

Abstract
Beta-lactam antibiotics kill Staphylococcus aureus bacteria by inhibiting the function of cell-wall penicillin binding proteins (PBPs) 1 and 3. However, β-lactams are ineffective against PBP2a, used by methicillin-resistant Staphylococcus aureus (MRSA) to perform essential cell wall crosslinking functions. PBP2a requires teichoic acid to properly locate and orient the enzyme, and thus MRSA is susceptible to antibiotics that prevent teichoic acid synthesis in the bacterial cytoplasm. As an alternative, we have used branched poly(ethylenimine), BPEI, to target teichoic acid in the bacterial cell wall. The result is restoration of MRSA susceptibility to the β-lactam antibiotic ampicillin with a MIC of 1 μg/mL, superior to that of vancomycin (MIC = 3.7 μg/mL). A checkerboard assay shows synergy of BPEI and ampicillin. Nuclear magnetic resonance (NMR) data show that BPEI alters the teichoic acid chemical environment. Laser scanning confocal microscopy (LSCM) images show BPEI residing on the bacterial cell wall where teichoic acids and PBPs are located.
Keywords: MRSA, β-lactam, penicillin binding protein, ampicillin, teichoic acid
INTRODUCTION
Methicillin-resistant Staphylococcus aureus (MRSA) is a current and growing risk to human health. It causes serious infections that show remarkable resistance to antibiotic treatment.1 Originally acquired exclusively in healthcare settings, MRSA is now regularly found outside the healthcare environment.2 Drug resistance hinders efforts to develop safe clinical treatments for MRSA infections.3 Fortunately, progress has been made towards developing new antibiotics such as oxadiazoles,4 tedizolid,5 and teixobactin.6 The timing coincides with a critical period in antibiotic research and development as MRSA is developing resistance to drugs of last resort.1, 7, 8 Therapeutic approaches to overcome resistance factors include efflux-pump inhibitors that increase the intracellular concentration of antibiotics.9 Bacteria can also use β-lactamase enzymes that degrade the antibiotics10 and thus treatment requires β-lactamase inhibitors.11
The cell envelope of Gram-positive bacteria is composed of a membrane, peptidoglycan, and teichoic acids (Figure 1A).6 Methicillin, a β-lactam antibiotic, occupies the active site of penicillin binding proteins (PBP) 1 and 3 to prevent the enzymatic cell-wall synthesis function (Figure 1B). Methicillin-resistant S. aureus performs cell-wall synthesis using PBP2a (Figure 1C), a transpeptidase enzyme with very low affinity for β-lactams. Hartman and Tomasz recognized and identified PBP2a in MRSA.12 The β-lactam/transpeptidase complex is stable; however, resistance arises because the rate of complex formation is much slower than the S. aureus cell division time. Thus, it is nearly impossible for the complex to form in vivo.13 Fuda et al. also presented a structure of PBP2a with no realistic access to the active site, suggesting there had to be a conformational change that took place as a result of binding non-crosslinked peptidoglycan at a location other than the active site, setting the groundwork for future investigations of allosteric regulation. Nevertheless, the cell wall remains an especially rich antimicrobial target, containing many opportunities for disruption, such as excess peptidoglycan,14 teichoic acids15, 16 and novel proteins.17 While these targets have shown promise, side-effects and slow progress towards clinical usage have hindered efforts to reduce the rate of MRSA infection and mortality.18 While it is possible to stop teichoic acid expression in the cytoplasm, thereby disabling the function of PBP2a,15 the quantity of drug required for activity prevent development into a clinical MRSA treatment.18
Figure 1.

Schematic6 of Gram-positive cell wall components (A) and crosslinking of peptidoglycan using PBPs 1 and 3, which can be inhibited by β-lactams (B), and PBP2a, which cannot (C). PBP2a requires WTA to be properly localized; BPEI can bind to WTA to prevent PBP2a from functioning properly (D).
The continued spread of resistance could be stemmed by re-sensitizing resistant strains of S. aureus to currently ineffective antibiotics, such as the β-lactam ampicillin. This approach can be viable by inhibiting the expression and/or functionality of proteins that contribute to resistance, such as PBP2a. PBPs are indispensable for cell growth as they create essential crosslinks between adjacent peptidoglycan segments. Targeting PBP2a with inhibitors has been shown to re-sensitize resistant strains to methicillin.19 In contrast, branched poly(ethylenimine) BPEI may indirectly disable PBP2a. Our work shows that BPEI, administered in concert with ampicillin, resensitizes MRSA to ampicillin. Laser scanning confocal microscopy (LSCM) images show that BPEI binds to the cell wall where it can interrupt the function of teichoic acids, inactivate PBP2a, and restore β-lactam antibiotic activity. Nuclear magnetic resonance (NMR) spectroscopy data demonstrate that BPEI binds to teichoic acids, likely through ionic attraction between the cationic polymer and the anionic teichoic acid. Ampicillin activity against MRSA was restored by low molecular weight (MW) BPEI. Checkerboard assays were used to measure the fractional inhibitory concentration (FIC) index and identify synergy between 1–8 μg/mL ampicillin and 16 μg/mL low-MW BPEI; or 8 μg/mL ampicillin and 8 μg/mL low-MW BPEI. However, BPEI does not increase the efficacy of vancomycin or novobiocin. Thus, BPEI’s synergistic properties arise by indirectly disabling PBP2a, rendering MRSA susceptible to ampicillin that disables PBP1 and PBP3.
MATERIALS AND METHODS
Materials
The bacteria used in this work were obtained from the American Type Culture Collection (methicillin-resistant Staphylococcus aureus (MRSA) strain ATCC 700787, Staphylococcus aureus ATCC 25923, Escherichia coli ATCC 11775, Bacillus subtilis (Ehrenberg) Cohn ATCC 23059). Chemicals from Sigma-Aldrich (DMSO, ampicillin, vancomycin, and novobiocin) were used as purchased.
Preparation and Characterization of Cationic Polymers
Branched and linear polyethylenimine was obtained from Sigma-Aldrich, Inc. as high molecular weight (~25 kDa) or low molecular weight (~0.5 kDa) polymers. The molecule has polycationic character from the protonation of its amine functional groups based on its protonation constant (pKa). Protonation constants for branched PEI molecules have been reported to be around 4.5 for primary amines, 6.7 for secondary amines, and 11.6 for tertiary amines.20 Thus, at pH = 7.2, the BPEI has many positively-charged primary amines to interact with teichoic acid.
Growth Inhibition Assays
Compounds were tested against methicillin-resistant Staphylococcus aureus (MRSA) strain ATCC 700787, which also exhibits reduced susceptibility to vancomycin. A stock culture was diluted in tryptic soy broth (TSB) and delivered into pre-sterilized 96-well plates. Stock solutions of BPEI and antibiotics, prepared in DMSO, were added to each well of a 96-well plate (final DMSO concentration was 1%), followed by inoculation with MRSA in TSB. Optical density measurements were performed with a Tecan Infinite M200 plate reader and an initial OD600 value recorded. The plates were incubated for 20 h in a humidified incubator at 37°C. Plates were removed, orbitally shaken, and a final OD600 value recorded. The final OD600 reading was subtracted from the initial OD600 reading to obtain the change in OD600 (recorded in the figures as ΔOD600). Antimicrobial activity was determined by the change in optical density. Duplicate measurements were performed and the average reported.
Using the average ΔOD600 values, separate MIC values for BPEI and ampicillin were determined by the lowest concentration of each that inhibited growth. From this, fractional inhibitory concentration indices (FIC) were calculated for all wells that showed inhibition.21, 22
Cytotoxicity Assay
Mammalian cell cytotoxicity assays were performed on NIH/3T3 mouse fibroblast cells by adding 5,000 cells per well into 96-well plates. The cells were allowed to adhere overnight at 37°C in a humidified incubator (5% CO2 atmosphere). Test compounds were diluted in DMSO and added to the wells so that the final concentration of DMSO per well did not exceed 1% by volume. The plates containing treated and control cells were incubated for 48 hours and cell viability was determined by MTT assay.23 Duplicate measurements were performed and the average reported.
Synthesis of the BPEI:Dye Conjugate
Low-molecular weight BPEI (Sigma-Aldrich) was added to Alexa Fluor 488 dye provided in the Alexa Fluor 488 Protein Labeling Kit (Life Technologies) at a ratio of 200 μL BPEI (3 mg/mL stock in Milli-Q H2O) per tube of powdered dye. After allowing the dye and BPEI to form the conjugate over 1.5 h at 25°C, the product was stored at 4°C and used without further purification.
Labeling MRSA Cells with the BPEI:Dye Conjugate
Cultures of E. coli ATCC 11775 and methicillin-resistant S. aureus ATCC 700787 at mid-log growth phase were pelleted by centrifugation at 2,000 × g for 5 minutes at room temperature, and the growth media supernatant was removed. 6 μM DAPI in phosphate-buffered saline (1X PBS, pH 7.2) was added to resuspend the cell pellet, which was allowed to incubate for 5 minutes at room temperature. The BPEI-dye conjugate was then added to a final concentration of 100 μg/mL. Two control samples were prepared with either unconjugated BPEI or Alexa Fluor 488 alone and added to MRSA cells as described above. The stained bacterial cells were immediately fixed by resuspension with 4% paraformaldehyde (PFA) followed by a 10-fold dilution in 1X PBS. Cells were added to a microscope slide immediately prior to imaging.
Confocal Microscopy
PFA-fixed cells were mounted in 1X PBS and imaged using a Leica SP8 LSCM with a 63x/1.4NA oil objective. A 405 nm GaN diode laser line was used to image DAPI, and a 488 nm argon laser line was used to observe Alexa Fluor 488 fluorescence. Single optical sections were acquired of cells that had adhered to the coverslip, for highest axial resolution, with a pixel resolution of 80 nm × 80 nm. Instrument settings were kept fixed for all imaging to allow for direct comparison of fluorescence intensity.
Image Processing
To visualize the relative localization of fluorescence, independent of intensity, images were processed (ImageJ v1.49m) such that the total fluorescence intensity within each image was visible (Figure S8, left column). To determine the relative fluorescence intensity between images, the fluorescence intensity of both the DAPI and the Alexa Fluor 488 were normalized to the respective intensities in the MRSA sample treated with BPEI conjugated with Alexa Fluor 488 (Figure S8, right column).
WTA Purification for NMR Studies
The NMR experiments require 1–5 mg WTA isolated from 1 L of bacterial cell culture. The poly(ribitol phosphate) WTA used in this work was isolated from B. subtilis W2324, 25 (Bacillus subtilis (Ehrenberg) Cohn ATCC® 23059™) rather than from MRSA, allowing high volumes (500–2000 mL) of culture to be processed with minimal risk to personnel. This also allows us to take the sample into the NMR facility room which is not rated for BSL-2 work. In the rare instance of sample breakage, using WTA from B. subtilis W23 does not present a clean-up hazard nor require decontamination of the expensive NMR analysis probes, magnets, or spectrometers.
B. subtilis W23 cells were grown in LB broth to an OD600 reading of approximately 0.8. After growth, the cells were harvested by centrifugation and physically disrupted using an Avestin® EmulsiFlex-C3 homogenizer. The insoluble cell wall was collected, placed in boiling 6% (w/v) SDS, washed with sterile water and EDTA then placed in a 10% trichloroacetic acid (TCA) treatment for 48 hours at 4°C. After allowing the TCA to remove the bound WTA from the cell wall, the WTA was collected in the supernatant and placed into a 500 Da molecular weight cutoff dialysis membrane. Membrane dialysis was performed at 4°C in 1.5 L of sterile water with continual water changes over a 24-hour period. The final 4 h of dialysis took place in a 1 kDa molecular weight cutoff membrane to assure sample purity. The sample was lyophilized and kept at −20°C until use.
NMR Spectroscopy
NMR samples were prepared in Eppendorf tubes by mixing teichoic acid with low-MW BPEI in water. The pH was measured with a microscopic pH probe and adjusted to 7.2 if necessary. A 3-mm NMR tube was filled with 160 μL of a 2 mM sample of WTA/D2O or a combination of 2 mM BPEI with 2 mM WTA in D2O. NMR data collection use Agilent VNMRS-500 MHz equipped with a PFG probe tuned to the 31P resonance frequency. Data acquisition and processing were performed using VNMRJ 2.2C software on a system running Red Hat Enterprise Linux.
RESULTS AND DISCUSSION
While there are numerous MRSA strains, Staphylococcus aureus subsp. aureus ATCC® 700787™ was isolated in Port Chester, NY from blood culture, exhibits vancomycin intermediate resistant (VISA)26 and also expresses mecA to produce PBP2a.27 The ability of BPEI to restore ampicillin effectiveness against MRSA is shown in Table 1. The growth of MRSA in vitro after 20 hours of incubation was inhibited by ampicillin when the antibiotic was co-administered with low molecular-weight branched poly(ethylenimine), low-MW BPEI. MRSA shows resistance towards ampicillin, with a minimum inhibitory concentration (MIC) of 32 μg/mL, but the presence of low-MW BPEI (16 μg/mL) rendered MRSA susceptible to ampicillin at a 32x lower dose (MIC = 1 μg/mL). At a reduced BPEI concentration of 8 μg/mL, the ampicillin MIC was still decreased, but only to 8 μg/mL. In the absence of ampicillin, BPEI itself inhibited growth of MRSA at a concentration of 64 μg/mL. With these values, it is possible to calculate the fractional inhibitory concentration (FIC) index21, 22 for each combination. When the FIC is equal to or below 0.5, ampicillin and low-MW BPEI stop MRSA growth through synergistic effects. Synergy occurred between 8 μg/mL ampicillin and 8 μg/mL low-MW BPEI (FIC = 0.375), as well as with combinations of 16 μg/mL low-MW BPEI with 1, 2, 4, and 8 μg/mL ampicillin (FIC = 0.281, 0.313, 0.275, 0.500 respectively). Although MRSA does not grow in the presence of 0.5 μg/mL ampicillin and 32 μg/mL low-MW BPEI, the FIC index is 0.516.
Table 1.
MRSA Growth Inhibition Assay in the Presence of Ampicillin and low-MW BPEI

|
MRSA cells (ATCC 700787) were used to inoculate growth media containing ampicillin and low-MW BPEI at 37 °C for 20 hours. Table entries are the optical density (OD) of the growth media measured at a wavelength of 600 nm. Each entry is the average of duplicate measurements. The bold line separates combinations that allowed MRSA growth (OD600 values above 0.05) from those combinations that prevented MRSA growth. The ampicillin MIC is 32 μg/mL demonstrating antibiotic resistance without the presence of BPEI. Values highlighted in bold text were determined to arise from synergy between ampicillin and BPEI.
BPEI’s potentiation of ampicillin to inhibit MRSA growth suggests that similar effects may be seen with other antibiotics. However, low-MW BPEI did not benefit vancomycin or novobiocin against MRSA (Figures S1 and S2, respectively). These data suggest that BPEI potentiation depends on the class of antibiotic used. Novobiocin, an aminocoumarin, works by inhibiting DNA gyrase.28 Vancomycin, a glycopeptide, inhibits peptidoglycan crosslinking by binding to the peptidoglycan stems.29 In contrast, ampicillin occupies the active sites of transpeptidase proteins that create the crosslinks between peptide stems.
The trend of lower ampicillin MICs with higher BPEI concentrations suggests therapeutic treatment of MRSA using FDA approved β-lactam antibiotics may be viable. To be clinically viable, BPEI must be safe for human use. Low-MW BPEI does not cause hemolysis 30 and is non-toxic31, as cytotoxicity and renal failure are more prevalent with high molecular weight polymers.30, 32, 33 Both linear and branched forms, LPEI and BPEI, are used as non-viral transfection agents for gene therapy.34–36 In this application, LPEI has undergone clinical trial testing and is awaiting final FDA approved for gene delivery to patients.37, 38 These studies also report that the cytotoxicity of PEI increases as its molecular weight increases35, 39 and that most branched PEIs are nontoxic below 25 kDa.40 To test the cytotoxicity of BPEI used in these experiments (MW ~0.5 kDa), mouse fibroblasts (NIH/3T3 cells) were incubated over three days in the presence of varying concentrations of BPEI (supplemental data, Figure S3). Viability was determined using the MTT assay, following the protocol of Hansen et al.23 At 2.65 μg/mL, only 1.5% of the fibroblasts became nonviable over the three-day incubation period compared against the control sample. This indicates negligible cytotoxicity from low-MW BPEI. From the checkerboard assay (Table 1), synergy occurs with 16 μg/mL low-MW BPEI which causes ~10% reduction in fibroblast viability (Figure S3). At 26.5 μg/mL low-MW BPEI, only a 16.3% reduction in viability compared to the control sample was observed. Thus, BPEI concentrations that induce synergy are also associated with low cytotoxicity; providing guarded optimism in conducting in vivo testing. Low-MW BPEI is non-toxic31 whereas high-MW BPEI penetrates mammalian cells leading to cytotoxicity and renal failure.30,32, 33 Low-MW BPEI, up to 1000 μg/mL, does not affect the membrane of human cells as judged by the lack of hemolysis in red blood cells and the lack of lactate hydrogenase leakage from HEp-2 cells.30 The BPEI concentration for this toxicity testing is ~25 times larger than the highest amount, 64 μg/mL, used for antibiotic potentiation.
Antibiotic potentiation by BPEI does not extend to Gram-negative Escherichia coli (ATCC 11775). Supplementary data show results of growing E. coli in the presence of ampicillin alone and with a fixed BPEI concentration (2.65 μg/mL, Figure S4), while Figure S8 shows the result of testing BPEI with a fixed ampicillin concentration (0.37 μg/mL). In the first data set, the observed ampicillin MIC was identical at 3.7 μg/mL with or without BPEI addition. In the second data set, BPEI did not show inhibitory effects on growth of E. coli up to 53 μg/mL, either by itself or with 0.37 μg/mL ampicillin. This is in contrast to the MRSA data in which the ampicillin MIC decreased by a factor of 32 when the BPEI concentration was 16 μg/mL. Additionally, our data differs from previously reported results showing PEI-induced antibiotic synergy against Gram-negative bacteria, including E. coli and P. aeruginosa.41, 42 This could be due to differences in branching or molecular weight between the polymers tested. Our tests use a low-MW BPEI whereas the prior reports use a high-MW LPEI.
The data in Table 1 suggests that BPEI may interact with PBP2a. This would prevent the enzyme from functioning properly while allowing the β-lactam to disable PBP1 and PBP3. If true, BPEI’s interaction with MRSA should be confined to the cell wall. By conjugating BPEI to a fluorescent marker, Alexa-Fluor 488, we were able to visualize BPEI localization in bacterial cultures using LSCM. Individual transverse optical sections clearly show BPEI interaction with the MRSA cell wall region (Figure 2A). Using DAPI, a DNA-binding fluorescent dye, as a marker for the cytoplasm within the cells (Figure 2B) the merged image (Figure 2C) confirms that BPEI was not detected within the cytoplasm, verifying that BPEI does not traverse the lipid bilayer membrane. Similar optical sections of E. coli cells treated with BPEI-conjugated Alexa-Fluor 488 revealed minimal fluorescence intensity within the cell envelope, indicating a weaker interaction between the E. coli cell envelope and BPEI (Figures 2D–F). This may explain the absence of antibiotic potentiation against E. coli in our present study. Additional LSCM images of the samples are provided as supplemental data (Figure S8).
Figure 2.
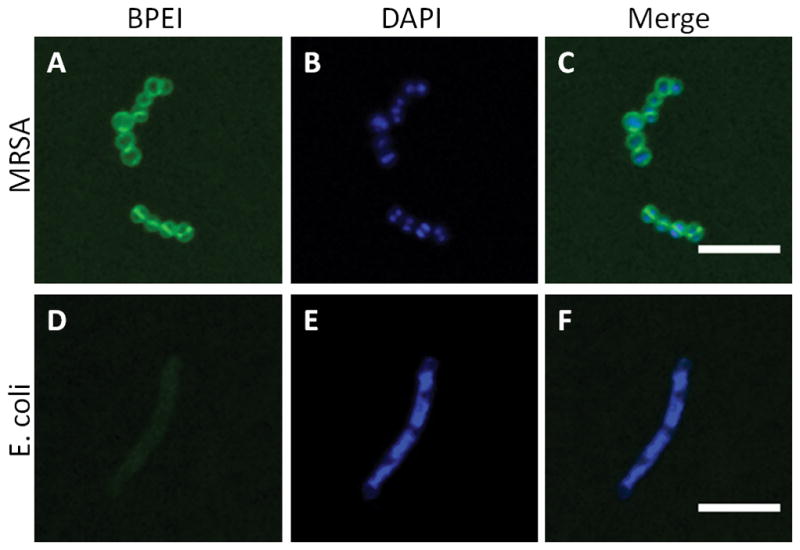
Optical sections of BPEI binding to MRSA and E. coli. Paraformaldehyde-fixed MRSA, stained with BPEI-Alexa Fluor 488 (A) and DAPI (B), is imaged by LSCM. The merged image (C) shows BPEI binding to the cell surface but not within the cytoplasm. In contrast, PFA-fixed E. coli stained with BPEI-Alexa Fluor 488 (D) and DAPI (E), and the merged image (F), shows a relatively low affinity between BPEI and E. coli. Scale bar = 5 μm.
The microscopy data, showing BPEI located in the cell wall region and not the cytoplasm, suggests that the observed antibiotic potentiation against MRSA is caused by an interaction of BPEI with some component of the bacterial cell wall. One major component of the Gram-positive cell wall is teichoic acid, a phosphodiester polymer whose anionic phosphate groups have been shown to interact strongly with metal cations.43–45 BPEI, with its polycationic properties, has the potential for very strong electrostatic interactions with the polyanionic WTA molecules. This interaction would involve the primary amines of BPEI and the phosphate groups of WTA (Figure 3). This interaction can be observed using nuclear magnetic resonance (NMR) studies of mixed BPEI-teichoic acid solutions when compared to NMR spectra of teichoic acid alone.
Figure 3.
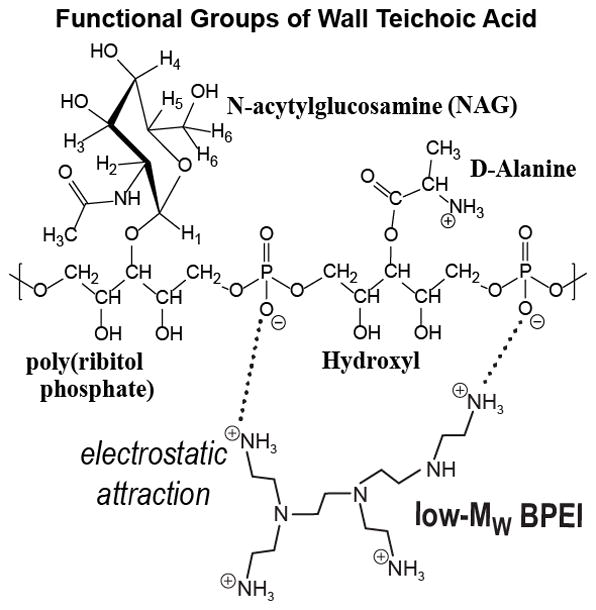
Chemical structure of the interaction between BPEI and the functional groups of wall teichoic acid. The cationic polymer is attracted to the anionic WTA. The result is a heterogeneous macromolecular complex that likely has increased steric bulk compared to WTA alone. These factors may prevent WTA from arranging PBP2a in its proper orientation required for crosslinking of peptidoglycan.
The 1-D 31P spectra (Figures 4A and B) show significant changes after mixing WTA with low-MW BPEI. WTA is a phosphodiester polymer with heterogeneous arrangement of N-acetylglucosamine (NAG), D-alanine, and hydroxyl groups. This creates variations in conformation of the poly(ribitol) backbone and differences in the phosphate conformations that generate distinct 31P NMR peaks. In the presence of low-MW BPEI, the 31P NMR peak at 1.3 ppm is very intense, demonstrating that a large fraction of the phosphates have similar conformations. However, signals near 4 ppm are produced by phosphates in a deshielded environment. The downfield shift arises from a loss of electron density around the phosphorous nucleus, an effect that could be caused by a hydrogen bond between the phosphate oxygen and a BPEI amine group. The addition of BPEI also increases the intensity of cross peaks in the 1H{31P} HMBC (heteronuclear multiple bond coupling) NMR data (Figures 4C and D). This experiment relies on strong through-bond coupling between the 1H and 31P nuclei. For flexible molecules, internal motion and dynamics causes relaxation of NMR signals46, 47 and thus the 1H{31P} HMBC signals are difficult to observe. When molecular motion is restricted, the signals are stronger.
Figure 4.

31P NMR spectra of WTA before and after the addition of low-MW BPEI (A and B) show significant changes in phosphate chemical shift caused by changes in the chemical environment. These changes are also manifested in the heteronuclear multiple-bond correlation (HMBC) spectra (C and D). The P-31 signals near 4 ppm are correlated with the proton signals of NAG sugar groups. However, clear identification of specific interactions is prevented by the heterogeneous nature of WTA functional groups, BPEI branching, and WTA:BPEI binding interactions.
Phosphate:amine binding from the WTA:BPEI interactions likely occurs through electrostatic attraction between the numerous cationic primary amines of BPEI and anionic phosphate groups of WTA (Figure 3). If this assumption is correct linear PEI, with only the 2 primary amines at its terminal ends, should not affect ampicillin’s MIC values. The 0.6 kDa form of LPEI (similar in mass to the 0.5 kDa BPEI) does not inhibit MRSA growth (supplemental data figure S7) until its concentration is very high (54 μg/mL). Low molecular weight quaternary ammonium compounds have recently been shown to overcome resistance if the number of cationic sites is increased.48 Therefore, the optimal cationic amine polymer should have a high number of primary amines while minimizing cytotoxicity with a low molecular weight.
Because low-MW BPEI binds to WTA, the cationic polymer has the ability to change WTA properties by altering molecular structure and/or creating steric bulk from the branched polymer (Figure 1D). This would change, or prevent, the interaction of WTA with PBP2a and thus disable the enzyme. The same effect can be created through WTA-deficient strains of MRSA, which are re-sensitized to amoxicillin, ampicillin, methicillin, nafcillin, and ceftizoxime.15 An inhibitor of WTA synthesis, tunicamycin, re-sensitizes MRSA to β-lactams such as methicillin, oxacillin, cefotaxime, and several others.49 Inhibition of another regulatory gene, tarG, also re-sensitizes MRSA strains to traditional β-lactams50–52 like imipenem.16 Thus WTA, while not essential to viability,50–52 is involved in β-lactam resistance.53 WTA helps to optimally localize PBP2a, and WTA-deficient mutants show a decreased functionality of the protein.15 It additionally localizes PBP4, which is essential for the highly cross-linked peptidoglycan exhibited by MRSA and for the expression of β-lactam resistance in community-acquired strains.54 Thus, restoration of β-lactam activity in therapeutic clinical usage could be achieved with antibiotics or other compounds that target WTA synthesis or interrupt the ability of WTA to localize PBP2a in the proper configuration required for peptidoglycan crosslinking. If this perspective is true, there should be little or no benefit when BPEI and ampicillin are used to treat non-resistant S. aureus strains that do not express PBP2a. Supplemental data (Figure S6) shows that the ampicillin MIC against methicillin-susceptible S. aureus ATCC 25923 was 50 ng/mL. When combined with 2.65 μg/mL of BPEI, the ampicillin MIC of S. aureus ATCC 25923 was not reduced.
Rather than developing new inhibitors which require exhaustive clinical testing, we have discovered that some FDA-approved β-lactam antibiotics, such as ampicillin, can regain their efficacy against MRSA. The benefits to human health could be dramatic if the ampicillin + BPEI combination, used as a routine antibiotic therapy, can eliminate S. aureus infections while simultaneously preventing the growth of ampicillin-resistant bacteria. Further experiments will be necessary to determine the extent of the BPEI-teichoic acid interaction and whether this interaction changes the structure of PBP2a. Importantly, the data for S. aureus ATCC 25923 provide a potential route to treat, and prevent, antibiotic resistant infections. By using a combination of BPEI and ampicillin to treat a non-resistant S. aureus infection, the emergence of β-lactam resistant strains in vivo could be slowed. This benefit would not be possible with ampicillin alone. Combination treatments of an antibiotic with a compound that blocks the resistance pathway have been shown to be a viable therapeutic strategy. For example, β-lactam antibiotics can be deactivated by bacteria that possess β-lactamases. The increasing incidence of β-lactamases has been problematic since the 1960s.55 Researchers discovered that amoxicillin could be co-administered with the β-lactamase inhibitor, clavulanic acid. In such combination therapies, commercialized as Augmentin®, the antibiotic is not deactivated, but rather can successfully inhibit cell wall synthesis and kill the bacterium.55, 56 The amoxicillin and clavulanic acid treatment provides a compelling precedent demonstrating that combination therapies can be a successful antibacterial approach. While this approach effectively treats methicillin-susceptible S. aureus (MSSA) infections, MRSA bacteria will endure.57 Our approach has the potential of simultaneously eliminating MSSA and MRSA to limit tissue damage from toxins, decreasing morbidity and mortality. Patients will not have to endure multiple treatments with an array of antibiotics to clear the infection, thereby improving quality of life.58 Fewer medical complications and courses of treatment will result in better patient outcomes at a lower cost to patients and providers.58
Colonies of MRSA bacteria invade host tissue to release toxins that cause tissue injury, leading to significant patient morbidity. The patient suffers while numerous first- and second-line antibiotics are prescribed to no avail. This increases the threat of MRSA to public health.59 Timely MRSA diagnosis58 and delivering drugs of last resort are essential to prevent mortality.60 In 2011, MRSA infected 80,500 people; nearly 1 in 7 cases resulted in death (11,300; 14%)61. Several antibiotics of last resort (vancomycin, linezolid, daptomycin) are effective at killing MRSA, and no MRSA strain is resistant to more than one of them.62, 63 Although there has never been a S. aureus isolate resistant to all approved antibiotics, patients still die from MRSA infections. MRSA infections are deadly because drugs of last resort are given after morbidity from staphylococcal toxins, too late to prevent mortality.58, 62 Vancomycin, a primary treatment option after MRSA diagnosis, presents additional barriers of high cost and toxicity.64–68 New antibiotics, such as oxadiazoles,4 tedizolid,5 and teixobactin,69 are awaiting FDA approval to meet the critical need for new treatments because S. aureus strains resistant to vancomycin and β-lactams have emerged.1, 7, 8 Although we envision improving human health with β-lactam plus polymer combinations, this represents a new pathway to develop other antibiotic treatments. Disabling PBP2a with cationic polymers enables advancement of antibiotic drug discovery by providing ways for other researchers to reinvigorate antibiotic development efforts that have stalled in the face of PBP2a.
Supplementary Material
Acknowledgments
This work is supported by the National Institutes of Health (CVR 1R01GM090064-01), (RHC 1R01AI085161), and the University of Oklahoma. DTG is grateful for partial support of this work by NSF (CBET 0967988) and Oklahoma Bioenergy Center (Project No. 100020) grants. The confocal microscope used in the work was purchased with support from the National Science Foundation (NSF 1126578). We appreciate feedback from Dr. Christopher Corbett, OU Director of Intellectual Property Management. CVR wishes to express our gratitude to Prof. Jacob Schaefer (Washington University in St. Louis) for insights regarding the drug resistance in MRSA and Prof. Cheryl Stevenson (Illinois State University) for helpful discussions.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interest.
SUPPORTING INFORMATION AVAILABLE
Additional data describing the 20-hour growth assays, cytotoxicity data, and confocal microscopy images provided in a supplementary data file. Supplementary Information accompanies the paper on The Journal of Antibiotics website (http://www.nature.com/ja).
References
- 1.Dhanalakshmi TA, Umapathy BL, Mohan DR. Prevalence of methicillin, vancomycin and multidrug resistance among Staphylococcus aureus. J Clin Diagn Res. 2012;6:974–977. [Google Scholar]
- 2.Veni EJK, Baliga S, Shenoy M, Gopalkrishna BK. Community-acquired methicillin resistant Staphylococcus aureus. Int J Curr Res Rev. 2014;6:1–10. [Google Scholar]
- 3.Keynan Y, Rubinstein E. Staphylococcus aureus bacteremia, risk factors, complications, and management. Crit Care Clin. 2013;29:547–562. doi: 10.1016/j.ccc.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 4.O’Daniel PI, et al. Discovery of a new class of non-β-lactam inhibitors of penicillin-binding proteins with gram-positive antibacterial activity. J Am Chem Soc. 2014;136:3664–3672. doi: 10.1021/ja500053x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhanel GG, et al. Tedizolid: A novel oxazolidinone with potent activity against multidrug-resistant gram-positive pathogens. Drugs. 2015;75:253–270. doi: 10.1007/s40265-015-0352-7. [DOI] [PubMed] [Google Scholar]
- 6.Ling LL, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Appelbaum PC. The emergence of vancomycin-intermediate and vancomycin-resistant Staphylococcus aureus. Clin Microbiol Infect. 2006;12:16–23. doi: 10.1111/j.1469-0691.2006.01344.x. [DOI] [PubMed] [Google Scholar]
- 8.Łęski TA, Tomasz A. Role of penicillin-binding protein 2 (pbp2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: Evidence for the cooperative functioning of PBP2, PBP4, and PBP2a. J Bacteriol. 2005;187:1815–1824. doi: 10.1128/JB.187.5.1815-1824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang W, et al. Artesunate has its enhancement on antibacterial activity of β-lactams via increasing the antibiotic accumulation within methicillin-resistant Staphylococcus aureus (mrsa) J Antibiot. 2013;66:339–345. doi: 10.1038/ja.2013.22. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhari K, Bajaj HK. The analysis of virulence factors and beta-lactamase production in clinical isolates of Staphylococcus aureus. Am J PharmTech Res. 2014;4:538–546. [Google Scholar]
- 11.Toussaint KA, Gallagher JC. B-lactam/β-lactamase inhibitor combinations: From then to now. Ann Pharmacother. 2015;49:86–98. doi: 10.1177/1060028014556652. [DOI] [PubMed] [Google Scholar]
- 12.Hartman BJ, Tomasz A. Low-affinity penicillin-binding protein associated with beta-lactam resistance in Staphylococcus aureus. J Bacteriol. 1984;158:513–516. doi: 10.1128/jb.158.2.513-516.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuda C, Suvorov M, Vakulenko SB, Mobashery S. The basis for resistance to beta-lactam antibiotics by penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. J Biol Chem. 2004;279:40802–40806. doi: 10.1074/jbc.M403589200. [DOI] [PubMed] [Google Scholar]
- 14.Gardete S, Tomasz A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J Clin Invest. 2014;124:2836–2840. doi: 10.1172/JCI68834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farha MA, et al. Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to β-lactams. ACS Chem Biol. 2013;8:226–233. doi: 10.1021/cb300413m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, et al. Discovery of wall teichoic acid inhibitors as potential anti-MRSA β-lactam combination agents. Chem Biol. 2013;20:272–284. doi: 10.1016/j.chembiol.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belcheva A, Golemi-Kotra D. A close-up view of the VraSR two-component system - a mediator of Staphylococcus aureus response to cell wall damage. J Biol Chem. 2008;283:12354–12364. doi: 10.1074/jbc.M710010200. [DOI] [PubMed] [Google Scholar]
- 18.Roemer T, Schneider T, Pinho MG. Auxiliary factors: A chink in the armor of MRSA resistance to beta-lactam antibiotics. Curr Opin Microbiol. 2013;16:538–548. doi: 10.1016/j.mib.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 19.Moreillon P. New and emerging treatment of Staphylococcus aureus infections in the hospital setting. Clin Microbiol Infect. 2008;14:32–41. doi: 10.1111/j.1469-0691.2008.01961.x. [DOI] [PubMed] [Google Scholar]
- 20.Demadis KD, Paspalaki M, Theodorou J. Controlled release of bis(phosphonate) pharmaceuticals from cationic biodegradable polymeric matrices. Ind Eng Chem Res. 2011;50:5873–5876. [Google Scholar]
- 21.Odds FC. Synergy, antagonism, and what the chequerboard puts between them. J Antimicrob Chemo. 2003;52:1–1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- 22.Johnson MD, MacDougall C, Ostrosky-Zeichner L, Perfect JR, Rex JH. Combination antifungal therapy. Antimicrob Agents Ch. 2004;48:693–715. doi: 10.1128/AAC.48.3.693-715.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 24.Swoboda JG, Campbell J, Meredith TC, Walker S. Wall teichoic acid function, biosynthesis, and inhibition. Chembiochem. 2010;11:35–45. doi: 10.1002/cbic.200900557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown S, Meredith T, Swoboda J, Walker S. Staphylococcus aureus and Bacillus subtilis W23 make polyribitol wall teichoic acids using different enzymatic pathways. Chem Biol. 2010;17:1101–1110. doi: 10.1016/j.chembiol.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brady MJ, Lisay CM, Yurkovetskiy AV, Sawan SP. Persistent silver disinfectant for the environmental control of pathogenic bacteria. Am J Infect Control. 2003;31:208–214. doi: 10.1067/mic.2003.23. [DOI] [PubMed] [Google Scholar]
- 27.Ubukata K, Nonoguchi R, Matsuhashi M, Konno M. Expression and inducibility in Staphylococcus-aureus of the mecA gene, which encodes a methicillin-resistant S aureus specific penicillin-binding protein. Journal of Bacteriology. 1989;171:2882–2885. doi: 10.1128/jb.171.5.2882-2885.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galm U, et al. Antimicrobial and DNA gyrase-inhibitory activities of novel clorobiocin derivatives produced by mutasynthesis. Antimicrob Agents Ch. 2004;48:1307–1312. doi: 10.1128/AAC.48.4.1307-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhanel GG, Schweizer F, Karlowsky JA. Oritavancin: Mechanism of action. Clin Infect Dis. 2012;54:S214–S219. doi: 10.1093/cid/cir920. [DOI] [PubMed] [Google Scholar]
- 30.Gibney KA, et al. Poly(ethylene imine)s as antimicrobial agents with selective activity. Macromol Biosci. 2012;12:1279–1289. doi: 10.1002/mabi.201200052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang GP, et al. Low molecular weight polyethylenimines linked by beta-cyclodextrin for gene transfer into the nervous system. J Gene Med. 2006;8:736–744. doi: 10.1002/jgm.874. [DOI] [PubMed] [Google Scholar]
- 32.Moghimi SM, et al. A two-stage poly(ethylenimine)-mediated cytotoxicity: Implications for gene transfer/therapy. Mol Ther. 2005;11:990–995. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 33.Zhou JY, Yockman JW, Kim SW, Kern SE. Intracellular kinetics of non-viral gene delivery using polyethylenimine carriers. Pharm Res. 2007;24:1079–1087. doi: 10.1007/s11095-006-9229-5. [DOI] [PubMed] [Google Scholar]
- 34.Fuente E, et al. Study of filler flocculation mechanisms and floc properties induced by polyethylenimine. Ind Eng Chem Res. 2005;44:5616–5621. [Google Scholar]
- 35.Boussif O, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc Nat Acad Sci. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taranejoo S, Liu J, Verma P, Hourigan K. A review of the developments of characteristics of PEI derivatives for gene delivery applications. J Appl Polym Sci. 2015:132. [Google Scholar]
- 37.Cubillos-Ruiz JR, et al. Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. J Clin Invest. 2009;119:2231–2244. doi: 10.1172/JCI37716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. [accessed 27 February 2016]; http://www.polyplus-transfection.com/products/cgmp-grade-in-vivo-jetpei/
- 39.Kunath K, et al. Low-molecular-weight polyethylenimine as a nonviral vector for DNA delivery: Comparison of physicochemical properties, transfection efficiency and in vivo distribution with high-molecular-weight polyethylenimine. J Controlled Release. 2003;89:113–125. doi: 10.1016/s0168-3659(03)00076-2. [DOI] [PubMed] [Google Scholar]
- 40.Wagner M, Rinkenauer AC, Schallon A, Schubert US. Opposites attract: Influence of the molar mass of branched poly(ethylene imine) on biophysical characteristics of siRNA-based polyplexes. RSC Adv. 2013;3:12774–12785. [Google Scholar]
- 41.Khalil H, Chen T, Riffon R, Wang R, Wang Z. Synergy between polyethylenimine and different families of antibiotics against a resistant clinical isolate of Pseudomonas aeruginosa. Antimicrob Agents Ch. 2008;52:1635–1641. doi: 10.1128/AAC.01071-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helander IM, Alakomi H-L, Latva-Kala K, Koski P. Polyethyleneimine is an effective permeabilizer of gram-negative bacteria. Microbiology. 1997;143:3193–3199. doi: 10.1099/00221287-143-10-3193. [DOI] [PubMed] [Google Scholar]
- 43.Thomas KJ, Rice CV. Revised model of calcium and magnesium binding to the bacterial cell wall. Biometals. 2014;27:1361–1370. doi: 10.1007/s10534-014-9797-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halye JL, Rice CV. Cadmium chelation by bacterial teichoic acid from solid-state nuclear magnetic resonance spectroscopy. Biomacromolecules. 2010;11:333–340. doi: 10.1021/bm9010479. [DOI] [PubMed] [Google Scholar]
- 45.Wickham JR, Halye JL, Kashtanov S, Khandogin J, Rice CV. Revisiting magnesium chelation by teichoic acid with phosphorus solid-state NMR and theoretical calculations. J Phys Chem B. 2009;113:2177–2183. doi: 10.1021/jp809313j. [DOI] [PubMed] [Google Scholar]
- 46.da Silva MW. NMR methods for studying quadruplex nucleic acids. Methods. 2007;43:264–277. doi: 10.1016/j.ymeth.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 47.Gotfredsen CH, Meissner A, Duus JO, Sorensen OW. New methods for measuring H-1-P-31 coupling constants in nucleic acids. Magn Reson Chem. 2000;38:692–695. [Google Scholar]
- 48.Jennings MC, Buttaro BA, Minbiole KPC, Wuest WM. Bioorganic investigation of multicationic antimicrobials to combat qac-resistant Staphylococcus aureus. ACS Infectious Disease. 2015;1:304–309. doi: 10.1021/acsinfecdis.5b00032. [DOI] [PubMed] [Google Scholar]
- 49.Campbell J, et al. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem Biol. 2011;6:106–116. doi: 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D’Elia MA, Millar KE, Beveridge TJ, Brown ED. Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J Bacteriol. 2006;188:8313–8316. doi: 10.1128/JB.01336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhavsar AP, Erdman LK, Schertzer JW, Brown ED. Teichoic acid is an essential polymer in Bacillus subtilis that is functionally distinct from teichuronic acid. J Bacteriol. 2004;186:7865–7873. doi: 10.1128/JB.186.23.7865-7873.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Swoboda JG, et al. Discovery of a small molecule that blocks wall teichoic acid biosynthesis in Staphylococcus aureus. ACS Chem Biol. 2009;4:875–883. doi: 10.1021/cb900151k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasquina LW, Santa Maria JP, Walker S. Teichoic acid biosynthesis as an antibiotic target. Curr Opin Microbiol. 2013;16:531–537. doi: 10.1016/j.mib.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Atilano ML, et al. Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc Natl Acad Sci. 2010;107:18991–18996. doi: 10.1073/pnas.1004304107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geddes AM, Klugman KP, Rolinson GN. Introduction: Historical perspective and development of amoxicillin/clavulanate. Int J Antimicrob Ag. 2007;30:S109–S112. doi: 10.1016/j.ijantimicag.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 56.Gill EE, Franco OL, Hancock REW. Antibiotic adjuvants: Diverse strategies for controlling drug-resistant pathogens. Chem Biol Drug Des. 2015;85:56–78. doi: 10.1111/cbdd.12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muller S, et al. Poorly cross-linked peptidoglycan in MRSA due to mecA induction activates the inflammasome and exacerbates immunopathology. Cell Host Microbe. 2015;18:604–612. doi: 10.1016/j.chom.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cosgrove SE, et al. The impact of methicillin-resistance in Staphylococcus aureus bacteremia on patient outcomes: Mortality, length of stay, and hospital charges. Infect Cont Hosp Ep. 2005;26:166–174. doi: 10.1086/502522. [DOI] [PubMed] [Google Scholar]
- 59.CDC. Antibiotic resistance threats in the United States 2013. Centers for Disease Control and Prevention, US Department of Health and Human Services; 2013. [Google Scholar]
- 60.Kopp BJ, Nix DE, Armstrong EP. Clinical and economic analysis of methicillin-susceptible and -resistant Staphylococcus aureus infections. Ann Pharmacother. 2004;38:1377–1382. doi: 10.1345/aph.1E028. [DOI] [PubMed] [Google Scholar]
- 61.Dantes R, et al. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. Jama Intern Med. 2013;173:1970–1978. doi: 10.1001/jamainternmed.2013.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pastagia M, Kleinman LC, de la Cruz EGL, Jenkins SG. Predicting risk for death from MRSA bacteremia. Emerg Infect Dis. 2012;18:1072–1080. doi: 10.3201/eid1807.101371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mangili A, Bica I, Snydman DR, Hamera DH. Daptomycin-resistant, methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis. 2005;40:1058–1060. doi: 10.1086/428616. [DOI] [PubMed] [Google Scholar]
- 64.Bosso JA, et al. Relationship between vancomycin trough concentrations and nephrotoxicity: A prospective multicenter trial. Antimicrob Agents Ch. 2011;55:5475–5479. doi: 10.1128/AAC.00168-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koppula S, Ruben S, Bangash F, Szerlip HM. Pitfalls in dosing vancomycin. Am J Med Sci. 2015;349:137–139. doi: 10.1097/MAJ.0000000000000368. [DOI] [PubMed] [Google Scholar]
- 66.Bruniera FR, et al. The use of vancomycin with its therapeutic and adverse effects: A review. Eur Rev Med Pharmaco. 2015;19:694–700. [PubMed] [Google Scholar]
- 67.Kurosu M, Siricilla S, Mitachi K. Advances in MRSA drug discovery: Where are we and where do we need to be? Expert Opin Drug Dis. 2013;8:1095–1116. doi: 10.1517/17460441.2013.807246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilke MH. Multiresistant bacteria and current therapy - the economical side of the story. Eur J Med Res. 2010;15:571–576. doi: 10.1186/2047-783X-15-12-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ling LL, et al. A new antibiotic kills pathogens without detectable resistance. Nature (London, UK) 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.