleotides are observed. One final point to note about the role of cyclic nucleotides in PE is that because they feed forward into Cx43 function and Ca2+ signaling in normal pregnancy long term, the effect of any small loss in circulating levels may have a profound impact on endothelial cell function as that feed forward support to adaptation is lost.
5.3 Hormones of Inflammation or Hormones of Wounding
Preeclampsia has been described by some as an exaggerated state of inflammation (Mihu et al. 2015) and others have made the case that the endocrine profile of PE is similar to that of a non-healing wound (Bird et al. 2013). In this section we will briefly review hormones of inflammation/wounding which have been described as altered in PE compared to normal pregnancy, and may have causative adverse effects on endothelial cell function. Other factors such as soluble endoglin (sEng), transforming growth factor (TGF)β, and endothelin (ET)-1 have been associated with PE and we refer to other recent reviews for more information on them (Liu et al. 2012; Saleh et al. 2016). We focus our attention instead on those factors which are known to signal through common pathways or interact with the factors discussed through crosstalk or stimulation of secretion of discussed factors.
5.3.1 VEGF/PlGF/sFlt-1
The role of VEGF family peptides and receptors in PE has been a controversial matter for some time and remains so today. Inconsistencies in gathering, measuring, and reporting circulating levels of VEGF has contributed greatly to the debate about just how much VEGF is present in normal and PE pregnancies, and how much is freely available to bind the plasma membrane VEGF receptors of the endothelial cells. Many early studies measured total VEGF levels, but the discovery that circulating levels of the VEGFR1 splice variant, sFlt-1, are increased in PE (Maynard et al. 2003) make interpretation of these early studies difficult since sFlt-1 binds VEGF and renders it unable to bind to endothelial surface bound VEGFR1 and VEGFR2. Even in those studies which measure total VEGF, there still remains no clear consensus as to whether circulating levels of total VEGF increase, decrease, or remain unchanged in PE. Some variation in the use of ELISA assays, sandwich assays or more recent multiplex assays has not helped the situation. In those studies which looked specifically at free VEGF levels, maternal plasma and serum levels are often decreased in PE pregnancies (Maynard et al. 2003), which is thought to be due to increased sFlt-1 levels. But even these studies often fail to take into account the complexity of VEGF splice variants (e.g. VEGFA, the predominant human VEGF coding gene, is often used interchangeably with its 165 amino acid variant VEGF165, but other variants with biological activity such as VEGF189 and VEGF121 are also present in substantial levels). One isoform that seems to be gaining more widespread acceptance at least as a predictive marker in PE is circulating PlGF (which preferentially binds VEGFR1). In PE, PlGF levels are decreased, whether free or sFlt-1 bound (Levine et al. 2004; Robinson et al. 2006) Certainly recent prospective trials suggest the sFlt:PlGF ratio may have particular value as a predictive marker (Zeisler et al. 2016) while another study suggests a single marker (PlGF, sFlt-1 or endoglin) may be effective (Duckworth et al. 2016). Others have reported the predictive value even at 20–34 weeks were limited (Andersen et al. 2016). The discovery of VEGF165b has further confused the picture regarding VEGF165 since VEGF165b has clearly been incorrectly assigned as VEGF165 in prior studies, and yet VEGF165b has different and even opposite effects on endothelial cell function (Bates 2011). Local concentrations of VEGF in PE can also vary in tissues beyond the placenta. For example, while histologic examination of PE placenta itself may show larger amounts of sFLT-1 and reduced PlGF and VEGF (Yong et al. 2015), decidua and decidual immune cells, and perhaps even decidual endothelial cells show higher levels of VEGF release (Sharma et al. 2016). So one limitation in dissecting this problem is that studies need to look beyond the placenta and circulation alone and look further at the uterine environment also.
There is still genomic and transcriptomic evidence for altered placental and decidual VEGF expression (Sharma et al. 2016; Soleymanlou et al. 2005; Yong et al. 2015), and this alone makes a case to further study abnormal VEGF biosynthesis and action in the development of PE. Beyond the disagreements in the literature surrounding real circulating levels of VEGF in PE, there are also seemingly contradictory results regarding the functional consequences of exogenously altered VEGF levels on endothelial regulation of vascular tone. On the one hand, treatment of myometrial resistance arteries with VEGF mimicked the decrease in endothelium-dependent vasorelaxation typically observed with treatment with serum from PE pregnancies (Brockelsby et al. 1999; Hayman et al. 2000). On the other hand, rat studies have also shown that manipulation of VEGF and PlGF cause vasorelaxation which can be antagonized by sFlt-1 administration (Maynard et al. 2003). While it may appear these studies are contradictory, we would offer that they may not be. Rather it may be that optimal VEGF signaling is critical to healthy endothelial function and any deviance from that, whether it be increased VEGF or decreased free VEGF, may cause changes in endothelial function which are similar to those observed in PE pregnancies. Both additional clinical studies and additional exogenous manipulation of VEGF and associated molecules will be necessary to unravel this problem in a meaningful way.
5.3.2 TNFα
Elevated circulating levels of TNFα are also thought to have a direct effect on endothelial cell function by increasing vascular leakiness and reducing cell responsiveness to vasodilators. Two recent meta-analyses have assessed the levels of TNFα in maternal circulation (plasma and serum) in PE and normal pregnancies (Lau et al. 2013; Xie et al. 2011). Both studies confirmed that TNFα is upregulated in PE in the 3rd trimester. Also according to the meta-analyses, studies on circulating TNFα in early and mid-pregnancy (1st and 2nd trimesters) show mixed results, with some claiming elevated TNFα at this early point while others show no difference, however these studies are limited by small amounts of data compared to late pregnancy studies. A few studies which looked at mild versus severe PE saw no difference between the two groups in TNFα levels. However, one non-parametric study did show a significantly higher level of TNFα in the severe preeclamptic group (Lau et al. 2013). Others who looked at both maternal and umbilical serum also observed increased TNFα concentrations in PE when compared to normal pregnancy for both blood sample types (Tosun et al. 2010). It should be noted that, while TNFα can directly cause endothelial dysfunction, it is also known to promote the release of other factors known to have effects on endothelial function, such as PDGF, ET-1, and IL-6 (Conrad and Benyo 1997).
5.3.3 Interleukins
Numerous interleukins have been associated with PE. Those which have been most well studied are IL-6, IL-8, and IL-10, although a few studies link other interleukins (i.e. IL-1α and IL-1β) with PE. Elevated maternal levels of both IL-6 and IL-8 have been extensively linked with PE. In two meta-analyses, IL-6 was shown to be significantly elevated in the maternal circulation in PE when compared to normal pregnancy (Lau et al. 2013; Xie et al. 2011). Tosun et al (Tosun et al. 2010) also confirmed elevated levels of IL-6 in maternal and umbilical serum. While no meta-analysis was readily available for circulating levels of IL-8 in PE, numerous studies are in agreement that IL-8 is elevated in maternal blood from PE pregnancies when compared to normal pregnancy (Jonsson et al. 2006; Pinheiro et al. 2015; Redman and Sargent 2003; Sharma et al. 2007; Tosun et al. 2010). Additionally, some studies have implicated IL-8 in recruitment and activation of immune cells such as neutrophils and t-lymphocytes (Mukaida et al. 1998; Sharma et al. 2007). A meta-analysis by Lau et al (Lau et al. 2013) reported conflicting IL-10 levels across the literature, with some reporting elevated IL-10 levels in PE vs normal pregnancy and others showing decreased levels. On balance, they concluded that slightly more studies favored increased IL-10 levels in PE. Another meta-analysis by Xie et al (Xie et al. 2011) indicated that there was a significant trend in the literature towards elevated IL-10 levels in maternal blood from PE pregnancies when compared to normal pregnancies.
5.3.4 INFγ
In a meta-analysis of 16 studies looking at maternal plasma and serum in normal and PE pregnancies by Yang et al (Yang et al. 2014), there was a significant trend in the literature for increased interferon (INF)γ levels in PE pregnancies. In 5 independent studies, however, there was no difference observed. Possible explanations for the heterogeneity of INFγ data sets could be differences in patient population or differences in sample collection and/or handling.
6. Effects of Hormones of PE on the Endothelium
The hormones outlined above which are altered in PE when compared to normal pregnancy with proper vascular adaptation to pregnancy can and do have consequences when it comes to endothelial cell function. The exact mechanism by which these hormones effect the maternal vascular endothelium are still being elucidated, but there is a growing body of evidence that aberrant growth factor and cytokine signaling in particular can bring about endothelial dysfunction in vivo and in vitro which could contribute to two key symptoms of PE: hypertension and vascular leakiness. In an acute sense, improper growth factor and cytokine signaling can inhibit Ca2+ signaling events that are critical for vasodilator production through closure of existing gap junctions (Bird et al. 2013). Long-term stimulation of the same signaling pathways can then have more sustained and profound effects on endothelial monolayer integrity, and some studies suggest this may be especially true in sensitive vascular beds such as the renal glomerulus (Turner et al. 2015; Xu et al. 2015). The proposed mechanisms for these effects are outlined in the following sections and are depicted in Figure 1.
Figure 1. Proposed Mechanism of Endothelial Dysfunction in Preeclampsia.
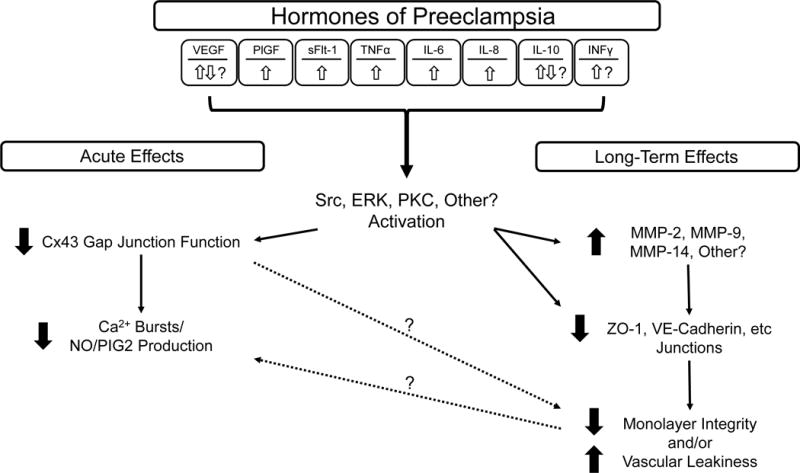
A diverse array of inflammatory hormones are altered in the circulation of late-term PE pregnancies compared to normal pregnancies. Together, these result in activation of kinase signaling pathways such as Src, ERK, PKC, and possibly others. Activation of these pathways results in both acute and long-term changes at the level of the plasma membrane. Acutely, closure of Cx43 alone results in immediate endothelial dysfunction by reducing the capacity of endothelial cells to coordinate Ca2+ responses needed to stimulate production of vasodilators (NO, PGI2). Longer term, further damage to cell junctional proteins such as ZO-1 and VECadherin (by altered turnover or degradation by MMPs) causes a reduction in cell endothelial monolayer integrity and even vessel leakage (edema).
6.1 Potential Acute Effects of Hormones of PE on the Endothelium
Momentary changes in vessel diameter are critical to allow for rapid changes in blood flow. If a vessel loses the ability to rapidly dilate when needed, hypertension may occur. A hallmark of PE is the inability of the vascular endothelium to produce the necessary vasodilators when called upon and in PE, gap junction dependent Ca2+ signaling events may be insufficient for production of the necessary amounts of vasodilators. Growing evidence now supports this theory. Studies on maternal hand vein endothelial cells and fetal umbilical vein endothelium from normal and PE pregnancies show a reduced capacity to sustain elevated [Ca2+]i and produce NO (Krupp et al. 2013; Mahdy et al. 1998; Steinert et al. 2002). Since PGI2 and some components of EDHF are Ca2+ sensitive, there is a strong likelihood this phenomena extends to vasodilators more universally. The correlation between PE and reduced Ca2+ and vasodilator production is likely due to a link between Ca2+ signaling, gap junction communication, and changes in the hormonal milieu during PE. Since sustained endothelial Ca2+ signaling is dependent on Cx43 gap junction function, Cx43 emerged as a likely target for aberrant cell signaling events downstream of membrane-bound growth factor and cytokine receptors. Many groups (Lampe and Lau 2000; Solan and Lampe 2005; Suarez and Ballmer-Hofer 2001; Warn-Cramer et al. 1996) have extensively shown that at least some of the growth factors and cytokines detailed above have the ability to phosphorylate Cx43 at resides on the c-terminus that render the protein less functional. Bird et al have reviewed the link between hormones of PE and Cx43 inhibition in detail (Bird et al. 2013). Common signaling ‘culprits’ that phosphorylate these inhibitory residues are Src, ERK, and PKC (Bird et al. 2013). This has been corroborated in the ovine uterine artery endothelial cell model, where administration of VEGF leads to both phosphorylation of Cx43 and reduced sustained phase Ca2+ signaling (Boeldt et al. 2015). Inhibitors of Src and ERK signaling reversed phosphorylation of Cx43 at their respective target residues and also rescued Ca2+ signaling to levels equivalent to control (Boeldt et al. 2015). More detailed studies on other hormones (growth factors and cytokines) associated with of PE are warranted, to assess the universality of these initial observations, and whether they are indeed occurring in PE.
6.2 Potential Long-term Effects of Hormones of PE on the Endothelium
Over time, sustained exposure of the endothelium to high levels of growth factors and cytokines can also result in breakdown of endothelial monolayer integrity itself by degrading and/or removing cell junctional proteins. It is perhaps no coincidence since monolayer breakdown of endothelial cells can often be initiated through the same ERK and Src signaling pathways that act acutely on Cx43 (Reviewed in (Bird et al. 2013)). Such monolayer breakdown could explain other common symptoms of PE including proteinuria, edema, and in extreme cases seizures due to blood-brain barrier breakdown. Cell-cell junctional proteins such as VE-Cadherin and zonula occludens (ZO)-1, which are commonly associated with plasma membrane Cx43 function, are commonly targeted by excessive growth factors and cytokines for internalization and possible breakdown. This is coupled with a net reduction in peptide trafficking and membrane placement as cyclic nucleotides decline would result in an inability of neighboring cells to remain anchored to each other. Without a strongly tethered junctional complex, cells begin to retract. While retraction is critical to initiate angiogenesis and wound healing in a non-pregnancy setting, in late pregnancy when the uterine vasculature in particular has switched from being angiogenic to vasodilatory function, this response can only contribute to endothelial dysfunction and lead to vascular leakiness. Glomerular endothelial cells and podocytes may be to be especially sensitive to these signals, potentially explaining the high incidence of proteinuria in PE (Turner et al. 2015; Xu et al. 2015).
Matrix metalloproteinases (MMP) are one class of enzymes involved in the cleaving of cell-cell junctional proteins, which are linked with PE. As with many factors linked with PE, timing and location of MMP expression is incredibly important. One case in point is MMP-14 (MT-MMP1), which can activate MMP2. Early in pregnancy MMP-14 is critical for trophoblast invasion (Onogi et al. 2011). In pregnancies which are destined to become PE, MMP-14 expression is decreased. However, in pregnancies already diagnosed with PE (by definition mid to late pregnancy), MMP-14 has been observed to be increased (Kaitu’u-Lino et al. 2012). Other have implicated both MMP-2 and MMP-9 as critical for trophoblast invasion. Trophoblasts initially secrete large amounts of MMP2 and then relatively less MMP2 and more MMP9 through early pregnancy with normal gestation (Xu et al. 2000), but secrete less MMP2 and less MMP9 with PE pregnancy associated with growth restriction (Zhu et al. 2014). Of interest studies in the reduced uterine perfusion pressure (RUPP) model of PE suggest that decreases in MMP-2 and MMP-9 may be seen in the systemic vasculature (aorta) as well as in the uterus and placenta and can be modulated by VEGF and sFlt-1 (Li et al. 2014). At the level of endothelial cells themselves, MMP-2, -9 and -14 all play a role in normal angiogenesis, and a part of that process can include the release of local surface bound growth factors (including VEGF) and TNFα (Pepper 2001). Abnormally elevated TNFα in turn can stimulate damage to cell monolayer integrity of blood brain barrier endothelial cells through ZO-1 breakdown and this effect is apparently mediated by MMP9 (Wiggins-Dohlvik et al. 2014).
6.3 Other Areas for Future Study
One remaining yet little explored consideration in discussing growth factors and cytokines linked with PE is that they often regulate the expression and secretion of each other. When this is coupled with immune cell migration to areas of inflammation or endothelial distress, which in turn secrete additional cytokines, the inflammatory condition at least locally, can become all the more severe. The study of such interacting cell types is limited in studies of pregnancy, but one example which illustrates this concept is the crosstalk between INFγ and IL-6 through signal transducer and activator of transcription (STAT) signaling in atherosclerosis. In this condition, T cell secretion of INFγ changes IL-6 from an anti-inflammatory signal to a pro inflammatory signal by shifting IL-6 from a STAT3 mediated response to a STAT1 pathway (Sikorski et al. 2011) IL-6 operating through STAT1 promotes immune cell adhesion to the endothelium, which could then have profound effects on local concentration of immune cell-secreted factors and inflammatory signal amplification. Clearly this is an area for further study and further review.
7. Integrated Discussion
Our ability to understand and treat PE depends heavily on our understanding of vascular adaptations in healthy pregnancies. Our understanding of the physiology of pregnancy adaptation quite often depends on the use of animal models which may or may not fully represent the human condition. Furthermore, our understanding of cellular and molecular regulation of pregnancy adaptation is in its infancy. But a common theme in both maternal vascular adaptation to pregnancy and the development and diagnosis of PE is the role of the vascular endothelium. It is apparent that many hormones related to pregnancy adaptation have profound effects on the endothelium, so perhaps it is no surprise these very same hormones are often dysregulated in PE. The more our attention is drawn to the hormones of pregnancy adaptation and those altered in PE, the more we appreciate the complex environment they exist within. There is a tendency in clinical medicine to seek out the simple blood test but to date the use of such an approach has failed to be of diagnostic value. It is becoming increasingly clear the reason lies in the fact adaptation to pregnancy and disorders of PE are not just confined to the placenta itself. Different tissues and vascular beds may experience altered local and circulating hormones in unique ways based upon local environment (hypoxia, cell state, status of invading immune cells). Multiple tissues and cell types such as trophoblasts, the decidua, the placenta, immune components, and the endothelium itself all interact with each other; sometimes cross-talking through known signaling pathways like Src or as yet unexplored pathways such as STAT are amplifying local signals to respond disproportionately to circulating levels of hormone. This coupled with redundant cell signaling among growth factors and cytokines linked with PE, means that looking for a single hormone cause or biomarker for PE may be futile. Indeed, there are many ways each of the hormones outline in this review could all contribute to the same clinical phenotype. While individual hormones could vary between human subjects, outcomes could be similar. However, working backwards from the symptoms of PE has also lead us to these very convergence points on which endothelial cell function depends. Vasodilator production depends on Ca2+ signaling and endothelial cell barrier function depends largely on monolayer integrity. These two endothelial functions are also linked, as Ca2+ signaling depends on cell-cell junctional proteins such as Cx43 gap junctions. Thus, if the complex and indeed variable cocktail of hormones which is dysregulated in PE could be pharmacologically targeted not at the level of hormone production, but at a convergence point of hormone signaling, heterogeneity in patient endocrine profiles may become less of an issue. Studies to better understand the functional consequences of complex hormone environments are warranted to test the efficacy of this approach.
Acknowledgments
This work was made possible with funding from NIH grants (R03HD079856 and P01HD38843) as well as NIHT32HD041921 for predoctoral training for DSB. Additional training support for DSB was from University of Wisconsin SMPH Herman I Shapiro Distinguished Graduate Fellowship.
Footnotes
Conflict of Interest: There is no conflict of interest to report.
Bibliogrophy
- Abrahams VM, Kim YM, Straszewski SL, Romero R, Mor G. Macrophages and apoptotic cell clearance during pregnancy. Am J Reprod Immunol. 2004;51:275–282. doi: 10.1111/j.1600-0897.2004.00156.x. [DOI] [PubMed] [Google Scholar]
- Acromite MT, Mantzoros CS, Leach RE, Hurwitz J, Dorey LG. Androgens in preeclampsia. Am J Obstet Gynecol. 1999;180:60–63. doi: 10.1016/s0002-9378(99)70150-x. [DOI] [PubMed] [Google Scholar]
- Akar F, Ark M, Uydes BS, Soysal ME, Saracoglu F, Abacioglu N, Van de Voorde J, Kanzik I. Nitric oxide production by human umbilical vessels in severe pre-eclampsia. J Hypertens. 1994;12:1235–1241. [PubMed] [Google Scholar]
- Andersen LB, Dechend R, Jorgensen JS, Luef BM, Nielsen J, Barington T, Christesen HT. Prediction of preeclampsia with angiogenic biomarkers. Results from the prospective Odense Child Cohort. Hypertens Pregnancy. 2016:1–15. doi: 10.3109/10641955.2016.1167219. [DOI] [PubMed] [Google Scholar]
- Ashkar AA, Di Santo JP, Croy BA. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J Exp Med. 2000;192:259–270. doi: 10.1084/jem.192.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PN, Davidge ST, Barankiewicz J, Roberts JM. Plasma of preeclamptic women stimulates and then inhibits endothelial prostacyclin. Hypertension. 1996;27:56–61. doi: 10.1161/01.hyp.27.1.56. [DOI] [PubMed] [Google Scholar]
- Barker DJ. The fetal and infant origins of adult disease. BMJ (Clinical research ed.) 1990;301:1111. doi: 10.1136/bmj.301.6761.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DO. An unexpected tail of VEGF and PlGF in pre-eclampsia. Biochem Soc Trans. 2011;39:1576–1582. doi: 10.1042/BST20110671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird IM, Boeldt DS, Krupp J, Grummer MA, Yi FX, Magness RR. Pregnancy, programming and preeclampsia: gap junctions at the nexus of pregnancy-induced adaptation of endothelial function and endothelial adaptive failure in PE. Curr Vasc Pharmacol. 2013;11:712–729. doi: 10.2174/1570161111311050009. [DOI] [PubMed] [Google Scholar]
- Bird IM, Sullivan JA, Di T, Cale JM, Zhang L, Zheng J, Magness RR. Pregnancy-dependent changes in cell signaling underlie changes in differential control of vasodilator production in uterine artery endothelial cells. Endocrinology. 2000;141:1107. doi: 10.1210/endo.141.3.7367. [DOI] [PubMed] [Google Scholar]
- Bird IM, Zhang L, Magness RR. Possible mechanisms underlying pregnancy-induced changes in uterine artery endothelial function. American journal of physiology.Regulatory, integrative and comparative physiology. 2003;284:R245. doi: 10.1152/ajpregu.00108.2002. [DOI] [PubMed] [Google Scholar]
- Boeldt DS, Grummer MA, Yi F, Magness RR, Bird IM. Phosphorylation of Ser-279/282 and Tyr-265 positions on Cx43 as possible mediators of VEGF-165 inhibition of pregnancy-adapted Ca2+ burst function in ovine uterine artery endothelial cells. Mol Cell Endocrinol. 2015;412:73–84. doi: 10.1016/j.mce.2015.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeldt DS, Yi FX, Bird IM. eNOS activation and NO function: pregnancy adaptive programming of capacitative entry responses alters nitric oxide (NO) output in vascular endothelium–new insights into eNOS regulation through adaptive cell signaling. The Journal of endocrinology. 2011;210:243. doi: 10.1530/JOE-11-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockelsby J, Hayman R, Ahmed A, Warren A, Johnson I, Baker P. VEGF via VEGF receptor-1 (Flt-1) mimics preeclamptic plasma in inhibiting uterine blood vessel relaxation in pregnancy: implications in the pathogenesis of preeclampsia. Laboratory investigation; a journal of technical methods and pathology. 1999;79:1101. [PubMed] [Google Scholar]
- Brown MA. The physiology of pre-eclampsia. Clinical and experimental pharmacology & physiology. 1995;22:781. doi: 10.1111/j.1440-1681.1995.tb01937.x. [DOI] [PubMed] [Google Scholar]
- Cale JM, Bird IM. Dissociation of endothelial nitric oxide synthase phosphorylation and activity in uterine artery endothelial cells. 2006;290:H1433. doi: 10.1152/ajpheart.00942.2005. [DOI] [PubMed] [Google Scholar]
- Canti V, Maggio L, Ramirez GA, Locatelli A, Cozzolino S, Ramoni V, Ruffatti A, Tonello M, Valsecchi L, Rosa S, et al. Hypertension negatively affects the pregnancy outcome in patients with antiphospholipid syndrome. Lupus. 2012;21:810. doi: 10.1177/0961203312441269. [DOI] [PubMed] [Google Scholar]
- Catchpole HR. Domestic Animals, Fourth Edition. Academic Press; 1991. Hormonal Mechanisms in Pregnancy and Parturition. [Google Scholar]
- Chen DB, Bird IM, Zheng J, Magness RR. Membrane estrogen receptor-dependent extracellular signal-regulated kinase pathway mediates acute activation of endothelial nitric oxide synthase by estrogen in uterine artery endothelial cells. Endocrinology. 2004;145:113–125. doi: 10.1210/en.2003-0547. [DOI] [PubMed] [Google Scholar]
- Clark SL, Cotton DB, Lee W, Bishop C, Hill T, Southwick J, Pivarnik J, Spillman T, DeVore GR, Phelan J. Central hemodynamic assessment of normal term pregnancy. American Journal of Obstetrics and Gynecology. 1989;161:1439. doi: 10.1016/0002-9378(89)90900-9. [DOI] [PubMed] [Google Scholar]
- Conrad KP, Benyo DF. Placental cytokines and the pathogenesis of preeclampsia. Am J Reprod Immunol. 1997;37:240–249. doi: 10.1111/j.1600-0897.1997.tb00222.x. [DOI] [PubMed] [Google Scholar]
- Corcoran JJ, Nicholson C, Sweeney M, Charnock JC, Robson SC, Westwood M, Taggart MJ. Human uterine and placental arteries exhibit tissue-specific acute responses to 17beta-estradiol and estrogen-receptor-specific agonists. Mol Hum Reprod. 2014;20:433–441. doi: 10.1093/molehr/gat095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Mantena SR, Kannan A, Evans DB, Bagchi MK, Bagchi IC. De novo synthesis of estrogen in pregnant uterus is critical for stromal decidualization and angiogenesis. Proc Natl Acad Sci U S A. 2009;106:12542–12547. doi: 10.1073/pnas.0901647106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekel N, Gnainsky Y, Granot I, Mor G. Inflammation and implantation. Am J Reprod Immunol. 2010;63:17–21. doi: 10.1111/j.1600-0897.2009.00792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing I, Shepherd GL, Lewis PJ. Reduced prostacyclin production in pre-eclampsia. Lancet. 1980;2:1374. doi: 10.1016/s0140-6736(80)92443-5. [DOI] [PubMed] [Google Scholar]
- Drost JT, Arpaci G, Ottervanger JP, de Boer MJ, van Eyck J, van der Schouw YT, Maas AH. Cardiovascular risk factors in women 10 years post early preeclampsia: the Preeclampsia Risk EValuation in FEMales study (PREVFEM) European journal of preventive cardiology. 2012;19:1138. doi: 10.1177/1741826711421079. [DOI] [PubMed] [Google Scholar]
- Duckworth S, Griffin M, Seed PT, North R, Myers J, Mackillop L, Simpson N, Waugh J, Anumba D, Kenny LC, et al. Diagnostic Biomarkers in Women With Suspected Preeclampsia in a Prospective Multicenter Study. Obstet Gynecol. 2016 doi: 10.1097/AOG.0000000000001508. [DOI] [PubMed] [Google Scholar]
- Dusse LM, Alpoim PN, Lwaleed BA, de Sousa LP, Carvalho M, Gomes KB. Is there a link between endothelial dysfunction, coagulation activation and nitric oxide synthesis in preeclampsia? Clin Chim Acta. 2013;415:226–229. doi: 10.1016/j.cca.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Fraser HM, Lunn SF. Angiogenesis and its control in the female reproductive system. British medical bulletin. 2000;56:787. doi: 10.1258/0007142001903364. [DOI] [PubMed] [Google Scholar]
- Gifford SM, Yi FX, Bird IM. Pregnancy-enhanced store-operated Ca2+ channel function in uterine artery endothelial cells is associated with enhanced agonist-specific transient receptor potential channel 3-inositol 1,4,5-trisphosphate receptor 2 interaction. The Journal of endocrinology. 2006;190:385. doi: 10.1677/joe.1.06773. [DOI] [PubMed] [Google Scholar]
- Gillespie SL, Porter K, Christian LM. Adaptation of the inflammatory immune response across pregnancy and postpartum in Black and White women. J Reprod Immunol. 2016;114:27–31. doi: 10.1016/j.jri.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RP, Killam AP, Brash AR, Branch RA. Prostacyclin production during pregnancy: comparison of production during normal pregnancy and pregnancy complicated by hypertension. Am J Obstet Gynecol. 1982;142:817–822. doi: 10.1016/s0002-9378(16)32525-x. [DOI] [PubMed] [Google Scholar]
- Goulopoulou S, Davidge ST. Molecular mechanisms of maternal vascular dysfunction in preeclampsia. Trends Mol Med. 2015;21:88–97. doi: 10.1016/j.molmed.2014.11.009. [DOI] [PubMed] [Google Scholar]
- Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br J Pharmacol. 2004;141:881–903. doi: 10.1038/sj.bjp.0705698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. 2009;104:288–291. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermehl DA, Janowiak MA, Vagnoni KE, Bird IM, Magness RR. Endothelial vasodilator production by uterine and systemic arteries. IV. Cyclooxygenase isoform expression during the ovarian cycle and pregnancy in sheep. Biology of reproduction. 2000;62:781. doi: 10.1095/biolreprod62.3.781. [DOI] [PubMed] [Google Scholar]
- Hammer ES, Cipolla MJ. Cerebrovascular Dysfunction in Preeclamptic Pregnancies. Curr Hypertens Rep. 2015;17:64. doi: 10.1007/s11906-015-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. 2006;12:1065–1074. doi: 10.1038/nm1452. [DOI] [PubMed] [Google Scholar]
- Hayman R, Warren A, Brockelsby J, Johnson I, Baker P. Plasma from women with pre-eclampsia induces an in vitro alteration in the endothelium-dependent behaviour of myometrial resistance arteries. BJOG: an international journal of obstetrics and gynaecology. 2000;107:108. doi: 10.1111/j.1471-0528.2000.tb11586.x. [DOI] [PubMed] [Google Scholar]
- Hermenegildo C, Oviedo PJ, Garcia-Martinez MC, Garcia-Perez MA, Tarin JJ, Cano A. Progestogens stimulate prostacyclin production by human endothelial cells. Hum Reprod. 2005;20:1554–1561. doi: 10.1093/humrep/deh803. [DOI] [PubMed] [Google Scholar]
- Irgens HU, Reisaeter L, Irgens LM, Lie RT. Long term mortality of mothers and fathers after pre-eclampsia: population based cohort study. BMJ (Clinical research ed.) 2001;323:1213. doi: 10.1136/bmj.323.7323.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabeen M, Yakoob MY, Imdad A, Bhutta ZA. Impact of interventions to prevent and manage preeclampsia and eclampsia on stillbirths. BMC public health. 2011;11(Suppl 3):S6. doi: 10.1186/1471-2458-11-S3-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowiak MA, Magness RR, Habermehl DA, Bird IM. Pregnancy increases ovine uterine artery endothelial cyclooxygenase-1 expression. Endocrinology. 1998;139:765. doi: 10.1210/endo.139.2.5739. [DOI] [PubMed] [Google Scholar]
- Jodkowska A, Martynowicz H, Kaczmarek-Wdowiak B, Mazur G. Thrombocytopenia in pregnancy - pathogenesis and diagnostic approach. Postepy Hig Med Dosw (Online) 2015;69:1215–1221. doi: 10.5604/17322693.1179649. [DOI] [PubMed] [Google Scholar]
- Jonsson Y, Ruber M, Matthiesen L, Berg G, Nieminen K, Sharma S, Ernerudh J, Ekerfelt C. Cytokine mapping of sera from women with preeclampsia and normal pregnancies. J Reprod Immunol. 2006;70:83–91. doi: 10.1016/j.jri.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Kaitu’u-Lino TJ, Palmer KR, Whitehead CL, Williams E, Lappas M, Tong S. MMP-14 is expressed in preeclamptic placentas and mediates release of soluble endoglin. Am J Pathol. 2012;180:888–894. doi: 10.1016/j.ajpath.2011.11.014. [DOI] [PubMed] [Google Scholar]
- Kazi AA, Koos RD. Estrogen-induced activation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor expression, and edema in the uterus are mediated by the phosphatidylinositol 3-kinase/Akt pathway. Endocrinology. 2007;148:2363–2374. doi: 10.1210/en.2006-1394. [DOI] [PubMed] [Google Scholar]
- Kenny LC, Baker PN, Kendall DA, Randall MD, Dunn WR. The role of gap junctions in mediating endothelium-dependent responses to bradykinin in myometrial small arteries isolated from pregnant women. Br J Pharmacol. 2002;136:1085–1088. doi: 10.1038/sj.bjp.0704817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharfi A, Giguère Y, Sapin V, Massé J, Dastugue B, Forest JC. Trophoblastic remodeling in normal and preeclamptic pregnancies: implication of cytokines. Clin Biochem. 2003;36:323–331. doi: 10.1016/s0009-9120(03)00060-2. [DOI] [PubMed] [Google Scholar]
- Kristiansson P, Wang JX. Reproductive hormones and blood pressure during pregnancy. Hum Reprod. 2001;16:13–17. doi: 10.1093/humrep/16.1.13. [DOI] [PubMed] [Google Scholar]
- Krupp J, Boeldt DS, Yi FX, Grummer MA, Bankowski Anaya HA, Shah DM, Bird IM. The loss of sustained Ca(2+) signaling underlies suppressed endothelial nitric oxide production in preeclamptic pregnancies: implications for new therapy. Am J Physiol Heart Circ Physiol. 2013;305:H969–979. doi: 10.1152/ajpheart.00250.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kublickiene KR, Lindblom B, Kruger K, Nisell H. Preeclampsia: evidence for impaired shear stress-mediated nitric oxide release in uterine circulation. Am J Obstet Gynecol. 2000;183:160–166. doi: 10.1067/mob.2000.105820. [DOI] [PubMed] [Google Scholar]
- Lampe PD, Lau AF. Regulation of gap junctions by phosphorylation of connexins. Archives of Biochemistry and Biophysics. 2000;384:205. doi: 10.1006/abbi.2000.2131. [DOI] [PubMed] [Google Scholar]
- Lau SY, Guild SJ, Barrett CJ, Chen Q, McCowan L, Jordan V, Chamley LW. Tumor necrosis factor-alpha, interleukin-6, and interleukin-10 levels are altered in preeclampsia: a systematic review and meta-analysis. Am J Reprod Immunol. 2013;70:412–427. doi: 10.1111/aji.12138. [DOI] [PubMed] [Google Scholar]
- Lawlor DA, Macdonald-Wallis C, Fraser A, Nelson SM, Hingorani A, Davey Smith G, Sattar N, Deanfield J. Cardiovascular biomarkers and vascular function during childhood in the offspring of mothers with hypertensive disorders of pregnancy: findings from the Avon Longitudinal Study of Parents and Children. European heart journal. 2012;33:335. doi: 10.1093/eurheartj/ehr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, et al. Circulating angiogenic factors and the risk of preeclampsia. The New England journal of medicine. 2004;350:672. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- Lewis PJ, Boylan P, Friedman LA, Hensby CN, Downing I. Prostacyclin in pregnancy. Br Med J. 1980;280:1581–1582. doi: 10.1136/bmj.280.6231.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Mata KM, Mazzuca MQ, Khalil RA. Altered matrix metalloproteinase-2 and -9 expression/activity links placental ischemia and anti-angiogenic sFlt-1 to uteroplacental and vascular remodeling and collagen deposition in hypertensive pregnancy. Biochem Pharmacol. 2014;89:370–385. doi: 10.1016/j.bcp.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Afink GB, Dijke PT. Soluble fms-like tyrosine kinase 1 and soluble endoglin are elevated circulating anti-angiogenic factors in pre-eclampsia. Pregnancy Hypertens. 2012;2:358–367. doi: 10.1016/j.preghy.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Lorentzen B, Endresen MJ, Hovig T, Haug E, Henriksen T. Sera from preeclamptic women increase the content of triglycerides and reduce the release of prostacyclin in cultured endothelial cells. Thromb Res. 1991;63:363–372. doi: 10.1016/0049-3848(91)90139-n. [DOI] [PubMed] [Google Scholar]
- Luksha L, Agewall S, Kublickiene K. Endothelium-derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis. 2009;202:330–344. doi: 10.1016/j.atherosclerosis.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Luksha L, Nisell H, Luksha N, Kublickas M, Hultenby K, Kublickiene K. Endothelium-derived hyperpolarizing factor in preeclampsia: heterogeneous contribution, mechanisms, and morphological prerequisites. Am J Physiol Regul Integr Comp Physiol. 2008;294:R510–519. doi: 10.1152/ajpregu.00458.2007. [DOI] [PubMed] [Google Scholar]
- Magness RR, Shaw CE, Phernetton TM, Zheng J, Bird IM. Endothelial vasodilator production by uterine and systemic arteries. II. Pregnancy effects on NO synthase expression. The American Journal of Physiology. 1997;272:H1730. doi: 10.1152/ajpheart.1997.272.4.H1730. [DOI] [PubMed] [Google Scholar]
- Magness RR, Shideman CR, Habermehl DA, Sullivan JA, Bird IM. Endothelial vasodilator production by uterine and systemic arteries. V. Effects of ovariectomy, the ovarian cycle, and pregnancy on prostacyclin synthase expression. Prostaglandins & other lipid mediators. 2000;60:103. doi: 10.1016/s0090-6980(99)00055-6. [DOI] [PubMed] [Google Scholar]
- Magness RR, Zheng J. Maternal Cardiovascular Alterations During Pregnancy. In: H MA, Gluckman PD, editors. Pediatric and Perinatal Perspectives. London: Edward Arnold Publishers; 1996. p. 762. [Google Scholar]
- Mahdy Z, Otun HA, Dunlop W, Gillespie JI. The responsiveness of isolated human hand vein endothelial cells in normal pregnancy and in pre-eclampsia. The Journal of physiology. 1998;508(Pt 2):609. doi: 10.1111/j.1469-7793.1998.00609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter F, S BM. Eclampsia. VIII. Risk factors for maternal morbidity. 2000;182:307–312. doi: 10.1016/s0002-9378(00)70216-x. [DOI] [PubMed] [Google Scholar]
- Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. The Journal of clinical investigation. 2003;111:649. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. American journal of physiology Cell physiology. 2007;292:C82. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- Mihu D, Razvan C, Malutan A, Mihaela C. Evaluation of maternal systemic inflammatory response in preeclampsia. Taiwan J Obstet Gynecol. 2015;54:160–166. doi: 10.1016/j.tjog.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Mills JL, DerSimonian R, Raymond E, Morrow JD, Roberts LJ, 2nd, Clemens JD, Hauth JC, Catalano P, Sibai B, Curet LB, et al. Prostacyclin and thromboxane changes predating clinical onset of preeclampsia: a multicenter prospective study. JAMA. 1999;282:356–362. doi: 10.1001/jama.282.4.356. [DOI] [PubMed] [Google Scholar]
- Morschauser TJ, Ramadoss J, Koch JM, Yi FX, Lopez GE, Bird IM, Magness RR. Local effects of pregnancy on connexin proteins that mediate Ca2+-associated uterine endothelial NO synthesis. Hypertension. 2014;63:589–594. doi: 10.1161/HYPERTENSIONAHA.113.01171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JS, Davidge ST. Arterial endothelium-derived hyperpolarization: potential role in pregnancy adaptations and complications. J Cardiovasc Pharmacol. 2013;61:197–203. doi: 10.1097/FJC.0b013e31827b6367. [DOI] [PubMed] [Google Scholar]
- Mukaida N, Harada A, Matsushima K. Interleukin-8 (IL-8) and monocyte chemotactic and activating factor (MCAF/MCP-1), chemokines essentially involved in inflammatory and immune reactions. Cytokine Growth Factor Rev. 1998;9:9–23. doi: 10.1016/s1359-6101(97)00022-1. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Matsui K, Ito M, Yoshimura T, Kawasaki N, Fujisaki S, Okamura H. Effects of pregnancy and hormone treatments on pressor response to angiotensin II in conscious rats. Am J Obstet Gynecol. 1988;159:989–995. doi: 10.1016/s0002-9378(88)80186-8. [DOI] [PubMed] [Google Scholar]
- Novak K, Kaufman S. Effects of pregnancy, estradiol, and progesterone on pressor responsiveness to angiotensin II. Am J Physiol. 1991;261:R1164–1170. doi: 10.1152/ajpregu.1991.261.5.R1164. [DOI] [PubMed] [Google Scholar]
- O’Leary P, Boyne P, Flett P, Beilby J, James I. Longitudinal assessment of changes in reproductive hormones during normal pregnancy. Clin Chem. 1991;37:667–672. [PubMed] [Google Scholar]
- Onogi A, Naruse K, Sado T, Tsunemi T, Shigetomi H, Noguchi T, Yamada Y, Akasaki M, Oi H, Kobayashi H. Hypoxia inhibits invasion of extravillous trophoblast cells through reduction of matrix metalloproteinase (MMP)-2 activation in the early first trimester of human pregnancy. Placenta. 2011;32:665–670. doi: 10.1016/j.placenta.2011.06.023. [DOI] [PubMed] [Google Scholar]
- Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology (Bethesda) 2009;24:58–71. doi: 10.1152/physiol.00033.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page EW, Vilee CA, Vilee DB. Physiological Adjustments in Pregnancy in Human Reproduction: The core content of Obstetrics, Gynecology, and Perinatal Medicine. WB Saunders Company; 1972. p. 253. [Google Scholar]
- Page NM. The endocrinology of pre-eclampsia. Clin Endocrinol (Oxf) 2002;57:413–423. doi: 10.1046/j.1365-2265.2002.01626.x. [DOI] [PubMed] [Google Scholar]
- Palmer SK, Zamudio S, Coffin C, Parker S, Stamm E, Moore LG. Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet Gynecol. 1992;80:1000–1006. [PubMed] [Google Scholar]
- Paulson AF, Lampe PD, Meyer RA, TenBroek E, Atkinson MM, Walseth TF, Johnson RG. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. Journal of cell science. 2000;113:3037. doi: 10.1242/jcs.113.17.3037. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Wang Q. Cyclic nucleotide crosstalk and the regulation of cerebral vasodilation. Prog Neurobiol. 1998;56:1–18. doi: 10.1016/s0301-0082(98)00009-4. [DOI] [PubMed] [Google Scholar]
- Pepper MS. Manipulating angiogenesis. From basic science to the bedside. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17:605. doi: 10.1161/01.atv.17.4.605. [DOI] [PubMed] [Google Scholar]
- Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21:1104–1117. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- Pinheiro MB, Gomes KB, Ronda CR, Guimaraes GG, Freitas LG, Teixeira-Carvalho A, Martins-Filho OA, Dusse LM. Severe preeclampsia: association of genes polymorphisms and maternal cytokines production in Brazilian population. Cytokine. 2015;71:232–237. doi: 10.1016/j.cyto.2014.10.021. [DOI] [PubMed] [Google Scholar]
- Plaks V, Birnberg T, Berkutzki T, Sela S, BenYashar A, Kalchenko V, Mor G, Keshet E, Dekel N, Neeman M, et al. Uterine DCs are crucial for decidua formation during embryo implantation in mice. J Clin Invest. 2008;118:3954–3965. doi: 10.1172/JCI36682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston L, McCarthy AL, Ritter JM. Control of vascular resistance in the maternal and feto-placental arterial beds. Pharmacol Ther. 1995;65:215–239. doi: 10.1016/0163-7258(94)00064-a. [DOI] [PubMed] [Google Scholar]
- Ray JG, Vermeulen MJ, Schull MJ, Redelmeier DA. Cardiovascular health after maternal placental syndromes (CHAMPS): population-based retrospective cohort study. Lancet. 2005;366:1797. doi: 10.1016/S0140-6736(05)67726-4. [DOI] [PubMed] [Google Scholar]
- Redman CW, Sargent IL. Pre-eclampsia, the placenta and the maternal systemic inflammatory response–a review. Placenta. 2003;24:S21. doi: 10.1053/plac.2002.0930. [DOI] [PubMed] [Google Scholar]
- Remuzzi G, Marchesi D, Zoja C, Muratore D, Mecca G, Misiani R, Rossi E, Barbato M, Capetta P, Donati MB, et al. Reduced umbilical and placental vascular prostacyclin in severe pre-eclampsia. Prostaglandins. 1980;20:105–110. doi: 10.1016/0090-6980(80)90010-6. [DOI] [PubMed] [Google Scholar]
- Robertson WB, Brosens I, Dixon G. Uteroplacental vascular pathology. European journal of obstetrics, gynecology, and reproductive biology. 1975;5:47. doi: 10.1016/0028-2243(75)90130-6. [DOI] [PubMed] [Google Scholar]
- Robinson CJ, Johnson DD, Chang EY, Armstrong DM, Wang W. Evaluation of placenta growth factor and soluble Fms-like tyrosine kinase 1 receptor levels in mild and severe preeclampsia. Am J Obstet Gynecol. 2006;195:255–259. doi: 10.1016/j.ajog.2005.12.049. [DOI] [PubMed] [Google Scholar]
- Rosing U, Carlstrom K. Serum levels of unconjugated and total oestrogens and dehydroepiandrosterone, progesterone and urinary oestriol excretion in pre-eclampsia. Gynecol Obstet Invest. 1984;18:199–205. doi: 10.1159/000299081. [DOI] [PubMed] [Google Scholar]
- Salamalekis E, Bakas P, Vitoratos N, Eleptheriadis M, Creatsas G. Androgen levels in the third trimester of pregnancy in patients with preeclampsia. Eur J Obstet Gynecol Reprod Biol. 2006;126:16–19. doi: 10.1016/j.ejogrb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Saleh L, Verdonk K, Visser W, van den Meiracker AH, Danser AH. The emerging role of endothelin-1 in the pathogenesis of pre-eclampsia. Ther Adv Cardiovasc Dis. 2016 doi: 10.1177/1753944715624853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandrim VC, Palei AC, Sertorio JT, Amaral LM, Cavalli RC, Tanus-Santos JE. Alterations in cyclic GMP levels in preeclampsia may reflect increased B-type natriuretic peptide levels and not impaired nitric oxide activity. Clin Biochem. 2011;44:1012–1014. doi: 10.1016/j.clinbiochem.2011.05.026. [DOI] [PubMed] [Google Scholar]
- Sathishkumar K, Balakrishnan M, Chinnathambi V, Chauhan M, Hankins GD, Yallampalli C. Fetal sex-related dysregulation in testosterone production and their receptor expression in the human placenta with preeclampsia. J Perinatol. 2012;32:328–335. doi: 10.1038/jp.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiessl B, Strasburger C, Bidlingmaier M, Mylonas I, Jeschke U, Kainer F, Friese K. Plasma- and urine concentrations of nitrite/nitrate and cyclic Guanosinemonophosphate in intrauterine growth restricted and preeclamptic pregnancies. Archives of Gynecology and Obstetrics. 2006;274:150. doi: 10.1007/s00404-006-0149-8. [DOI] [PubMed] [Google Scholar]
- Schneider F, Lutun P, Baldauf JJ, Quirin L, Dreyfus M, Ritter J, Tempe JD. Plasma cyclic GMP concentrations and their relationship with changes of blood pressure levels in pre-eclampsia. Acta Obstet Gynecol Scand. 1996;75:40–44. doi: 10.3109/00016349609033281. [DOI] [PubMed] [Google Scholar]
- Sharifzadeh F, Kashanian M, Fatemi F. A comparison of serum androgens in pre-eclamptic and normotensive pregnant women during the third trimester of pregnancy. Gynecol Endocrinol. 2012;28:834–836. doi: 10.3109/09513590.2012.683061. [DOI] [PubMed] [Google Scholar]
- Sharma A, Satyam A, Sharma JB. Leptin, IL-10 and inflammatory markers (TNF-alpha, IL-6 and IL-8) in pre-eclamptic, normotensive pregnant and healthy non-pregnant women. Am J Reprod Immunol. 2007;58:21–30. doi: 10.1111/j.1600-0897.2007.00486.x. [DOI] [PubMed] [Google Scholar]
- Sharma S, Godbole G, Modi D. Decidual Control of Trophoblast Invasion. Am J Reprod Immunol. 2016;75:341–350. doi: 10.1111/aji.12466. [DOI] [PubMed] [Google Scholar]
- Shaul PW, Farrar MA, Magness RR. Prostacyclin synthesis and stimulation of cyclic AMP production in ovine fetal vasculature: heterogeneity in pulmonary and systemic arteries. Dev Pharmacol Ther. 1992;18:89–99. [PubMed] [Google Scholar]
- Shimada S, Nishida R, Takeda M, Iwabuchi K, Kishi R, Onoe K, Minakami H, Yamada H. Natural killer, natural killer T, helper and cytotoxic T cells in the decidua from sporadic miscarriage. Am J Reprod Immunol. 2006;56:193–200. doi: 10.1111/j.1600-0897.2006.00417.x. [DOI] [PubMed] [Google Scholar]
- Sibai BM, Caritis S, Hauth J, National Institute of Child Health and Human Development Maternal-Fetal Medicine Units N What we have learned about preeclampsia. Seminars in perinatology. 2003;27:239. doi: 10.1016/s0146-0005(03)00022-3. [DOI] [PubMed] [Google Scholar]
- Sibai BM, Caritis SN, Thom E, Klebanoff M, McNellis D, Rocco L, Paul RH, Romero R, Witter F, Rosen M, et al. Prevention of preeclampsia with low-dose aspirin in healthy, nulliparous pregnant women. The National Institute of Child Health and Human Development Network of Maternal-Fetal Medicine Units. N Engl J Med. 1993;329:1213–1218. doi: 10.1056/NEJM199310213291701. [DOI] [PubMed] [Google Scholar]
- Sikorski K, Czerwoniec A, Bujnicki JM, Wesoly J, Bluyssen HA. STAT1 as a novel therapeutical target in pro-atherogenic signal integration of IFNgamma, TLR4 and IL-6 in vascular disease. Cytokine Growth Factor Rev. 2011;22:211–219. doi: 10.1016/j.cytogfr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Simoncini T, Fu XD, Caruso A, Garibaldi S, Baldacci C, Giretti MS, Mannella P, Flamini MI, Sanchez AM, Genazzani AR. Drospirenone increases endothelial nitric oxide synthesis via a combined action on progesterone and mineralocorticoid receptors. Hum Reprod. 2007;22:2325–2334. doi: 10.1093/humrep/dem109. [DOI] [PubMed] [Google Scholar]
- Sladek SM, Magness RR, Conrad KP. Nitric oxide and pregnancy. The American Journal of Physiology. 1997;272:R441. doi: 10.1152/ajpregu.1997.272.2.R441. [DOI] [PubMed] [Google Scholar]
- Smith R, Smith JI, Shen X, Engel PJ, Bowman ME, McGrath SA, Bisits AM, McElduff P, Giles WB, Smith DW. Patterns of plasma corticotropin-releasing hormone, progesterone, estradiol, and estriol change and the onset of human labor. J Clin Endocrinol Metab. 2009;94:2066–2074. doi: 10.1210/jc.2008-2257. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochimica et biophysica acta. 2005;1711:154. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, Zamudio S, Post M, Caniggia I. Molecular evidence of placental hypoxia in preeclampsia. The Journal of clinical endocrinology and metabolism. 2005;90:4299. doi: 10.1210/jc.2005-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steier JA, Ulstein M, Myking OL. Human chorionic gonadotropin and testosterone in normal and preeclamptic pregnancies in relation to fetal sex. Obstet Gynecol. 2002;100:552–556. doi: 10.1016/s0029-7844(02)02088-4. [DOI] [PubMed] [Google Scholar]
- Steinert JR, Wyatt AW, Poston L, Jacob R, Mann GE. Preeclampsia is associated with altered Ca2+ regulation and NO production in human fetal venous endothelial cells. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2002;16:721. doi: 10.1096/fj.01-0916fje. [DOI] [PubMed] [Google Scholar]
- Suarez S, Ballmer-Hofer K. VEGF transiently disrupts gap junctional communication in endothelial cells. Journal of cell science. 2001;114:1229. doi: 10.1242/jcs.114.6.1229. [DOI] [PubMed] [Google Scholar]
- Sullivan JA, Grummer MA, Yi FX, Bird IM. Pregnancy-enhanced endothelial nitric oxide synthase (eNOS) activation in uterine artery endothelial cells shows altered sensitivity to Ca2+, U0126, and wortmannin but not LY294002–evidence that pregnancy adaptation of eNOS activation occurs at multiple levels of cell signaling. Endocrinology. 2006;147:2442. doi: 10.1210/en.2005-0399. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Hattori T, Kajikuri J, Yamamoto T, Suzumori K, Itoh T. Reduced function of endothelial prostacyclin in human omental resistance arteries in pre-eclampsia. J Physiol. 2002;545:269–277. doi: 10.1113/jphysiol.2002.022384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, Pang Y. Protective actions of progesterone in the cardiovascular system: potential role of membrane progesterone receptors (mPRs) in mediating rapid effects. Steroids. 2013;78:583–588. doi: 10.1016/j.steroids.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Tosun M, Celik H, Avci B, Yavuz E, Alper T, Malatyalioglu E. Maternal and umbilical serum levels of interleukin-6, interleukin-8, and tumor necrosis factor-alpha in normal pregnancies and in pregnancies complicated by preeclampsia. J Matern Fetal Neonatal Med. 2010;23:880–886. doi: 10.3109/14767051003774942. [DOI] [PubMed] [Google Scholar]
- Tran QK, Leonard J, Black DJ, Nadeau OW, Boulatnikov IG, Persechini A. Effects of combined phosphorylation at Ser-617 and Ser-1179 in endothelial nitric-oxide synthase on EC50(Ca2+) values for calmodulin binding and enzyme activation. The Journal of biological chemistry. 2009;284:11892. doi: 10.1074/jbc.M806205200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troisi R, Potischman N, Johnson CN, Roberts JM, Lykins D, Harger G, Markovic N, Siiteri P, Hoover RN. Estrogen and androgen concentrations are not lower in the umbilical cord serum of pre-eclamptic pregnancies. Cancer Epidemiol Biomarkers Prev. 2003a;12:1268–1270. [PubMed] [Google Scholar]
- Troisi R, Potischman N, Roberts JM, Ness R, Crombleholme W, Lykins D, Siiteri P, Hoover RN. Maternal serum oestrogen and androgen concentrations in preeclamptic and uncomplicated pregnancies. Int J Epidemiol. 2003b;32:455–460. doi: 10.1093/ije/dyg094. [DOI] [PubMed] [Google Scholar]
- Tropea T, De Francesco EM, Rigiracciolo D, Maggiolini M, Wareing M, Osol G, Mandala M. Pregnancy Augments G Protein Estrogen Receptor (GPER) Induced Vasodilation in Rat Uterine Arteries via the Nitric Oxide - cGMP Signaling Pathway. PloS one. 2015;10:e0141997. doi: 10.1371/journal.pone.0141997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RJ, Bloemenkamp KW, Penning ME, Bruijn JA, Baelde HJ. From Glomerular Endothelium to Podocyte Pathobiology in Preeclampsia: a Paradigm Shift. Curr Hypertens Rep. 2015;17:54. doi: 10.1007/s11906-015-0566-9. [DOI] [PubMed] [Google Scholar]
- Wallis AB, Saftlas AF, Hsia J, Atrash HK. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension, United States, 1987–2004. Am J Hypertens. 2008;21:521–526. doi: 10.1038/ajh.2008.20. [DOI] [PubMed] [Google Scholar]
- Wang JD, Fu Y, Shi WL, Zhu PD, Cheng J, Qiao GM, Wang YQ, Greene GL. Immunohistochemical localization of progesterone receptor in human decidua of early pregnancy. Hum Reprod. 1992;7:123–127. doi: 10.1093/oxfordjournals.humrep.a137545. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gu Y, Loyd S, Jia X, Groome LJ. Increased urinary levels of podocyte glycoproteins, matrix metallopeptidases, inflammatory cytokines, and kidney injury biomarkers in women with preeclampsia. Am J Physiol Renal Physiol. 2015;309:F1009–1017. doi: 10.1152/ajprenal.00257.2015. [DOI] [PubMed] [Google Scholar]
- Warn-Cramer BJ, Lampe PD, Kurata WE, Kanemitsu MY, Loo LW, Eckhart W, Lau AF. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. The Journal of biological chemistry. 1996;271:3779. doi: 10.1074/jbc.271.7.3779. [DOI] [PubMed] [Google Scholar]
- Wiggins-Dohlvik K, Merriman M, Shaji CA, Alluri H, Grimsley M, Davis ML, Smith RW, Tharakan B. Tumor necrosis factor-alpha disruption of brain endothelial cell barrier is mediated through matrix metalloproteinase-9. Am J Surg. 2014;208:954–960. doi: 10.1016/j.amjsurg.2014.08.014. discussion 960. [DOI] [PubMed] [Google Scholar]
- Xie C, Yao MZ, Liu JB, Xiong LK. A meta-analysis of tumor necrosis factor-alpha, interleukin-6, and interleukin-10 in preeclampsia. Cytokine. 2011;56:550–559. doi: 10.1016/j.cyto.2011.09.021. [DOI] [PubMed] [Google Scholar]
- Xu C, Wu X, Hack BK, Bao L, Cunningham PN. TNF causes changes in glomerular endothelial permeability and morphology through a Rho and myosin light chain kinase-dependent mechanism. Physiol Rep. 2015;3 doi: 10.14814/phy2.12636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Wang YL, Zhu SJ, Luo SY, Piao YS, Zhuang LZ. Expression of matrix metalloproteinase-2, -9, and -14, tissue inhibitors of metalloproteinase-1, and matrix proteins in human placenta during the first trimester. Biol Reprod. 2000;62:988–994. doi: 10.1095/biolreprod62.4.988. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Suzuki Y, Kojima K, Suzumori N, Suzuki T. The biological investigation of prostacyclin in preeclamptic women seen reduced endothelial function. Hypertens Pregnancy. 2010;29:484–491. doi: 10.3109/10641950903322873. [DOI] [PubMed] [Google Scholar]
- Yang W, Ahn H, Hinrichs M, Torry RJ, Torry DS. Evidence of a novel isoform of placenta growth factor (PlGF-4) expressed in human trophoblast and endothelial cells. J Reprod Immunol. 2003;60:53–60. doi: 10.1016/s0165-0378(03)00082-2. [DOI] [PubMed] [Google Scholar]
- Yang Y, Su X, Xu W, Zhou R. Interleukin-18 and interferon gamma levels in preeclampsia: a systematic review and meta-analysis. Am J Reprod Immunol. 2014;72:504–514. doi: 10.1111/aji.12298. [DOI] [PubMed] [Google Scholar]
- Yi FX, Boeldt DS, Gifford SM, Sullivan JA, Grummer MA, Magness RR, Bird IM. Pregnancy enhances sustained Ca2+ bursts and endothelial nitric oxide synthase activation in ovine uterine artery endothelial cells through increased connexin 43 function. Biology of reproduction. 2010;82:66. doi: 10.1095/biolreprod.109.078253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi FX, Magness RR, Bird IM. Simultaneous imaging of Ca2+ i and intracellular NO production in freshly isolated uterine artery endothelial cells: effects of ovarian cycle and pregnancy. American journal of physiology. Regulatory, integrative and comparative physiology. 2005;288:R140. doi: 10.1152/ajpregu.00302.2004. [DOI] [PubMed] [Google Scholar]
- Yong HE, Melton PE, Johnson MP, Freed KA, Kalionis B, Murthi P, Brennecke SP, Keogh RJ, Moses EK. Genome-wide transcriptome directed pathway analysis of maternal pre-eclampsia susceptibility genes. PloS one. 2015;10:e0128230. doi: 10.1371/journal.pone.0128230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisler H, Llurba E, Chantraine F, Vatish M, Staff AC, Sennstrom M, Olovsson M, Brennecke SP, Stepan H, Allegranza D, et al. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N Engl J Med. 2016;374:13–22. doi: 10.1056/NEJMoa1414838. [DOI] [PubMed] [Google Scholar]
- Zhu J, Zhong M, Pang Z, Yu Y. Dysregulated expression of matrix metalloproteinases and their inhibitors may participate in the pathogenesis of pre-eclampsia and fetal growth restriction. Early Hum Dev. 2014;90:657–664. doi: 10.1016/j.earlhumdev.2014.08.007. [DOI] [PubMed] [Google Scholar]