

Abstract
Objective
Spinocerebellar ataxia type 1 is an autosomal dominant fatal neurodegenerative disease caused by a polyglutamine expansion in the coding region of ATXN1. We showed previously that partial suppression of mutant ataxin‐1 (ATXN1) expression, using virally expressed RNAi triggers, could prevent disease symptoms in a transgenic mouse model and a knockin mouse model of the disease, using a single dose of virus. Here, we set out to test whether RNAi triggers targeting ATXN1 could not only prevent, but also reverse disease readouts when delivered after symptom onset.
Methods
We administered recombinant adeno‐associated virus (rAAV) expressing miS1, an artificial miRNA targeting human ATXN1 mRNA (rAAV.miS1), to a mouse model of spinocerebellar ataxia type 1 (SCA1; B05 mice). Viruses were delivered prior to or after symptom onset at multiple doses. Control B05 mice were treated with rAAVs expressing a control artificial miRNA, or with saline. Animal behavior, molecular phenotypes, neuropathology, and magnetic resonance spectroscopy were done on all groups, and data were compared to wild‐type littermates.
Results
We found that SCA1 phenotypes could be reversed by partial suppression of human mutant ATXN1 mRNA by rAAV.miS1 when delivered after symptom onset. We also identified the therapeutic range of rAAV.miS1 that could prevent or reverse disease readouts.
Interpretation
SCA1 disease may be reversible by RNAi therapy, and the doses required for advancing this therapy to humans are delineated. Ann Neurol 2016;80:754–765
Spinocerebellar ataxia type 1 (SCA1) is an autosomal dominant progressive neurodegenerative disease caused by a polyglutamine repeat expansion in exon 8 of the ataxin‐1 (ATXN1) gene.1 Healthy individuals have from 6 to 42 CAG repeats in ATXN1 interspersed by 1 to 3 CAT codons. A pure CAG expansion exceeding 40 glutamines causes disease pathogenesis by a toxic gain‐of‐function mechanism. Although ataxin‐1 protein (ATXN1) is ubiquitously expressed, specific cerebellar Purkinje cells (PCs) and brainstem neurons are more susceptible to expanded ATXN1 expression and carry the bulk of pathology. As a result, patients with SCA1 develop a prominent cerebellar syndrome with brainstem features, including gait and limb ataxia, dysarthria, or dysphagia, among others. To date, SCA1 remains a fatal disease with no known disease‐modifying therapies.
The initial mouse model for SCA1 is a transgenic line expressing mutant human ATXN1 from a PC‐specific promoter (Pcp2).2, 3 This mouse, known as the B05 model, develops progressive disease, with many features similar to SCA1 including measurable gait deficits by 7 weeks of age. Additionally, there are transcriptional changes by postnatal day 25 and PC loss by 24 weeks of age.3 Earlier, a doxycycline‐inducible form of the B05 model revealed that preexisting phenotypes were reversible after many weeks of mutant ATXN1 expression.4 This suggests that a window of opportunity exists not only for halting the disease, but perhaps also to reverse the presence or severity of symptoms once present.
We previously assessed the utility of gene‐silencing strategies as a putative therapy for SCA1 in this and a knockin (KI) model of disease. Delivery of recombinant adeno‐associated viruses (rAAVs) expressing RNAi triggers in the form of shRNAs provided clinical benefit in B05 mice after direct injection into the cerebellar cortex.5 More recently, we showed that delivery of rAAVs expressing artificial miRNAs directed at mutant ATXN1 mRNA, to the deep cerebellar nuclei (DCN) for transport to PCs and other brainstem neurons, also improved pathologic and behavioral phenotypes in this model.6 Similarly, rAAVs expressing miRNAs directed at mutant mouse Atxn1 mRNA was beneficial when applied to the KI SCA1 model.7 Importantly, the DCN‐directed approach is scalable from mice to larger mammals; clinically relevant biodistribution and silencing were observed when tested in nonhuman primates.8 Although these studies provided the necessary proof of principle for advancing RNAi therapy for SCA1, the treatment was applied before symptom onset.
Here, we set out to test the clinically relevant hypothesis stating that viral‐expressed RNAi provides therapeutic benefit when delivered after symptom onset. Furthermore, before advancing this treatment to patients it is critical to define the dosing window between the minimal effective doses and the toxicity threshold. We therefore evaluated the effects of increasing doses of a clinical SCA1 gene therapy product directed at silencing mutant ATXN1 mRNA on the prevention or reversal of clinical and neuropathological signs in B05 mice.
Materials and Methods
Plasmids and Viral Vectors
The therapeutic miRNA sequence targeting human and rhesus ataxin‐1 (miS1) has been previously described.7 The original therapeutic vector rAAV1.miS1.eGFP was modified to no longer express enhanced green fluorescence protein (eGFP) and instead contain a “safe stuffer” sequence.9 Recombinant rAAV serotype 2/1 vectors (rAAV1.miS1 and rAAV1.miControl) were generated at the Children's Hospital of Philadelphia Research Vector Core. AAV vectors were resuspended in Diluent Buffer (the Children's Hospital of Philadelphia Research Vector Core), and titers (viral genomes/ml) were determined by quantitative polymerase chain reaction.
Cell Culture and Transfection
HEK293 cells were transfected (Lipofectamine 2000) in quadruplicate in 24‐well plates per manufacturer's instructions with 500ng of plasmid containing pAAV.miS1.eGFP, pAAV.miS1, pAAV, or no plasmid. Total RNA was harvested at 24 hours with TRIzol.
Animals
All animal protocols were approved by the Children's Hospital of Philadelphia Animal Care and Use Committee. Wild‐type FVB mice were obtained from Jackson Laboratory (Bar Harbor, ME). B05 transgenic mice were previously provided by Dr H. T. Orr and rederived by Jackson Laboratory. The B05 line was maintained on the FVB background. Mice were genotyped using primers specific for the mutant human ataxin‐1 transgene.2 Hemizygous and age‐matched wild‐type littermates were used for the indicated experiments. Treatment groups comprised approximately equal numbers of male and female mice. Mice were housed in a controlled temperature environment on a 12‐hour light/dark cycle. Food and water were provided ad libitum.
AAV Injections and Brain Tissue Isolation
B05 mice were injected with rAAV1 vectors expressing miS1 or a control scrambled miRNA sequence (miC). Mice were stereotaxically injected bilaterally to the deep cerebellar nuclei (coordinates: −6.0mm caudal to bregma, ±2.0mm from midline, and −2.2mm deep from the cerebellar surface) with 4μl of rAAV1 virus at doses of 1 × 107 vector genomes (vg), 1 × 108 vg, 6 × 108 vg, 1 × 109 vg, 6 × 109 vg, or 1 × 1010 vg/hemisphere or saline (Diluent Buffer). Mice were anesthetized with 4% isoflurane/oxygen mixtures and transcardially perfused with 20ml of ice‐cold saline. Mice were decapitated, and for histological analyses, brains were removed and postfixed overnight in 4% paraformaldehyde. Brains were stored in 30% sucrose/0.05% azide solution at 4°C until cut on a sledge microtome at 40μm thickness and stored at −20°C in a cryoprotectant solution. For RNA and metabolite analyses, brains were removed and cerebellar hemispheres were flash frozen in liquid nitrogen and stored at −80°C. RNA was isolated from whole cerebellum using 1ml of Trizol; RNA quantity and quality were measured using a NanoDrop 2000. For metabolite analysis, tissues were subjected to perchloric acid extraction. Frozen samples were weighed and homogenized using beads in a TissueLyzer LT (Qiagen, Valencia, CA). Ice‐cold 3.6% HClO4 was added, further homogenized, and centrifuged at 4°C. Supernatant was buffered to a pH of ∼7.0 with KOH, centrifuged, lyophilized, and again weighed.
Immunohistochemical Analysis
Free‐floating sagittal cerebellar sections (40μm thick) were washed in 1 × Tris‐buffered (TBS) with 0.05% Triton X‐100 at room temperature and blocked for 1 hour in 5% serum, 0.05% Triton X‐100, in 1 × TBS. Sections were incubated with primary antibody in 3% serum and 0.05% Triton X‐100 in TBS overnight at room temperature. Primary antibodies used were polyclonal rabbit anticalbindin (1:2,000; Sigma, St Louis, MO), polyclonal anti‐Iba1 (1:1,000; Wako Chemicals, Richmond, VA), polyclonal anti–glial fibrillary acidic protein (1:2,000; Dako, Carpinteria, CA), and polyclonal rabbit 12NQ (1:1,000; Orr laboratory10). For fluorescent immunohistochemistry (IHC), sections were incubated with goat antirabbit Alexa Fluor 488 or 568 (1:200; Life Technologies, Carlsbad, CA) in 3% serum and 0.05% Triton X‐100 in 1 × TBS for 1 hour at room temperature. For 3,3′‐diaminobenzidine IHC, sections were incubated in goat antirabbit biotin‐labeled secondary antibody (1:200; Jackson ImmunoResearch, West Grove, PA) in 3% serum and 0.05% Triton X‐100 in 1 × TBS for 1 hour at room temperature. Tissues were developed with Vectastain ABC Elite Kit (Vector Laboratories, Burlingame, CA), according to the manufacturer's instructions. All sections were mounted onto Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA) and cover‐slipped with Fluoro‐Gel (Electron Microscopy Sciences, Hatfield, PA) or dehydrated and cover‐slipped with 1,3,‐diethyl‐8‐phenylxanthine. Images were captured on a Leica (Wetzlar, Germany) DM6000B fluorescence microscope using LAS X software.
Semiquantitative Polymerase Chain Reaction
Reverse transcription (High Capacity cDNA Reverse Transcription Kit; Applied Biosystems, Foster City, CA) was performed on 1μg total RNA collected from cerebellum using a standard stem‐loop polymerase chain reaction (PCR) primer designed to identify miS1 previously described.7 Semiquantitative RT‐cDNA was subjected to reverse transcriptase (RT)‐PCR with a standard reverse primer and a forward primer specific to miS1.
Quantitative PCR
Random‐primer first‐strand cDNA synthesis was performed using 2μg total RNA (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems) per manufacturer's instructions. Assays were performed on a Bio‐Rad (Hercules, CA) CFX384 Real Time System using TaqMan (Thermo Fisher Scientific, Waltham, MA) primer/probe sets specific for human ataxin‐1, mouse Pcp2, mouse Grm1, or mouse β‐actin (TaqMan 2X Universal Master Mix, Life Technologies).
1H‐Magnetic Resonance Spectroscopy
Analysts were blinded to the treatment groups. Nuclear magnetic resonance (NMR) spectroscopy was performed at 400MHz on a Bruker (Billerica, MA) Avance III 400 wide‐bore spectrometer. Each lyophilized tissue extract was dissolved in 0.4ml of D2O, the pH was adjusted to 7.0, and the solution was introduced into a 5mm NMR tube. An external standard made of a sealed capillary containing a solution of trimethylsilyl propionic acid in D2O was centered in the NMR tube and used as a chemical shift reference and quantitation standard. Fully relaxed proton spectra were acquired with a 5mm proton probe. Standard acquisition conditions were as follows: PW = 5 microseconds (45°), repetition time = 8.84 seconds (AQ = 4.84 seconds, D1 = 4 seconds), SW = 6,775Hz, TD = 64,000, 128 scans, 4 DS. A soft water saturation pulse was applied during the 4‐second relaxation delay.
Rotarod Analysis
Mice were tested by a tester blinded to the treatment groups on an accelerated rotarod apparatus (model 47600; Ugo Basile, Varese, Italy). For distribution to groups of equal abilities at baseline, mice were first tested at 5 weeks of age prior to treatment. Mice were habituated to the rotarod for 4 minutes, then subjected to three trials per day (with at least 30 minutes of rest between trials) for 4 consecutive days. For each trial, acceleration was from 4 to 40rpm over 5 minutes, and then speed was maintained at 40rpm. Latency to fall (or if mice hung on for 2 consecutive rotations without running) was recorded for each mouse per trial. Trials were stopped at 500 seconds, and mice remaining on the rod at that time were scored as 500 seconds. Two‐way analysis of variance followed by a Tukey post hoc analysis was used to assess for significant differences. Variables were time and treatment.
Statistical Analysis
For all studies, probability values were obtained by using 1‐way analysis of variance followed by Tukey post hoc analysis to assess for significant differences between individual groups. In all statistical analysis, p < 0.05 was considered significant.
Figure Preparation
All photographs were formatted with Adobe (San Jose, CA) Photoshop software. All graphs were made with GraphPad (San Diego, CA) Prism software. All figures were constructed with Adobe Illustrator software.
Results
AAV1.miS1 Prevents the Development of Rotarod Deficits in B05 Mice
We first modified the former rAAV1.miS17 wherein the eGFP reporter expression cassette was replaced with a stuffer sequence. The stuffer sequence ensured appropriate length of the expression cassette required for optimal AAV packaging (Fig 1A). To assess that this modification did not impact the silencing potency of the miRNA, we transfected HEK 293 cells with the shuttle plasmid pAAV.miS1.eGFP, pAAV.miS1, or a control plasmid. Compared to control transduced cells, both pAAV.miS1.eGFP and pAAV.miS1 significantly reduced ATXN1 mRNA expression 24 hours post‐transfection (see Fig 1B); replacing eGFP with stuffer sequence did not alter the potency of the artificial miRNA.
Figure 1.

Experimental design and rotarod analysis. (A) Diagram of therapeutic viral construct. Inverted terminal repeats (ITR) surround murine U6 promoter driving expression of an artificial miRNA targeting human ATXN1 (miS1) followed by a noncoding stuffer sequence. (B) Comparative silencing efficiency of pAAV.miS1.eGFP and pAAV1.miS1 in HEK293 cells (n = 4 replicates; ***p < 0.0001). (C) Timeline of disease progression in B05 mice and study design. (D) Rotarod performance over 4 days at 30 weeks of age. Treatment groups consisted of untreated wild‐type mice, B05 mice injected with saline, B05 mice injected with 8 × 108 vector genomes (vg) of rAAV2/1.miControl (denoted as miC), B05 mice injected with 8 × 107 vg (denoted as 8E7) of rAAV2/1.miS1, B05 mice injected with 8 × 108 vg (denoted as 8E8) of rAAV2/1.miS1, B05 mice injected with 8 × 109 vg (denoted as 8E9) of rAAV2/1.miS1, and B05 mice injected with 8 × 1010 vg (denoted as 8E10) of rAAV2/1.miS1 (*p < 0.05, ***p < 0.0001, difference from rAAV1.miC‐ and saline‐treated B05 animals).
We used the B05 model2 for this work, as the RNAi trigger expressed from rAAV1.miS1 targets human ATXN1. B05 transgenic mice show progressive disease, with transcriptional changes evident prior to noted behavioral deficits (see Fig 1C). To identify the efficacy and toxicity thresholds to prevent disease progression, mice were injected bilaterally into the DCN with increasing doses of rAAV1.miS1, rAAV1.miC, or saline after baseline behavior testing (Table 1). Twenty‐four weeks after injection (30 weeks of age), animals were reassayed by rotarod and then euthanized, and tissues were collected for postnecropsy analysis. As seen in Figure 1D, at 30 weeks of age control‐treated transgenic mice could not remain on the rotarod apparatus after 98.8 ± 22 seconds. B05 mice treated with rAAV1.miS1 at doses of 8 × 108 vg and 8 × 109 vg performed significantly better than control‐treated transgenic animals and were not statistically differently than their wild‐type littermates.
Table 1.
Treatment Groups for Preventative Study
| Genotype | Injectate | Dose, vg |
|---|---|---|
| B05 | rAAV1.miS1 | 8 × 107 |
| B05 | rAAV1.miS1 | 8 × 108 |
| B05 | rAAV1.miS1 | 8 × 109 |
| B05 | rAAV1.miS1 | 8 × 1010 |
| B05 | rAAV1.miC | 8 × 108 |
| B05 | Saline | — |
| Wild‐type | — | — |
vg = vector genomes.
rAAV1.miS1 Reduces ATXN1 mRNA in SCA1 Mouse in a Dose‐Dependent Manner, Preventing Changes in Cerebellar Metabolites
Semiquantitative PCR on whole cerebellar lysates confirmed miS1 expression (Fig 2A), with a clear dose response. Quantitative RT‐PCR for mutant human ATXN1 mRNA (see Fig 2B) showed a similar dose response that inversely correlated with miS1 levels. B05 mice treated with rAAV1.miC or rAAV1.miS1 at a dose of 8 × 107 vg had similar levels of ATXN1 mRNA (98 ± 4% and 100 ± 3%, respectively) relative to saline‐treated animals. B05 mice administered 8 × 108 vg or 8 × 109 vg of rAAV1.miS1 had increasingly reduced levels of ATXN1 mRNA (77 ± 3% and 47 ± 7%, respectively). B05 mice in the high‐dose group had almost complete reduction of human ATXN1 mRNA levels (4 ± 1) relative to the control‐treated animals.
Figure 2.

Semiquantitative polymerase chain reaction (PCR), quantitative reverse transcriptase (qRT)‐PCR, and nuclear magnetic resonance analyses of cerebellar extracts from treated B05 mice and untreated wild‐type (WT) littermates. (A) Semiquantitative analysis of miS1 from whole cerebellar extracts. (B) qRT‐PCR for human ATXN1 mRNA levels from whole cerebellar extracts (n ≥ 3; **p < 0.01, ***p < 0.0001, difference from saline‐injected SCA1 mice). (C) Ratio of N‐acetylaspartate (NAA)/inositol levels from whole cerebellar extracts (n = 3; ***p < 0.0001, difference from control‐treated B05 mice). ns = not significant; Sal = saline.
High‐field proton magnetic resonance spectroscopy (1H MRS) allows quantitation of biomarkers in SCA1 patients in a noninvasive manner. In 2010, Oz and colleagues showed that SCA1 patients have reduced N‐acetylaspartate (NAA) levels and elevated inositol levels.11 The same observations have been made in SCA1 mouse models. We therefore assayed metabolite levels in cerebellar lysates from all groups using NMR. Similar to results on untreated B05 mice,12 control‐treated mice had reduced NAA/inositol ratios compared to wild‐type littermates (see Fig 2C). However, this difference was normalized to wild‐type levels in B05 mice treated with 8 × 109 vg or 8 × 1010 vg of rAAV1.miS1.
rAAV.miS1 at 8 × 109 vg Prevents Cerebellar Pathology
In SCA1, PC dendrites progressively retract, resulting in cerebellar molecular layer (ML) thinning.13 To investigate the potential protective effect of rAAV1.miS1 on this phenotype, brain sections were evaluated by anticalbindin staining of sagittal sections, and ML widths were quantified. In control‐treated B05 animals, lobules III–IV/V have marked thinning, as do sections from animals injected with 8 × 108 vg of rAAV1.miS1 compared to wild‐type littermates (Fig 3A). However, there was no significant difference between the ML widths of wild‐type animals and B05 mice treated with 8 × 108 vg, 8 × 109 vg, or 8 × 1010 vg of rAAV1.miS1. In lobules IV/V–VI, all groups of B05 treated mice, except those treated with rAAV1.miS1 at 8 × 109 vg, were significantly reduced relative to their wild‐type littermates (see Fig 3B).
Figure 3.

Cerebellar pathology. (A) Molecular layer (ML) widths of lobules III and IV/V from sagittal cerebellar sections 0.5mm from midline (n ≥ 3; **p < 0.01, difference from wild‐type [WT]). (B) ML widths of lobules IV/V and VI from sagittal cerebellar sections 0.5mm from midline (n ≥ 3; *p < 0.05, **p < 0.01, ***p < 0.0001, difference from WT). (C) Representative photomicrographs of sagittal cerebellar sections immunostained for human ataxin‐1. Scale bars = 100μm. (D) Representative photomicrographs of sections from deep cerebellar nuclei (DCN) immunostained for Gfap. Scale bars = 100μm. (E) Representative photomicrographs of sagittal cerebellar cortices immunostained for Iba1. Scale bars = 100μm. (F) Representative photomicrographs of sagittal cerebellar DCN immunostained for Iba1. Scale bars = 100μm. ns = not significant; Sal = saline.
IHC for human ATXN1 in PCs of control‐treated mice showed, as expected, expression of the transgene in B05 but not wild‐type mice (see Fig 3C). B05 mice treated with increasing doses of rAAV1.miS1 had progressively less ATXN1‐positive PCs. At a dose of 8 × 1010 vg, no ATXN1‐positive PCs were detectable. Histological staining for glial fibrillary acidic protein (Gfap; a marker of astroglial activation) revealed enhanced immunoreactivity at the site of injection (the DCN) in all injected animals, and those treated at 8 × 1010 vg had robust enhancement (see Fig 3D).
Histological staining for ionized calcium‐binding adapter molecule 1 (Iba1), a marker for microglial activation, did not show differences among any experimental groups except for those receiving the highest dose of rAAV1.miS1 (see Fig 3E, F).
rAAV2/1.miS1 Is Effective after Disease Onset
Two doses (8 × 108 vg and 8 × 109 vg) were effective and nontoxic in our preonset treatment design. Because most patients with SCA1 present to the clinic with some disease manifestations, we tested the effects of miS1 therapy after disease onset.
B05 mice have deficits on the rotarod by 10 to 11 weeks of age (Fig 4A). We baseline tested B05 mice and wild‐type littermates on the rotarod at 11 weeks, to confirm deficits (see Fig 4B), and then performed dosing studies as before, except that 5 doses (rather than 4) of rAAV1.miS1 or control were injected at 12 weeks of age. Additionally, the doses were stepped up by one‐half log, and the lowest dose in the predisease onset treatment paradigm, which resulted in no silencing, was omitted (Table 2). End‐study rotarod was conducted at 20 weeks of age (see Fig 4C), and 2 weeks later tissue was collected for postnecropsy analysis. Nine weeks after injection, wild‐type mice performed significantly better than B05 mice receiving saline, those receiving rAAV1.miC, and the low‐dose and 2 high‐dose groups. In contrast to wild‐type mice, treated B05 mice in these groups had poorer performance relative to their baseline. However, B05 mice treated with 2.6 × 109 vg or 8 × 109 vg of rAAV1.miS1 performed significantly better than they did at 11 weeks of age, and also significantly better than the other B05 treatment groups. Cumulatively, the data support the hypothesis that rAAV1.miS1 delivery into the DCN of symptomatic ataxic mice improves motor symptoms in the B05 model of SCA1.
Figure 4.
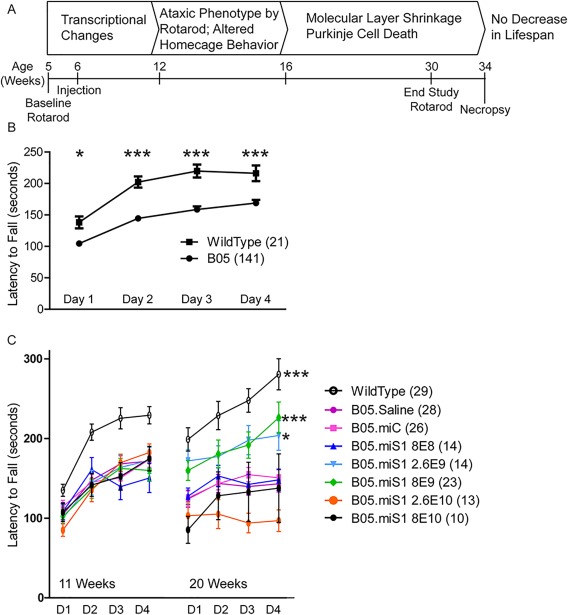
Experimental design of reversal study and rotarod data. (A) Timeline of disease progression in B05 mice and study design. (B) Eleven‐week‐old baseline rotarod. Rotarod performance over 4 days at 11 weeks of age (*p < 0.05, ***p < 0.0001). (C) Rotarod performance over 4 days at 11 and 20 weeks of age (*p < 0.05, ***p < 0.0001, difference from rAAV1.miC‐ and saline‐treated B05 animals).
Table 2.
Treatment Groups for Reversal Study
| Genotype | Injectate | Dose, Total vg |
|---|---|---|
| B05 | rAAV1.miS1 | 8 × 108 |
| B05 | rAAV1.miS1 | 2.6 × 109 |
| B05 | rAAV1.miS1 | 8 × 109 |
| B05 | rAAV1.miS1 | 2.6 × 1010 |
| B05 | rAAV1.miS1 | 8 × 1010 |
| B05 | rAAV1.miC | 8 × 108 |
| B05 | rAAV1.miC | 8 × 109 |
| B05 | Saline | — |
| Wild‐type | — | — |
vg = vector genomes.
rAAV1.miS1 Reduces ATXN1 and Improves Molecular Readouts in Symptomatic B05 Mice
miS1 expression was detected in B05 mice treated with rAAV1.miS1 (Fig 5A), and there was a clear dose‐dependent reduction of human ATXN1 mRNA levels in cerebellar lysates (seeFig 5B). ATXN1 mRNA levels were not different between B05 mice treated with saline, rAAV1.miC, and 8 × 108 vg rAAV1.miS1. B05 mice treated with 2.6 × 109 vg, 8 × 109 vg, 2.6 × 1010 vg, or 8 × 1010 vg of rAAV1.miS1 had progressively greater levels of knockdown relative to saline‐treated B05 mice.
Figure 5.

Semiquantitative polymerase chain reaction (PCR), quantitative reverse transcriptase (qRT)‐PCR, and nuclear magnetic resonance analyses of cerebellar extracts. (A) Semiquantitative expression of miS1 from whole cerebellar extracts. (B) qRT‐PCR for human ATXN1 mRNA levels from whole cerebellar extracts (n ≥ 3; ***p < 0.0001, difference from saline injected B05 mice). (C) qRT‐PCR for mouse Pcp2 mRNA levels from whole cerebellar extracts (n = 3–4; *p < 0.05, **p < 0.01, ***p < 0.0001, difference from wild‐type [WT] mice). (D) qRT‐PCR for mouse Grm1 mRNA levels from whole cerebellar extracts (n = 3–4; *p < 0.05, **p < 0.01, difference from wild‐type mice). (E) Ratio of N‐acetylaspartate (NAA)/inositol levels from whole cerebellar extracts (***p < 0.0001, difference from wild‐type mice). ns = not significant; Sal = saline.
Evaluation of transcripts from Purkinje cell protein 2 (Pcp2) and the metabotropic glutamate receptor type 1 (Grm1), two transcripts downregulated in this model, were also done.14, 15 B05 mice treated with rAAV1.miC had significantly lower levels of Pcp2 mRNA than wild‐type littermates (see Fig 5C). Although B05 mice treated with 8 × 108 vg or 8 × 1010 vg of rAAV1.miS1 had significantly lower levels of Pcp2, B05 mice given 8 × 109 vg of rAAV1.miS1 had Pcp2 levels that were not different from wild‐type. Similar to Pcp2, we detected significantly reduced Grm1 mRNA levels in control‐treated B05 mice or B05 mice treated with rAAV1.miS1 at 8 × 108 vg or 8 × 1010 vg (see Fig 5D). Of note, SCA1 mice treated with 8 × 109 vg of rAAV1.miS1 expressed Grm1 at levels not significantly different from wild‐type littermates.
Consistent with the results shown earlier (see Fig 2C), cerebellar lysates of control‐treated B05 mice had abnormal NAA/inositol ratios as measured by NMR that improved with treatment (see Fig 5E). B05 mice treated with 8 × 109 vg of rAAV1.miS1 had an NAA/inositol ratio that was not significantly different from their wild‐type littermates.
rAAV1.miS1 Improves Cerebellar Pathology in Symptomatic B05 Mice
Sagittal sections were processed to quantify the ML widths in medial cerebellar regions of lobules IV/V and VI. The data show marked thinning in control‐treated B05 mice, and mice treated with 8 × 108 vg or 8 × 1010 vg of rAAV.miS1 relative to those regions in wild‐type mice (Fig 6A). However, there was no significant difference between wild‐type mice and B05 mice treated with 8 × 109 vg of rAAV1.miS1. ML widths in the caudal medial cerebellar sections between lobules VIII and IX also show significant thinning in control‐treated B05 mice and B05 mice administered 8 × 1010 vg rAAV1.miS1 (see Fig 6B). B05 mice treated with 8 × 108 vg or 8 × 109 vg of rAAV1.miS1 had ML widths that were not significantly different from wild‐type littermates.
Figure 6.

Cerebellar pathology in mice treated after disease onset. (A) Molecular layer (ML) widths of lobules IV/V and VI from sagittal cerebellar sections 0.5mm from midline (n ≥ 3; ***p < 0.0001 difference from wild‐type [WT]). (B) ML widths of lobules VIII and IX from sagittal cerebellar sections 0.5mm from midline (n ≥ 3; **p < 0.01, ***p < 0.0001, difference from WT). (C) Representative photomicrographs of sagittal cerebellar sections immunostained for human ataxin‐1. Scale bars = 100μm. (D) Representative photomicrographs of sagittal deep cerebellar nuclei (DCN) immunostained for Gfap. Scale bars = 100μm. (E) Representative photomicrographs of sagittal cerebellar cortex immunostained for Iba1. Scale bars = 100μm. (F) Representative photomicrographs of sagittal cerebellar DCN immunostained for Iba1. Scale bars = 100μm.
Similar to the results observed in the presymptomatic dosing experiment, B05 mice treated with saline or rAAV1.miC are immunoreactive for human ATXN1 in most PCs. B05 mice treated postsymptomatically with rAAV1.miS1 show decreasing levels of ATXN1‐positive PCs that correlate inversely to the dose injected (see Fig 6C). B05 mice treated with rAAV1.miS1 at 8 × 108 vg had fewer ATXN1‐positive PCs than control‐treated mice, whereas those treated with rAAV1.miS1 at 8 × 109 vg or 8 × 1010 vg have few to no detectable ATXN1‐positive PCs. In the DCN, the site of injection, there were similar amounts of Gfap+ immunoreactive cells in all sections, with the exception of enhanced immunoreactivity in B05 mice treated with 8 × 1010 vg of rAAV1.miS1 (see Fig 6D). B05 mice treated with saline or rAAV1.miC showed slightly higher levels of Iba1 immunoreactivity in the cortex and DCN than those treated with 8 × 108 vg or 8 × 109 vg rAAV1.miS1, or wild‐type mice. B05 mice treated with rAAV1.miS1 at 8 × 1010 vg showed elevated Iba1 immunoreactivity in both the cortex and the DCN (see Fig 6E, F).
Discussion
This work provides solid experimental evidence demonstrating that RNAi‐mediated suppression of ATXN1 mRNA alters disease progression, reverses symptoms, and normalizes cerebellar pathology and disease biomarkers in an SCA1 model. In addition, it identifies doses within the efficacy–toxicity window to guide clinical development of RNAi for the treatment of SCA1. We identified the least effective dose, the toxicity threshold, and several effective doses that could either prevent or improve SCA1 readouts. Importantly, the doses used in prior work6, 7 showing the efficacy of the approach for preventing mutant (human or mouse) ataxin‐1–induced symptoms were recapitulated here.
Cumulatively, our data in symptomatic mice corroborate and extend earlier work demonstrating that eliminating mutant human ataxin‐1 expression is therapeutic, even after cerebellar pathology and neurological deficits are evident.4, 16 In earlier studies by Orr and colleagues, mutant ataxin‐1 was completely eliminated and aggregates resolved quickly with recovery of cerebellar pathology.4 Our results are similar but contrasting in important ways. The similarities are that RNAi trigger expression, even when initiated after symptom onset, was therapeutic and improved symptomatology. The major difference is that our suppression was partial rather than complete, and mutant ATXN1 was not suppressed in every PC. This result is exciting and suggests that (1) partial suppression after disease onset can be beneficial; and (2) limiting coverage of the RNAi therapy to even a portion of the cerebellum, most notably the medial regions, can improve behavioral outcomes. We found that 2.6 × 109 vg was associated with 48% and 8 × 109 vg with ∼71% reduction of mutant ATXN1 mRNA, and both provided benefit.rAAV.miS1 not only prevented further disease progression, but also improved disease readouts (eg, see rotarod studies). At baseline, B05 animals were significantly impaired (see Fig 4B, C). After receiving miS1 at doses of 2.6 × 109 vg and 8 × 109 vg, rotarod performance at 20 weeks of age demonstrated a reversal of preexisting impairment, and mice performed no differently from their wild‐type littermates. The difference between a 28% reduction in ATXN1 produced by 8 × 108 vg and a 48% reduction in ATXN1 produced by 2.6 × 109 vg of rAAV1.miS1 delineates the threshold for reversal of rotarod performance when delivered to postsymptomatic B05 mice. This is the first time, to our knowledge, that delivery of an RNAi vector has been shown to quantifiably reverse disease pathology in B05 SCA1 mice. Of note, presymptomatic B05 mice receiving 8 × 108 vg at 6 weeks of age failed to develop the phenotypic rotarod deficit by 30 weeks of age, suggesting that earlier treatment with less viral load can be beneficial.
PC dysfunction occurs prior to cell loss in SCA1 and in SCA1 mice models. One measure of this is a reduction in ML width due to PC dendritic retraction. The anterior lobe (rostral lobules) of the cerebellum is key to maintain balance, and dysfunction causes truncal ataxia.17 It is also a region affected early in the pathogenesis of SCA1.18, 19 The posterior lobe (caudal) plays an important role in motor coordination.20 In mice that received 8 × 108 vg, ML widths were similar in width to wild‐type mice in caudal lobules, with measurable thinning in rostral lobules. However, mice treated with 8 × 109 vg of rAAV.miS1 retained ML widths similar to wild‐type in both rostral and caudal lobules. This suggests that this dose, with scaling for human use, may provide preservation of both rostral and caudal aspects of the MLs, lending improved balance and motor coordination, respectively, as well as rescue motor symptoms.
It is worth noting that in the B05 model, the human transgene is expressed in the PCs only. However, assays for transduction efficiency, molecular readouts of efficacy, and transgene (miS1) levels were performed on whole cerebellar lysates. These data demonstrate the PC‐targeting efficiency of this vector system. Molecular indicators of efficacy include Pcp2, the mRNA of which is trafficked to the dendrites,21 and Grm1, a metabotropic glutamate receptor 1 located in the postsynaptic termini of dendrites.21 The latter is critical for coordinated motor function.22 Both Pcp2 and Grm1 deficits were reversed in mice by 67% reduction in ATXN1.
It has been previously shown that B05 mice have elevated Iba1+ immunoreactivity that is reversible in the SCA1 conditional model.23 We also noted enhanced Iba1+ immunoreactivity in control‐treated B05 mice relative to wild‐type animals, and qualitatively more in the 8 × 1010 vg rAAV1.miS1 treatment group. In B05 mice treated with 8 × 108 vg or 8 × 109 vg, Iba1+ immunoreactivity was reduced. Thus, even a 28% reduction in ATXN1 (8 × 108 vg dose), which does not result in behavioral rescue, can abate the phenotypic increase in Iba1+ signal.
Noninvasive biomarkers will provide critical tools for assessing efficacy in SCA1. 1H MRS indicates that SCA1 patients have lower levels of NAA, and elevated levels of inositol.11 These data have been recapitulated in transgenic SCA1 mice12 and were reversed in a conditional mouse model of SCA1.16 NAA reduction usually precedes neuronal loss, and is used as a marker of neuronal dysfunction.24 In this work, NAA levels were modestly reduced in control‐treated SCA1 mice at 20 weeks of age, a time prior to significant PC loss. Importantly, mice treated with 8 × 109 vg of rAAV.miS1 had an NAA/inositol ratio similar to their wild‐type littermates. This, together with previous studies, suggests that the NAA/inositol ratio may be a sensitive, noninvasive measure of efficacy and a possible biomarker in disease‐modifying clinical trials for SCA1.
Motor deficits quantified by the Scale for Assessment and Rating of Ataxia (SARA) correlate with altered neurochemical levels quantified by MRS.25 We see a similar relationship in our untreated B05 mice, with improved “scores” upon treatment. These data suggest that rAAV1.miS1 could provide therapeutic benefit and prevention of further pathogenesis in SCA1 patients if administered prior to disease onset. Moreover, rAAV1.miS1 could halt or even reverse preexisting motor deficits in early, symptomatic SCA1 patients. Thus, SARA scores could stabilize or improve with treatment, along with concomitant improvements in neurochemical levels.
Although the B05 model was useful for assessing the utility of our clinical product, a more genetically relevant model would be a humanized KI that would allow testing of miS1. This would allow efficacy testing in a setting more biologically similar to SCA1 patients, as compared to the transgenic model where the transgene is only expressed in PCs. Of note is that RNAi was tested in mice expressing an expanded CAG repeat in mouse ataxin‐1,26 with efficacy at the same E9 dose (using an RNAi trigger that targeted mouse ataxin‐1). Whether the doses that reverse disease are similar to what we found in the B05 model remains to be tested, but these doses are important to determine in future work. Additionally, we cannot assume that miS1 (targets human) and miSCA1 (targets mice) are equivalent in safety, as they are different sequences. Nonetheless, it is encouraging that similar doses prevented disease in the current models that are available.
Although we noted prevention or reversal of disease at several doses, we also noted toxicity at the highest dose tested. The reasons for this are unclear. It could be due to the near complete absence of ATXN1 in transduced cells, although we think this is not likely due to chronic, 100% loss of Atxn1 in mice causing transcriptional misregulation but no noted neuropathology or ataxia.27, 28 A second possibility is that the high‐level expression of miS1 somehow causes toxicity due to abnormal processing of endogenous miRNAs. This can be evaluated in future work to examine endogenous miRNA processing and miRNA activity. We think this is improbable, however, as we used miS1 in an older vector platform and reversed aberrant miRNA expression in the B05 model.29 A third possibility is that the high capsid dose is toxic. Testing for this could be done using empty capsid preparations as controls at the doses evaluated here. We are investigating the exact mechanisms underlying the toxicity as part of a follow‐up study. Although these data inform us of a toxicity ceiling and the maximum tolerable dose, understanding the mechanism for that toxicity is important moving forward.
In summary, we show that AAV‐mediated delivery of RNAi triggers can reverse neuropathological phenotypes, transcriptional changes, and behavioral phenotypes in a mouse model of SCA1. We also identify the minimal effective and maximally tolerated doses that will guide our clinical application for SCA1 therapy. Together with earlier work demonstrating the scalability of our approach to nonhuman primates,8 our studies will guide the initial application to SCA1 and other cerebellar diseases in which PCs, brainstem neurons, and the DCN are important therapeutic targets.
Author Contributions
Study concept and design: all authors; data acquisition and analysis: M.S.K. R.C., P.G.‐A., B.L.D.; drafting the manuscript and figures: all authors.
Potential Conflicts of Interest
B.L.D. is a founder of Spark Therapeutics, a gene therapy company, that has licensed the technology described in this study. B.L.D. is also on the scientific advisory board of Intellia Therapeutics and Serepta Therapeutics.
Acknowledgment
This work was funded by the NIH National Institute of Neurological Disorders and Stroke (UH2NS094355), the National Ataxia Foundation Pioneer Award, and the Children's Hospital of Philadelphia Research Institute.
We thank the Children's Hospital of Philadelphia Research Vector Core for the viruses used in this work; and S. L. Wehrli, the Children's Hospital of Philadelphia Small Animal Imaging Facility; and M. Sowada for assistance with behavioral experiments.
References
- 1. Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Ann Rev Neurosci 2007;30:575–621. [DOI] [PubMed] [Google Scholar]
- 2. Burright EN, Clark HB, Servadio A, et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995;82:937–948. [DOI] [PubMed] [Google Scholar]
- 3. Clark HB, Burright EN, Yunis WS, et al. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci 1997;17:7385–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zu T, Duvick LA, Kaytor MD, et al. Recovery from polyglutamine‐induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci 2004;24:8853–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xia H, Mao Q, Eliason SL, et al. RNAi suppresses polyglutamine‐induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med 2004;10:816–820. [DOI] [PubMed] [Google Scholar]
- 6. Keiser MS, Boudreau RL, Davidson BL. Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: implications for spinocerebellar ataxia type 1 therapy. Mol Ther 2014;22:588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keiser MS, Geoghegan JC, Boudreau RL, et al. RNAi or overexpression: alternative therapies for spinocerebellar ataxia type 1. Neurobiol Dis 2013;56:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Keiser MS, Kordower JH, Gonzalez‐Alegre P, Davidson BL. Broad distribution of ataxin 1 silencing in rhesus cerebella for spinocerebellar ataxia type 1 therapy. Brain 2015;138:3555–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Monteys AM, Spengler RM, Dufour BD, et al. Single nucleotide seed modification restores in vivo tolerability of a toxic artificial miRNA sequence in the mouse brain. Nucleic Acids Res 2014;42:13315–13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Servadio A, Koshy B, Armstrong D, et al. Expression analysis of the ataxin‐1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat Genet 1995;10:94–98. [DOI] [PubMed] [Google Scholar]
- 11. Oz G, Hutter D, Tkác I, et al. Neurochemical alterations in spinocerebellar ataxia type 1 and their correlations with clinical status. Mov Disord 2010;25:1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oz G, Nelson CD, Koski DM, et al. Noninvasive detection of presymptomatic and progressive neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci 2010;30:3831–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klement IA, Skinner PJ, Kaytor MD, et al. Ataxin‐1 nuclear localization and aggregation: role in polyglutamine‐induced disease in SCA1 transgenic mice. Cell 1998;95:41–53. [DOI] [PubMed] [Google Scholar]
- 14. Serra HG, Byam CE, Lande JD, et al. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet 2004;13:2535–2543. [DOI] [PubMed] [Google Scholar]
- 15. Skinner PJ, Vierra‐Green CA, Clark HB, et al. Altered trafficking of membrane proteins in Purkinje cells of SCA1 transgenic mice. Am J Pathol 2001;159:905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oz G, Vollmers ML, Nelson CD, et al. In vivo monitoring of recovery from neurodegeneration in conditional transgenic SCA1 mice. Exp Neurol 2011;232:290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Purves D, Augustine GJ, Fitzpatrick D, et al. Neuroscience. 3rd ed. Sunderland, MA: Sinauer Associates, 2004. [Google Scholar]
- 18. Robitaille Y, Schut L, Kish SJ. Structural and immunocytochemical features of olivopontocerebellar atrophy caused by the spinocerebellar ataxia type 1 (SCA‐1) mutation define a unique phenotype. Acta Neuropathol 1995;90:572–581. [DOI] [PubMed] [Google Scholar]
- 19. Robitaille Y, Lopes‐Cendes I, Becher M, et al. The neuropathology of CAG repeat diseases: review and update of genetic and molecular features. Brain Pathol 1997;7:901–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cicirata F, Angaut P, Panto MR, Serapide MF. Neocerebellar control of the motor activity: experimental analysis in the rat. Comparative aspects. Brain Res Brain Res Rev 1989;14:117–141. [DOI] [PubMed] [Google Scholar]
- 21. Vassileva G, Smeyne RJ, Morgan JI. Absence of neuroanatomical and behavioral deficits in L7/pcp‐2‐null mice. Brain Res Mol Brain Res 1997;46:333–337. [DOI] [PubMed] [Google Scholar]
- 22. Knopfel T, Grandes P. Metabotropic glutamate receptors in the cerebellum with a focus on their function in Purkinje cells. Cerebellum 2002;1:19–26. [DOI] [PubMed] [Google Scholar]
- 23. Cvetanovic M, Ingram M, Orr H, Opal P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience 2015;289:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Demougeot C, Garnier P, Mossiat C, et al. N‐Acetylaspartate, a marker of both cellular dysfunction and neuronal loss: its relevance to studies of acute brain injury. J Neurochem 2001;77:408–415. [DOI] [PubMed] [Google Scholar]
- 25. Adanyeguh IM, Henry PG, Nguyen TM, et al. In vivo neurometabolic profiling in patients with spinocerebellar ataxia types 1, 2, 3, and 7. Mov Disord 2015;30:662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lorenzetti D, Watase K, Xu B, et al. Repeat instability and motor incoordination in mice with a targeted expanded CAG repeat in the Sca1 locus. Hum Mol Genet 2000;9:779–785. [DOI] [PubMed] [Google Scholar]
- 27. Goold R, Hubank M, Hunt A, et al. Down‐regulation of the dopamine receptor D2 in mice lacking ataxin 1. Hum Mol Genet 2007;16:2122–2134. [DOI] [PubMed] [Google Scholar]
- 28. Matilla A, Roberson ED, Banfi S, et al. Mice lacking ataxin‐1 display learning deficits and decreased hippocampal paired‐pulse facilitation. J Neurosci 1998;18:5508–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodriguez‐Lebron E, Liu G, Keiser M, et al. Altered Purkinje cell miRNA expression and SCA1 pathogenesis. Neurobiol Dis 2013;54:456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]