

Abstract
HIV/HCV co-infection accelerates progressive liver fibrosis, however the mechanisms remain poorly understood. HCV and HIV independently induce profibrogenic markers TGFβ1 (mediated by reactive oxygen species (ROS)) and NFκB in hepatocytes and hepatic stellate cells (HSC) in monoculture, however, they do not account for cellular cross-talk that naturally occurs. We created an in vitro co-culture model and investigated the contributions of HIV and HCV to hepatic fibrogenesis. GFP reporter cell lines driven by functional ROS (ARE), NFκB, and SMAD3 promoters were created in Huh7.5.1 and LX2 cells, using a transwell to generate co-cultures. Reporter cells lines were exposed to HIV, HCV or HIV/HCV. Activation of the 3 pathways were measured, and compared according to infection status. Extracellular matrix products (Col1A1 and TIMP1) were also measured. Both HCV and HIV independently activate TGFβ1 signaling via ROS (ARE), NFκB, and SMAD3 in both cell lines in co-culture. Activation of these profibrotic pathways was additive following HIV/HCV co-exposure. This was confirmed when examining Col1A1 and TIMP1, where mRNA and protein levels were significantly higher in LX2 cells in co-culture following HIV/HCV co-exposure compared with either virus alone. In addition, expression of these profibrotic genes was significantly higher in the co-culture model compared to either cell type in monoculture, suggesting an interaction and feedback mechanism between Huh7.5.1 and LX2 cells. We conclude that HIV accentuates an HCV-driven profibrogenic program in hepatocyte and HSC lines through ROS, NFκB and TGFβ1 upregulation. Furthermore, co-culture of hepatocyte and HSC lines significantly increased expression of Col1A1 and TIMP1. Our novel co-culture reporter cell model represents an efficient and more authentic system for studying transcriptional fibrosis responses, and may provide important insights into hepatic fibrosis.
Keywords: HIV/HCV co-exposure; hepatic fibrosis; TGFβ1; reactive oxygen species, NFκB/RelA; SMAD3; Col1A1; TIMP1; antioxidant response elements; co-culture
Introduction
Of the approximately one million people infected with human immunodeficiency virus (HIV) in the United States, thirty percent are co-infected with the hepatitis C virus (HCV) due to shared routes of transmission [1]. Chronic infection with HCV is well recognized to be associated with significant liver-related morbidity and mortality. In the highly active anti-retroviral treatment (HAART) era, liver disease is now the third leading cause of mortality among HIV-infected patients after acquired immunodeficiency syndrome (AIDS)-related causes and non-AIDS-defining cancers [1]. HIV co-infection with concomitant HCV infection has been shown to accelerate HIV/HCV-related fibrosis progression compared to HCV monoinfection [2–4]. In this context, HCV-related liver disease has rapidly contributed to a dramatic rise in the proportion of patients with advanced fibrosis or cirrhosis, an increase in the number of co-infected patients who have been referred for liver transplantation, a rise in the wait-list mortality among those awaiting liver transplantation, and a sharp rise in liver-related mortality [5].
Hepatic fibrosis is a wound-healing response to chronic liver injury that results in the accumulation of extracellular matrix (ECM) products, including collagen, fibronectin and proteoglycans. Hepatic stellate cells (HSC) are the primary source of ECM, and therefore are the main cell type within the liver that drive hepatic fibrogenesis[6]. Overexpression of the cytokine transforming growth factor beta-1 (TGFβ1) has been widely demonstrated following liver injury. Induction of TGFβ1 occurs in multiple cell types through several pathways, including reactive oxygen species (ROS), extracellular signal-regulated kinases (ERK), Jun amino-terminal kinases (JNK), and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) [7–9]. TGFβ1 also can activate HSC [10], which in turn further upregulates TGFβ1 production [11], and induces type I alpha 1 collagen (Col1A1) and tissue inhibitor of metalloproteinase 1 (TIMP1) expression, leading to deposition of ECM resulting in hepatic fibrogenesis [12].
Both HCV and HIV can establish infection within the liver through parenchymal and non-parenchymal cells, respectively. Previously, we have demonstrated that HCV and HIV each contribute to fibrogenesis via the induction of profibrogenic cytokines, such as TGFβ1, in monoculture models of hepatocyte or HSC lines infected with HCV or HIV [7]. Several other groups have also demonstrated that HCV induces mitochondrial oxidative stress and ROS [13–15]. In a follow-on study, we found that HCV upregulates TGFβ1 via the induction of ROS, as well as through a p38 mitogen-activated protein kinases, JNK, and ERK1/2-dependent pathway [16]. We also identified that the induction of TGFβ1 by ROS is NFκB dependent [16].
In contrast to HCV, which establishes infection within hepatocytes, HIV cannot infect hepatocytes. However recent evidence supports that HIV is able to infect HSC through a chemokine receptor-independent mechanism [17]. In addition, HIV-1 is also capable of interacting with and can signal through cysteine-X-cysteine receptor 4 (CXCR4) and chemokine motif receptor 5 (CCR5), the co-receptors required for HIV entry, which are expressed on both HSCs and hepatocytes [18–20]. Furthermore, infection with HIV has been shown to induce oxidative stress and upregulate TGFβ1 expression [17]. Therefore, HIV-1 may act both directly and indirectly to contribute to hepatic fibrosis in HIV/HCV co-infected patients. Although HIV itself and its envelope glycoprotein gp120 can each increase TGFβ1 levels, they also augment HCV-induced TGFβ1 expression in infectious JFH1 HCV tissue culture models of HIV/HCV co-infection [17]. In addition, TGFβ1 produced by hepatocytes and other resident liver cell populations further upregulate HCV replication in hepatocytes, thereby augmenting responses to HCV infection [18].
Previously, we provided evidence that both HIV and HCV independently regulate hepatic fibrosisby regulating the production and deposition of components of ECM, including Col1A1 and TIMP1 via induction of ROS [7]. This regulation occurs in an NFκB-dependent fashion in both hepatocytes and HSCs. In addition, we have shown that HIV/HCV co-exposure enhances the production of these profibrogenic cytokines, leading to greater induction of Col1A1 and TIMP1 expression in both hepatocytes and HSCs. These data demonstrate that HIV and HCV both contribute to hepatic fibrogenesis in several cell types within the liver, and that HIV and HCV act cooperatively to induce hepatic fibrogenesis in the context of HIV/HCV co-exposure compared to exposure with either virus.
Cell-cell interactions are central to the function and responses of many organ systems, and the liver is a prime example of where cross talk between cell types is critical. This is further complicated by the lack of an adequate small animal model to study HIV/HCV co-infection; chimpanzees are the only robust animal model that supports HIV and HCV infection, which is costly and inaccessible to many. Therefore, there is a great need for cell culture systems that more realistically recapitulate the in vivo liver milieu during HIV/HCV co-infection, which could also apply more broadly to other liver diseases. The transwell co-culture system provides the unique ability to assess cell-to-cell interactions within specific cell types of interest in a real-time, high throughput manner. Understanding how cell-to-cell interactions modulate or amplify tissue responses to viral infection may provide further insight into the complex cellular processes that accelerate liver disease and identify novel targets that may be amenable for therapeutic applications. To further explore the mechanisms by which HIV and HCV collaborate to stimulate liver fibrogenesis using conditions more closely approximating the in vivo liver environment, we developed reporter cell lines for hepatocytes and HSCs and placed them in co-culture.
Material and Methods
Cell culture
Experiments we performed using Huh7.5.1 cells (kindly provided by Dr Francis Chisari, Scripps Institute, La Jolla, CA, USA) – a sub-line of Huh7 human hepatoma cells that are highly permissive for HCV replication [21], LX2 cells – immortalized human hepatic stellate cells [22], and 293T Human Embryonic Kidney cells. Primary hepatic stellate cells were isolated from the non-parenchymal cell fraction from HCV/HIV seronegative donor livers (Triangle Research Labs, Durham, NC, USA). HSCs were isolated as previously described [23]. Cells were cultured in Dulbecco's modified Eagle's medium (Mediatech, Manassas, VA, USA) supplemented with 10% fetal bovine serum (Mediatech, Manassas, VA, USA), 100 U/mL of penicillin and 100 µg/mL of streptomycin (Lonza/BioWhittaker, Walkersville, MD, USA) and were maintained at 37°C in humidified air containing 5% carbon dioxide.
Viral stocks
The infectious JFH1 genotype 2a clone [24], a generous gift from Dr Takaji Wakita, was used to prepare cell-culture derived HCV particles as previously described [24]. The HIV NL4-3 virus clone, a molecularly cloned highly cytopathic CXCR4-tropic virus, was obtained from the Ragon Institute (MGH Harvard, Boston, MA, USA). HIV stock virus was generated as previously described [25].
Construction of reporter plasmids
Reporter plasmids for the transcription factor response elements of the three major pathways of interest were constructed as previously described [26,27]. The first reporter plasmid is antioxidant response elements (ARE, representing ROS response), second is NFκB, and third is SMAD3 representing TGFβ1 response. In brief, lentiviral expression reporter plasmids were each generated using a green fluorescent protein (copGFP) lentiviral vector (SBI, Mountain View, CA, USA). This vector contains a minimal CMV promoter upstream of the copGFP gene. The sequence of the transcription factor response elements (TRE) for ARE, NFκB, and SMAD3 were TTTGCTGAGTCACTGTGA, GGGACTTTCC, and CAGACA, respectively. ARE, NFκB, and SMAD3 TREs were synthesized at the Center for Computational and Integrative Biology (Boston, MA, USA). These inserts were sub-cloned using EcoRI and SpeI restriction sites in the copGFP lentiviral vector. Three plasmids were constructed containing multiple copies of the specific TRE for each reporter to improve the efficiency. DNA sequences were verified following generation of the plasmid. Binding of the specific transcription factor to the TRE regulates copGFP expression.
Production of lentiviral reporter system particles
Lentiviral reporter system particles were generated as previously described [28]. Briefly, lentiviral particles were produced by co-transfection of 293T cells with the packaging vectors psPAX2 (Addgene plasmid 12260), pRSV-REV (Addgene plasmid 12253), and pMD2.G (Addgeneplasmid 12259). Lentiviral supernatants were harvested at 48 and 72 hours post-transfection, filtered through a 0.45 µm filter, and aliquoted and stored at −80°C until further use.
siRNA transfection
siRNAs were transfected into cells using Lipofectamine RNAiMAX reagent (Invitrogen). Dharmacon On-Target Plus Smart Pool human siRNAs (Fisher Scientific Life Science Research) were used for gene knockdown for RelA (siRelA), NRF2 (siNRF2), SMAD3 (siSMAD3), and a non-targeting negative control (siNeg). Protein levels for each gene knockdown were confirmed by western blot.
Generation of stable reporter cell lines
The three reporter systems were stably transduced into Huh7.5.1 and LX2 cell lines for 4 hours in the presence of polybrene at 8 mg/mL (Sigma Life Science and Biochemicals, St. Louis, MO, USA). Reporter cell lines were then assessed for basal GFP activity by flow cytometry. GFP-negative cells were selected and sorted for subsequent experiments. After 48 hours, transduced reporter cell lines were stimulated with the appropriate inducer for each TRE (tert-butylquinone (tBHQ) for ARE, TNFα for NFκB, and TGFβ1 for SMAD3) for 48 hours. The total number of GFP-positive cells was less than 10%. Therefore, these GFP-positive cells were positively selected and sorted using flow cytometry at the Center for Regenerative Medicine and Technology (MGH, Boston, MA, USA), allowing this functional GFP-positive population with minimal background GFP activity to be expanded for our studies.
Assessment of efficiency of reporter cell lines
The efficiency of each reporter cell line was assessed by determining the number of GFP-positive cells following stimulation with the appropriate inducer. This was performed using flow cytometry using the BD FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA, USA), calculated as the percentage of the total number of cells positive for GFP and the geometric mean fluorescence intensity (geoMFI) of the GFP-positive population (reported as fold change in geoMFI following infection with HCV, HIV or HIV/HCV compared to mock treated cells). Data were analyzed using Flowing software (version 2.5.1, Turku Centre for Biotechnology, Turku, Finland), and confirmed with fluorescence microscopy. Briefly, cells were washed in phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS for 15 minutes. Following fixation, cells were washed again in PBS for 5 minutes. Reporter cells were then incubated with DAPI diluted 1:10000 in PBS and GFP fluorescence was evaluated using an EVOS FL digital inverted fluorescence microscope (Life technologies, Grand Island, NY, USA). Quantitation of GFP-positive cells was performed using ImageJ v1.47 (NIH, Bethesda, MD, USA). The optimal threshold was calculated using Otsu’s method, and the proportion of GFP-positive cells was calculated per high power field at ×40 magnification [29].
Co-culture of Huh7.5.1 and LX2 cells in a transwell system
A co-culture transwell system of Huh7.5.1 and LX2 cells was developed (Figure 2), permitting free exchange of proteins (cross-talk) between cell types across a porous membrane, but still allowing compartmentalization of cell types in order for cell-specific analyses to be performed. The co-culture transwell system was prepared as follows: Huh7.5.1 cells were plated at 2×105 cells/well in 12-well plates, and LX2 cells were seeded at 1×105 on 0.4µM transwells (Costar Corp. Kennebunk, ME, USA) placed over cell-free 12-well plates. After overnight culture, the LX2 seeded transwells were then loaded into the Huh7.5.1 containing wells. After a further 24 hours, the cells were washed with the serum-free media. Huh7.5.1 cells were plated at a 2:1 ratio to HSCs to account for the faster cell multiplication rate of Huh7.5.1 cells compared to HSCs, aiming for a 10:1 ratio at the time the assay was performed [30]. All experiments were performed in triplicate.
Figure 2. Schematic representation of the co-culture transwell model.
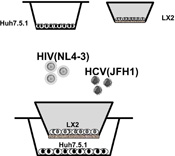
Huh7.5.1 cells supportive of HCV infection were seeded into the larger bottom transwell and hepatic stellate cells (LX2) were seeded into the smaller upper transwell. The membrane separating the two wells is porous at 0.4 microns to allow free exchange of secreted molecules between the two cell types. HCV and/or HIV were then added to the culture media at an MOI of 3.
MOI = multiplicity of infection
HCV and HIV exposure of monoculture and co-culture cell lines
Cells were incubated with HCV JFH1 and / or HIV NL4-3 virus at a multiplicity of infection (MOI) of 3 at 37°C in humidified air with 5% CO2. After a 4-hour adsorption period, the virus containing medium was removed, the cells were washed with PBS, and replaced with fresh growth medium for a further 72 hours at which time cells were harvested for RNA and protein.
Quantitative RT-PCR
Total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen, Hilden, Germany). A total of 1 µg of extracted RNA was used in the reverse transcriptase reaction to generate complimentary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit according to the manufacturer's instructions (Applied Biosystems, Foster City, CA, USA). CoL1A1 and TIMP1 mRNA expression were then measured by quantitative PCR using 1µl of the synthesized cDNA using the DyNAmo HS SYBR Green qPCR kit (Thermo Scientific, Waltham, MA, USA). The primer sequences are listed in Table 1. CoL1A1 and TIMP1 mRNA expression was normalized to GAPDH to calculate a fibrosis gene/GAPDH arbitrary unit (fold-change expression).
TABLE 1.
Primer sequences used for quantitative PCR
| Forward primer | Reverse primer | |
|---|---|---|
| CoL1A1 | ATGTTCAGCTTTGTGGACCTC | TTCGTCTTGGCCCTCGACTT |
| GAPDH | ACCTTCCCCATGGTGTCTGA | GCTCCTCCTGTTCGACAGTCA |
| TIMP1 | ACTTCCACAGGTCCCACAAC | ACTTCCACAGGTCCCACAAC |
| JFH-1 | TCTGCGGAACCGGTGAGTA | TCAGGCAGTACCACAAGGC |
Protein quantification by Western Blot
Proteins from whole cell lysates were separated by SDS-polyacrylamide gel electrophoresis (PAGE) with NuPAGE Novex Bis-Tris precast 4 to 12% gradient gels (Invitrogen, Carlsbad, CA, USA) and transferred to nitrocellulose membranes. The primary antibodies anti-actin (Mouse mAB; Sigma Life Science and Biochemicals), anti-TIMP1 (Rabbit pAB; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-CoL1A1 (Rabbit pAB;Thermo Fisher, Rockford, IL, USA), phosphorylated and unphosphorylated NFκB (RelA) (Rabbit pAB; Cell Signaling Technology, Inc., Danvers, MA, USA), phosphorylated NRF2 (Rabbit mAB; abcam, Cambridge, MA, USA), unphosphorylated NRF2 (Rabbit pAB; abcam), and phosphorylated and unphosphorylated SMAD3 (Rabbit mAB; Cell Signaling Technology, Inc.) were incubated overnight at 4°C. The secondary antibody, anti-mouse IgG (Donkey mAB; amersham Biosciences, Piscataway, NJ, USA), and anti-rabbit IgG (Sheep mAB; amersham Biosciences) were incubated for 60 minutes at room temperature.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism v6 (La Jolla, CA, USA) and Microsoft Excel 2013. Continuous data are presented as medians + interquartile ranges and comparisons were made using the Mann-Whitney U test, with results considered significant at P<0.05. Quantification analyses for western blots were performed using ImageJ software and normalized to β-actin. Means plus the standard error of the mean are presented for three independent experiments.
Results
Generation of stable cell lines expressing a functional ARE, NFκB or SMAD3 driven reporter transgene
Efficiency of the three reporter cell lines
Following stimulation, GFP signal was observed to increase in all three of the transduced reporter cell lines. In Huh7.5.1 cells, the percentage of GFP-positive cells following stimulation with the appropriate stimulant were 44.4% for ARE, 76.7% for NFκB and 59.1% for SMAD3 (Figure 1A). Similarly, in LX2 transduced reporter cell lines GFP signal increased following stimulation (83.1% for ARE, 68.5% for NFκB and 86.6% for SMAD3) (Figure 1A). The reporter cell lines were also tested using fluorescence microscopy, with similar efficiency of GFP fluorescence signal activation noted following stimulation for all reporter cell lines (Figure 1B).
Figure 1. Efficiency of the reporter cell lines.


Efficiency of the newly generated Huh7.5.1 and LX2 reporter cell lines for ARE, NFκB and SMAD3 following stimulation were tested using (A) flow cytometry and (B) fluorescence microscopy. Following stimulation for 48 hours with the appropriate stimulant (tBHQ for ARE, TNFα for NFκB and TGFβ1 for SMAD3), the number of GFP-positive cells increased significantly compared to mock treated cells as measured by flow cytometry and fluorescence microscopy. Nuclei were counterstained with DAPI (blue).(C) NRF2, RelA and SMAD3 siRNA pre-treatment significantly reduces the effects of tHBQ, TNFα, and TGFβ on reporter cell lines (LX2, and Huh7.5.1). Reporter cell lines were transfected with siRNA. At 48 following transfection, cells were stimulated with appropriate stimulant for a further 48 hours. Cells were tested by flow cytometry. siRNA pre-treatment significantly reduced the percentage of GFP-positive cells following stimulation.
ARE = Antioxidant Response Elements; tBHQ = tert-Butylhydroquinone; NFκB = Nuclear Factor-κB; TNFα = Tumor necrosis factor alpha; SMAD3 = SMAD family member 3; TGFβ1 = Transforming Growth Factor Beta 1.
We then demonstrated we could effectively block the downstream activation of the transcriptional response elements after stimulation with the suitable stimulator by using siRNA. We used NRF2 siRNA for the ARE reporter, RelA siRNA for the NFκB reporter, and SMAD3 siRNA for the SMAD3 reporter. The percentage of GFP positive cells were significantly reduced following siRNA transfection (26.8% for ARE, 9.4% for NFκB and 20.4% for SMAD3) (Figure 1C). Similarly, in LX2 reporter cell lines GFP positive cells were significantly reduced after siRNA pre-treatment (14.7% for ARE, 14.1% for NFκB and 17.9% for SMAD3) (Figure 1C) corroborating the efficiency of these reporter cell lines.
Validation of the novel transwell reporter co-culture model
Co-exposure with HIV augments HCV induction of ARE responsive genes
ROS induces NRF2 phosphorylation, which in turn drives ARE-dependent transcription of antioxidant genes. To more adequately dissect the relationship between HCV and HIV in a more authentic in vitro system, we explored the possibility that co-exposure with HIV augments HCV-related fibrogenesis but that cross-talk between cells within the liver are required to fully activate these profibrotic pathways. First we validated our new reporter co-culture model in whichHuh7.5.1 and LX2 cells are separated by a transwell as shown in Figure 2. This configuration facilitates communication between the two cell types while also permitting recovery of the individual cell types for further individual analysis. To confirm cooperative interaction between HCV and HIV in our co-culture reporter model system, we first evaluated the induction of ARE responsive genes in the hepatocyte and HSC lines harboring the ROS-regulated ARE reporters. These cell lines were co-cultured and exposed to HIV, HCV and HIV/HCV.
HCV monoinfection significantly induced ARE compared to mock infection in Huh7.5.1 cells as measured by the percentage of GFP-positive cells (37% vs. 15.5%GFP-positive cells for HCV and mock infection, respectively, P<0.005; Figure 3A), geoMFI of GFP signal (1.5 fold vs. 1-fold induction for HCV and mock infection, respectively, P<0.005; Figure 3B) and the amount of ARE-regulated transcription factor phosphor-NRF2 (P-NRF2) by Western blot (Figure 3C). Similar findings were noted in LX2 cells following exposure to HCV compared to mock infection (30% vs. 5.2% ARE activation as measured by the percentage of GFP positive cells; 1.5 fold vs. 1-fold for geoMFI of GFP signal; and by Western blot; Figure 3). HIV exposure also induced ARE activation in Huh7.5.1 cells compared to mock treated cells with a significantly higher proportion of GFP-positive cells (22% vs. 15.6%, P<0.05), higher geoMFI (1.2 fold vs. 1-fold, P<0.05) and greater P-NRF2 in Huh7.5.1 cells exposed to HIV (Figure 3). However, no significant difference was noted in LX2cells following HIV exposure compared to mock treated cells (Figure 3).
Figure 3. Induction of ARE responsive genes in LX2 cells and Huh7.5.1 cells following mock treatment, HCV exposure, HIV exposure and HIV/HCV co-exposure for 72 hours.

Activation of ARE responsive genes were measured by (A) percentage of GFP-positive cells by flow cytometry; (B) geoMFI of GFP-positive cells by flow cytometry; and (C) phoshphorylated NRF2. The upper panel of part C shows representative western blots of the protein levels of phosphorylated NRF2 (P-NRF2), total NRF2, and β Actin. The lower panel of part C depicts densitometry quantification of the ratio of P-NRF2 to total NRF2 after exposure to HCV and/or HIV for 72 hours.
* P<0.05
** P<0.005
HCV = hepatitis C virus; HIV = human immunodeficiency virus; GFP = green fluorescence protein; geoMFI = geometric fluorescence intensity; ARE = antioxidant response element; P-NRF2 = phosphorylated NRF2
The greatest degree of ARE activation was observed in the context of HCV/HIV co-exposure of Huh7.5.1 cells in co-culture, where the highest percentage of GFP-positive cells (41.6%, P<0.05), highest geoMFI of GFP signal intensity (1.8 fold induction, P<0.005) and greatest induction of P-NRF2 were observed (Figure 3), validating our previous findings in the monoculture model. A similar trend was observed in LX2 cells co-cultured with Huh7.5.1 cells (Figure 3).
Cooperative induction of NFκB responsive genes by HCV and HIV in the co-culture model
NFκB is a transcription factor that regulates the expression of multiple pro-inflammatory genes [31] and also plays a central role in hepatic fibrogenesis [32]. We have previously shown that both HCV and HIV independently regulate fibrogenesis through NFκB induction [7].
We therefore evaluated NFκB reporter cells in our co-culture model system. Again, we validated that HCV or HIV mono-exposure significantly induced NFκB activation compared to mock treatment in both cell lines (Figure 4). This was observed when considering all parameters tested: percentage of GFP-positive cells (42.5% vs. 1.5% for HCV and mock treatment in LX2 cells, respectively; 37% vs. 7.5% for HCV and mock treatment in Huh7.5.1 cells, respectively; Figure 4A), GFP signal intensity (2.2 fold vs. 1-fold for HCV and mock treatment in LX2 cells, respectively, P<0.05; 3.0 fold vs. 1-fold for HCV and mock treatment in Huh7.5.1 cells, respectively, P<0.05; Figure 4B) and amount of phosphorylated RelA (P-RelA), a subunit of the NFκB complex (Figure 4C). As expected, HIV/HCV co-exposure had an additive effect on NFκB responsive gene induction in the reporter co-culture model system, particularly in LX2 cells, confirming that co-exposure to HIV and HCV induces NFκB responsive genes to a greater extent than either HCV or HIV infection alone (Figure 4B).
Figure 4. Co-exposure with HCV and HIV induces NFκB responsive genes in LX2 cells and Huh7.5.1 cells following mock treatment, HCV exposure, HIV exposure and HIV/HCV co-exposure for 72 hours.

Induction of NFκB responsive genes were as measured by (A) percentage of GFP-positive cells by flow cytometry; (B) geoMFI of GFP-positive cells by flow cytometry; and (C) phosphorylated RelA. The upper panels in part C shows representative western blots of the protein levels of phosphorylated RelA, total RelA, and β Actin. The lower panel of part C depicts densitometry quantification of the ratio of P-RelA to total RelA after exposure to HCV and/or HIV for 72 hours.
* P<0.05
HCV = hepatitis C virus; HIV = human immunodeficiency virus; GFP = green fluorescence protein; geoMFI = geometric fluorescence intensity; P-RelA = phosphorylated RelA
HIV augments HCV induction of SMAD3 responsive genes in our novel reporter co-culture model
Next we validated our SMAD3 GFP reporter cell lines, which evaluate changes in TGFβ1 responsive genes, in the co-culture model. HCV and HIV mono-exposure each activated TGFβ1 in both cell lines as demonstrated by the increase in the percentage of GFP-positive cells (32.8% vs. 12.6 for HCV and mock exposure in Huh7.5.1 cells, and28.3% vs. 3.7% for in LX2 cells, P<0.05; Figure 5A), geoMFI of GFP signal (1.5 fold vs. 1-fold for HCV and mock exposure in Huh7.5.1 cells, and 1.6 fold vs. 1-fold in LX2 cells; Figure 5B) and phosphorylated SMAD3 (Figure 5C). Again, we confirmed that HIV/HCV co-exposure significantly increased TGFβ1 activation in the reporter co-culture model compared to mono-exposure with either virus. It was as expected, the highest in percentage of GFP-positive cells (44.2%, and 59%in LX2 and Huh7.5.1respectively, p<0.05), and in geoMFI of GFP signal (2.2 and 2.3 fold induction in LX2 and Huh7.5.1respectively, p<0.005; Figure 5).
Figure 5.

HIV augments HCV induction of SMAD3 responsive genes in co-culture model following mock treatment, HCV exposure, HIV exposure and HIV/HCV co-exposure for 72 hours. SMAD3 responsive gene activation was measured by (A) percentage of GFP-positive cells by flow cytometry; (B) geoMFI of GFP-positive cells by flow cytometry; and (C) phosphorylated SMAD3. The upper panel of part C shows a representative western blot of protein levels of phosphorylated SMAD3, total SMAD3, and β Actin. The lower panel of part C depicts the densitometry quantification of the ratio of P-SMAD3 to total SMAD3 after exposure to HCV and/or HIV for 72 hours.
* P<0.05
HCV = hepatitis C virus; HIV = human immunodeficiency virus; GFP = green fluorescence protein; geoMFI = geometric fluorescence intensity; P-SMAD3 = phosphorylated SMAD3
HIV/HCV co-exposure additively promote ECM production in the co-culture model
We have previously demonstrated that HIV increases CoL1A1 and TIMP1 expression in both HSCs and Huh7.5.1 cell lines in monoculture, and that the addition of HCV further increased the expression of these ECM products [7]. This additive effect of both viruses on induction of these fibrogenic gene products may underlie the more aggressive disease phenotype observed in HIV/HCV co-infected patients. However, these data were generated in monoculture systems that do not accurately reflect the multicellular human in vivo liver environment, and therefore do not take into consideration changes in activation of genes that may result from the natural cross-talk between cell types.
Therefore, we sought to evaluate the difference in activation of the ECM products COL1A1 and TIMP1 in our novel reporter co-culture model in the context of HIV and/or HCV infection and directly compare these results with our previous monoculture model.
Monoculture model
In monoculture of LX2 cells or Huh7.5.1 cells, we again found that both HIV and HCV individually induced mRNA expression of CoL1A1 and TIMP1 in each cell line (2.0 to 4.0 fold induction, P<0.05; Figure 6).
Figure 6.


Comparison of induction of (A) CoL1A1 mRNA expression; (B) TIMP1 mRNA expression; (C) CoL1A1 and TIMP1 protein levels. The upper panel of part C shows a representative western blot of the intracellular protein levels of CoL1A1 and TIMP1 in LX2 and Huh7.5.1 cells following exposure to HCV and/or HIV infection for 72 hours. The lower panel of part C shows the densitometry quantification of the normalized CoL1A1 and TIMP1 protein levels to β-actin after exposure to HCV and/or HIV for 72 hours; (D) Comparison of induction of CoL1A1 and TIMP1 mRNA expression in pHSCs in monoculture and in co-culture with Huh7.5.1 cells.
* P<0.05
** P<0.005
CoL1A1 = Collagen, type I, alpha 1; TIMP1 =Tissue inhibitor of metalloproteinase-1; HCV = hepatitis C virus; HIV = human immunodeficiency virus
HIV/HCV co-exposure resulted in significantly greater induction of CoL1A1 mRNA in both LX2 and Huh7.5.1 reporter cells in monoculture compared to HIV infection alone (3.5 and3.6 fold induction for HIV/HCV co-exposure vs. 2.1, and 2.3 fold for HIV mono-exposure in LX2 and Huh7.5.1 respectively, p<0.05; Figure 6A). HIV/HCV co-exposure also significantly increased CoL1A1 expression in LX2 cells compared to HCV mono-exposure (3.5 fold vs 2.2 fold, p<0.005; Figure 6A). In contrast, no further increase in CoL1A1 mRNA expression was noted in Huh7.5.1 cells following HIV/HCV co-exposure compared to HCV mono-exposure (Figure 6A).
HIV exposure was noted to have a weaker effect on stimulating TIMP1 mRNA expression in Huh7.5.1 cells compared to HCV infection (1.4 fold vs. 3.7 fold; Figure 6B).
In monoculture, HCV and HIV exposure each upregulated TIMP1 expression in LX2 and Huh7.5.1 reporter cell lines (Figure 6B). However, co-exposure with HIV/HCV in LX2 did not appear to further induce TIMP1 expression in monoculture (0.5 fold induction; Figure 6B).
Co-culture model
We then evaluated our co-culture model under the same experimental conditions to determine if co-operative cross talk between LX2 cells co-cultured with Huh7.5.1 cells alter the degree to which the profibrotic ECM markers, CoL1A1 and TIMP1, are induced following HCV, HIV and HIV/HCV co-exposure compared to the mono-exposure model system.
We again found that both HCV and HIV mono-exposure each upregulate CoL1A1 and TIMP1 mRNA expression in both LX2 and Huh7.5.1 reporter cell lines in co-culture (3.9–4.2 fold vs. 1.1–3.0 fold for HCV and HIV exposure, respectively in both LX2 and Huh7.5.1, P<0.05; Figure 6). While in Huh7.5.1 and LX2 co-culture and co-exposure with HIV and HCV did not consistently increase mRNA induction of these ECM products in Huh7.5.1, we observed significant upregulation of both profibrotic ECM markers in LX2 cells co-cultured with Huh7.5.1 cells compared to that observed in our LX2 reporter cells in monoculture. CoL1A1 mRNA expression was increased 4.4 fold in LX2 cells co-cultured with Huh7.5.1 cells following HIV/HCV co-exposure but only 3.5 fold in LX2 cells in monoculture (P<0.05; Figure 6A). The results for TIMP1 mRNA expression were even more striking, with a 5.3 fold induction in LX2 cells co-cultured with Huh7.5.1 cells compared to 3.0 fold induction in LX2 cells in the previous monoculture model (p<0.005; Figure 6B). We then confirmed the effect of HIV/HCV co-exposure on CoL1A1 and TIMP1 protein levels from total cell lysates in our new co-culture model compared to our previous monoculture model of LX2 cells (Figure 6C). Therefore, these data strongly highlight the importance of cooperative activation of profibrotic pathways in HSCs in the pathogenesis of HCV and HIV-related liver fibrosis.
To validate our findings, we performed identical experiments in primary HSC (pHSC) isolated from healthy HIV/HCV seronegative human liver donors. pHSCs were cultured and exposed to HCV, and/or HIV for 72 hours either in monoculture or in the transwell co-culture system together with Huh7.5.1 cells. A significant increase in the pHSC activation markers Col1A1 and TIMP1 mRNA expression was observed following exposure to HCV and/or HIV in both monoculture and co-culture models (2 to 6 fold induction; p<0.05; Figure 6D). As observed in the LX2 cell lines, the expression of these profibrotic markers was noted to be significantly higher in the pHSC in co-culture with Huh7.5.1 cells compared to pHSCs in monoculture (p<0.05; Figure 6D). These data valid our findings in our reporter cell line co-culture model, demonstrating greater activation of pHSCs profibrotic markers in vitro following exposure to both viruses compared to either virus alone, suggesting that both viruses are additive in activating these pathways and may contribute to the accelerated fibrogenesis seen in HCV/HIV coinfection.
HIV increases permissiveness to HCV infection in Huh7.5.1 cells
It has been well established that JFH1 infects Huh7.5.1 cells, but not HSCs. However, despite the fact that HSCs are not directly infected by HCV, hepatic non-parenchymal cells, including HSC, may play a central role in the maintenance of normal hepatocyte function. In addition, in vitro exposure to the HIV envelope protein gp120 increases HCV replication 2–3 fold in hepatocytes. We have previously shown that HIV has direct and indirect effects on two major hepatic cell types (hepatocytes and HSCs) leading to increased HCV replication[7]. However, because these studies were performed in monoculture models, it remains unknown what the contribution of cellular cross-talk is to cells in co-culture and its possible impact upon HCV permissiveness.
To determine whether HCV permissiveness is further altered in the context of a model of LX2 cells co-cultured with Huh7.5.1 cells following HIV/HCV co-exposure, we infected two groups of co-cultured cells with HCV (JFH1) alone or, HCV co-incubated with HIV for 72 hours. We performed parallel experiments in Huh7.5.1 cells monoculture.
In monoculture, we observed a significantly greater increase in HCV RNA in the Huh7.5.1 cells incubated with HIV and HCV compared to HCV alone (1.7 fold vs. 1-fold, P<0.005; Figure 7A). This was also confirmed by a 2.5 fold increase in intracellular HCV core detected by Western blot in HIV/HCV co-exposure compared to infection with HCV alone (Figure 7B).
Figure 7. HIV increases permissiveness to HCV infection in Huh7.5.1 cells in monoculture and co-culture models, but HCV permissiveness was further increased in the co-culture model.

HCV permissiveness was measured by (A) intracellular HCV RNA levels by RT-PCR and (B) HCV core protein levels measured by western blot. The upper panel of part B shows a representative western blot of intracellular HCV core protein levels in Huh7.5.1 cells. The lower panel of part B shows the densitometry quantification of the normalized HCV core protein levels to β-actin, where HCV monoinfection monoculture was considered 1 fold change. Huh7.5.1 cells were exposed to HCV and/or HIV for 72 hours in monoculture or with the addition of LX2 reporter cell lines in the transwell co-culture system.
HCV = hepatitis C virus; HIV = human immunodeficiency virus
In the co-culture model, when exposed to HCV alone, we found that permissiveness to HCV infection was even greater in Huh7.5.1 cells co-cultured with LX2 cells as measured by HCV RNA levels (1.8 fold increase; Figure 7A)and amount of HCV core (3.0 fold increase; Figure 7B). In addition, co-exposure with HIV and HCV further enhanced HCV permissiveness in the co-culture model compared to theHuh7.5.1 monoculture model (3.2 increase in HCV RNA and 3.7 fold increase in HCV core protein; Figure 7). These data indicate that HIV alters and increases permissiveness of Huh7.5.1 cells to HCV infection, and further underscore the importance of the co-culture model in providing an even richer context for disease pathogenesis in HIV/HCV co-exposure.
Discussion
HCV co-infection is highly prevalent among HIV-infected individuals and is associated with a more aggressive liver disease phenotype driven by accelerated fibrosis progression. This has been reflected in recent studies that have shown that liver disease is now the third highest cause of mortality among HIV infected persons in the post-HAART era. Currently there are no therapeutic options to halt or reverse hepatic fibrosis, apart from addressing the underlying cause of liver disease. Although the treatment landscape for both HIV and more recently HCV have advanced significantly over time, these therapies may still not meaningfully reverse the natural history of liver disease in HIV/HCV co-infected patients who have established significant fibrosis or cirrhosis. Therefore, there has been much effort directed towards understanding hepatic fibrogenesis and fibrosis progression in these patients in order to identify potential new approaches to intervention.
In our previous monoculture studies, we demonstrated that HIV, HCV, and HIV/HCV co-exposure lead to induction of ROS, TGFβ1, and NFκB. Activation of these profibrotic pathways was observed to be greater in the setting of HIV/HCV co-exposure compared to exposure to either HIV or HCV. However, since these studies were performed in monoculture models, they do not take into consideration further modulation of these pathways mediated by cross-talk between cell subsets. Therefore, these models do not faithfully recapitulate the in vivo liver environment, so caution must be applied when interpreting these data.
In an effort to more accurately dissect the mechanistic contributions of HIV, HCV, and HIV/HCV infection to hepatic fibrogenesis, we created a novel, reproducible in vitro reporter co-culture model. We generated reporter plasmids for the response elements of the key postulated transcription factor pathways associated with hepatic fibrogenesis in two major cell types within the liver that are activated by HIV and HCV; Huh7.5.1 cells (supportive of HCV infection and signaled by HIV), and LX2 HSCs (signaled by HIV and HCV). This co-culture model utilizes a transwell approach that allows for cell-to-cell communication, thereby permitting assessment of the contribution of cross-talk between cell types in the activation of profibrotic pathways (ARE (ROS), NFκB, and SMAD3) in the context of exposure with either HIV or HCV, and co-exposure with HIV/HCV. In addition, the transwell approach allows compartmentalization of the cell types so that cell-specific analyses can be performed, but still permits the free exchange of secreted factors between cell types.
Using this co-culture system, we confirmed that HIV and HCV exposure each induce oxidative stress, which upregulates TGFβ1 expression and induces NFκB responsive genes in a multicellular environment. Again, we observed that in the context of HIV/HCV co-exposure, there was greater induction of these profibrotic pathways compared to either HIV or HCV infection alone. However, when compared the old monoculture model, we demonstrated greater upregulation of the ECM genes COL1A1 and TIMP1 in LX2 cells in the co-culture model. These data not only confirm the additive effect of HIV/HCV co-exposure on hepatic fibrogenesis but also demonstrates that cross-talk between Huh7.5.1 and LX2 cells occurs and results in further activation of profibrotic pathways in LX2 cells. These data support that our co-culture reporter model system is an effective method to dissect the transcriptional activation of profibrotic pathways and confirm that HCV and HIV significantly induce these pathways in both reporter cells lines compared to mock exposure. Thus, our data strongly highlights the critical importance of co-culture model systems in evaluating hepatic fibrosis in the context of HCV, HIV or HIV/HCV exposure in order to more accurately study and mimic the in vivo liver milieu to understand liver disease pathogenesis. Our data also validate our previous findings from our monoculture system, and demonstrate that HIV and HCV have an additive effect in driving the TGFβ1 pro-fibrotic pathway in co-culture.
This novel co-culture reporter system offers several advantages over the previous monoculture model. Using a transwell approach, two different cell lines can be studied with the added benefit that the polyester membrane allows for cross-talk between the two cell types, becausethey remain compartmentalized their ability to be isolated and analyzed individually is preserved, neither of which is possible in monoculture or in conditioned media models. Furthermore, the transwell approach allows for pure cell populations to be seeded without contamination from other cell types, as may occur with percoll density gradient cell fraction isolation, and is simple and easy to use if FACS sorting is not available. Therefore, it is possible to examine in greater depth the relative roles of the key drivers of HIV/HCV-related liver disease pathogenesis that previously was not possible. Furthermore, the introduction of stable fluorescent reporter constructs into each of the cell lines allowed direct live cell analysis of the transcriptional activation of the profibrotic pathways of interest (ARE (ROS), TGFβ1, and NFκB) activated by HIV, HCV, and HIV/HCV infection. The degree of induction could be assessed and quantified using flow cytometry or fluorescent microscopy. However, the latter may be partly limited due to impedance from the transwell system itself and the background fluorescence associated with the culture medium. These improvements to the in vitro models to study liver disease pathogenesis may in turn, identify novel targets or pathways for therapeutic development that have the potential to alter disease progression, as well as provide a more realistic system to evaluate the effect of HAART therapy in an in vitro context. Since hepatic fibrogenesis is a common pathway that leads to cirrhosis irrespective of the etiology of liver disease, this novel reporter co-culture model system has applications beyond viral hepatitis and HIV liver disease pathogenesis. In addition, theco-culture model can be adapted to include additionalcell types of interest, including macrophage cell lines, and therefore has the potential to moreaccurately simulate in vivo conditions. In addition, this new co-culture model could be used to more accurately study drug metabolism and toxicity.
Although the use of primary cells are preferred to more accurately mimic in vivo conditions, Huh7.5.1 and LX2 cells were chosen to test our co-culture model system for several reasons, including limited permissiveness for HCV infection and genetic heterogeneity of PHHs based on donor source. However, it will be possible to adapt our co-culture model to include PHHs in addition to the primary HSCs to evaluate non-viral diseases. Furthermore, techniques to generate hepatocyte-like cells derived from inducible pluripotent stem cells have been developed and show significant promise as a superior cell line for studying hepatotropic viruses that could be readily substituted in our co-culture model.
In summary, we have shown that HIV/HCV co-exposure in hepatocytes and stellate cells reveals cooperative transcriptional activation of profibrotic pathways. We also show the importance of cross-talk between liver cell types in order to maximally activate these profibrotic pathways, as highlighted by the augmented profibrotic responses observed when hepatocytes and HSC that are co-cultured together, compared to HSC monoculture. Our novel, reproducible and easy to use co-culture model therefore provides a more authentic cell-culture system to study hepatic fibrogenesis and is able to evaluate the contribution of cell-specific responses in driving HIV/HCV-related liver disease. Furthermore, our co-culture model has wider applicability beyond the scope of HIV/HCV-related liver disease pathogenesis and can be adapted to include other cells and diseases of interest, and therefore has the potential to unravel new insights into liver disease pathogenesis that can be targeted for novel therapeutic development.
Acknowledgments
We are grateful to these investigators and institutes for supplying the following: Drs. Ralf Bartenschlager and Takaji Wakita (infectious HCV virus JFH-1 DNA construct); Dr. Francis Chisari (Huh7.5.1 cell line); and Dr. Scott L. Friedman (HSC LX-2 cells). We would also like to thank Dr. Todd Allen (Ragon Institute, MGH Harvard, Boston, MA, USA) for providing the HIV NL4-3 virus clone, obtained through the NIH AIDS Research and Reference Reagent Program.
Funding Source: NIH DK098079 (RTC), DK108370 (RTC)
Abbreviations
- AIDS
acquired immune deficiency syndrome
- ARE
antioxidant response elements
- CCR5
chemokine motif receptor 5
- cDNA
complimentary DNA
- CXCR4
cysteine-X-cysteine receptor 4
- Col1A1
collagen type 1 alpha 1
- ECM
extracellular matrix
- ERK
extracellular signal-regulated kinases
- GFP
green fluorescence protein
- HAART
highly active anti-retroviral treatment
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- HSCs
hepatic stellate cells
- JNK
jun amino-terminal kinases (JNK)
- MOI
multiplicity of infection
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- P-NRF2
phosphorylated NRF2
- P-RelA
phosphorylated RelA
- PAGE
SDS-polyacrylamide gel electrophoresis
- PHH
primary human hepatocyte
- pHSCs
primary hepatic stellate cells
- ROS
reactive oxygen species
- tBHQ
tert-butylquinone
- TGFβ1
transforming growth factor beta-1
- TIMP1
tissue inhibitor of metalloproteinase 1
- TRE
transcription factor response elements
References
- 1.Smith CJ, Ryom L, Weber R, Morlat P, Pradier C, et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet. 2014;384:241–248. doi: 10.1016/S0140-6736(14)60604-8. [DOI] [PubMed] [Google Scholar]
- 2.Thein HH, Yi Q, Dore GJ, Krahn MD. Estimation of stage-specific fibrosis progression rates in chronic hepatitis C virus infection: a meta-analysis and meta-regression. Hepatology. 2008;48:418–431. doi: 10.1002/hep.22375. [DOI] [PubMed] [Google Scholar]
- 3.Kim AY, Chung RT. Coinfection with HIV-1 and HCV--a one-two punch. Gastroenterology. 2009;137:795–814. doi: 10.1053/j.gastro.2009.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen JY, Feeney ER, Chung RT. HCV and HIV co-infection: mechanisms and management. Nat Rev Gastroenterol Hepatol. 2014;11:362–371. doi: 10.1038/nrgastro.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ly KN, Xing J, Klevens RM, Jiles RB, Ward JW, et al. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann Intern Med. 2012;156:271–278. doi: 10.7326/0003-4819-156-4-201202210-00004. [DOI] [PubMed] [Google Scholar]
- 6.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 7.Lin W, Wu G, Li S, Weinberg EM, Kumthip K, et al. HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NFkappaB. J Biol Chem. 2011;286:2665–2674. doi: 10.1074/jbc.M110.168286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002;115:3193–3206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- 9.Thein HH, Yi Q, Dore GJ, Krahn MD. Natural history of hepatitis C virus infection in HIV-infected individuals and the impact of HIV in the era of highly active antiretroviral therapy: a meta-analysis. AIDS. 2008;22:1979–1991. doi: 10.1097/QAD.0b013e32830e6d51. [DOI] [PubMed] [Google Scholar]
- 10.Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 11.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188–1198. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 12.Wang K, Lin B, Brems JJ, Gamelli RL. Hepatic apoptosis can modulate liver fibrosis through TIMP1 pathway. Apoptosis. 2013;18:566–577. doi: 10.1007/s10495-013-0827-5. [DOI] [PubMed] [Google Scholar]
- 13.Korenaga M, Wang T, Li Y, Showalter LA, Chan T, et al. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem. 2005;280:37481–37488. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Boehning DF, Qian T, Popov VL, Weinman SA. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007;21:2474–2485. doi: 10.1096/fj.06-7345com. [DOI] [PubMed] [Google Scholar]
- 15.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, et al. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 16.Lin W, Tsai WL, Shao RX, Wu G, Peng LF, et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology. 2010;138:2509–2518. 2518, e2501. doi: 10.1053/j.gastro.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuyama AC, Hong F, Saiman Y, Wang C, Ozkok D, et al. Human immunodeficiency virus (HIV)-1 infects human hepatic stellate cells and promotes collagen I and monocyte chemoattractant protein-1 expression: implications for the pathogenesis of HIV/hepatitis C virus-induced liver fibrosis. Hepatology. 2010;52:612–622. doi: 10.1002/hep.23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin W, Weinberg EM, Tai AW, Peng LF, Brockman MA, et al. HIV increases HCV replication in a TGF-beta1-dependent manner. Gastroenterology. 2008;134:803–811. doi: 10.1053/j.gastro.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 19.Rotman Y, Liang TJ. Coinfection with hepatitis C virus and human immunodeficiency virus: virological, immunological, and clinical outcomes. J Virol. 2009;83:7366–7374. doi: 10.1128/JVI.00191-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruno R, Galastri S, Sacchi P, Cima S, Caligiuri A, et al. gp120 modulates the biology of human hepatic stellate cells: a link between HIV infection and liver fibrogenesis. Gut. 2010;59:513–520. doi: 10.1136/gut.2008.163287. [DOI] [PubMed] [Google Scholar]
- 21.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitani H, Takenouchi T, Sato M, Yoshioka M, Yamanaka N. A simple and efficient method to isolate macrophages from mixed primary cultures of adult liver cells. J Vis Exp. 2011 doi: 10.3791/2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedroza-Martins L, Gurney KB, Torbett BE, Uittenbogaart CH. Differential tropism and replication kinetics of human immunodeficiency virus type 1 isolates in thymocytes: coreceptor expression allows viral entry, but productive infection of distinct subsets is determined at the postentry level. J Virol. 1998;72:9441–9452. doi: 10.1128/jvi.72.12.9441-9452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janorkar AV, King KR, Megeed Z, Yarmush ML. Development of an in vitro cell culture model of hepatic steatosis using hepatocyte-derived reporter cells. Biotechnol Bioeng. 2009;102:1466–1474. doi: 10.1002/bit.22191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wieder KJ, King KR, Thompson DM, Zia C, Yarmush ML, et al. Optimization of reporter cells for expression profiling in a microfluidic device. Biomed Microdevices. 2005;7:213–222. doi: 10.1007/s10544-005-3028-3. [DOI] [PubMed] [Google Scholar]
- 28.Salloum S, Wang H, Ferguson C, Parton RG, Tai AW. Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog. 2013;9:e1003513. doi: 10.1371/journal.ppat.1003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao PS, Chen TS, Chung PC. A fast algorithm for multilevel thresholding. Journal of Information Science and Engineering. 2001;17:713–727. [Google Scholar]
- 30.Narmada BC, Kang Y, Venkatraman L, Peng Q, Sakban RB, et al. Hepatic stellate cell-targeted delivery of hepatocyte growth factor transgene via bile duct infusion enhances its expression at fibrotic foci to regress dimethylnitrosamine-induced liver fibrosis. Hum Gene Ther. 2013;24:508–519. doi: 10.1089/hum.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luedde T, Schwabe RF. NF-kappaB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]