

Abstract
Background
Mutations in leucine-rich repeat kinase 2 (LRRK2) contribute to both familial and idiopathic forms of Parkinson’s disease (PD). Neuroinflammation is a key event in neurodegeneration and aging, and there is mounting evidence of LRRK2 involvement in inflammatory pathways. In a previous study, we described an alteration of the inflammatory response in dermal fibroblasts from PD patients expressing the G2019S and R1441G mutations in LRRK2.
Methods
Taking advantage of cellular reprogramming, we generated induced pluripotent stem cell (iPSC) lines and neurons thereafter, harboring LRRK2G2019S and LRRK2R1441G mutations. We used gene silencing and functional reporter assays to characterize the effect of the mutations. We examined the temporal profile of TNFα-induced changes in proteins of the NF-κB pathway and optimized western blot analysis to capture α-synuclein dynamics. The effects of the mutations and interventions were analyzed by two-way ANOVA tests with respect to corresponding controls.
Results
LRRK2 silencing decreased α-synuclein protein levels in mutated neurons and modified NF-κB transcriptional targets, such as PTGS2 (COX-2) and TNFAIP3 (A20). We next tested whether NF-κB and α-synuclein pathways converged and found that TNFα modulated α-synuclein levels, although we could not detect an effect of LRRK2 mutations, partly because of the individual variability. Nevertheless, we confirmed NF-κB dysregulation in mutated neurons, as shown by a protracted recovery of IκBα and a clear impairment in p65 nuclear translocation in the LRRK2 mutants.
Conclusions
Altogether, our results show that LRRK2 mutations affect α-synuclein regulation and impair NF-κB canonical signaling in iPSC-derived neurons. TNFα modulated α-synuclein proteostasis but was not modified by the LRRK2 mutations in this paradigm. These results strengthen the link between LRRK2 and the innate immunity system underscoring the involvement of inflammatory pathways in the neurodegenerative process in PD.
Electronic supplementary material
The online version of this article (doi:10.1186/s12974-016-0761-x) contains supplementary material, which is available to authorized users.
Keywords: Parkinson’s disease, LRRK2, Inflammation, iPSCs, NF-κB, α-Synuclein
Background
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by a progressive and relatively selective death of dopamine (DA) neurons within the substantia nigra of the midbrain [1]. The neuropathological hallmark of PD is the Lewy body fibrillar aggregates in which α-synuclein is the major constituent [2]. The great majority of PD cases are sporadic, with only 5–10% being familial. Mutations in the leucine-rich repeat kinase 2 (LRRK2, PARK8) gene are the most common cause of monogenic PD [3]. Furthermore, both common and uncommon variants are associated with an increase odd risk in GWAS analyses [4]. The precise physiological function of LRRK2 has yet to be defined due to its involvement in multiple pathways, but we and others have proposed an active role in the immune response (reviewed in [5]). Indeed, in a previous study, we demonstrated a defective NF-κB activation in response to a pro-inflammatory stimulus in dermal fibroblasts from PD patients [6]. NF-κB activation is responsible for the intracellular regulation of age-related inflammation which appears to play a major role in neurodegeneration.
LRRK2 is a large multi-domain protein with two enzymatic activities, a serine/threonine kinase and a ROC (Ras of complex)-GTPase [3]. The G2019S substitution in the kinase activation loop is by far the most common pathogenic mutation and increases the kinase activity [7, 8]. The R1441G/C/H/S substitutions in the ROC-GTPase domain generally result in lower GTPase activity, with more inconsistent effects on kinase activity [3]. Despite these differences, most pathogenic mutants display an increase in the (auto)phosphorylation at Ser1292 [9] and also in the phosphorylation of at least one other substrate, the Rab GTPases [10]. Unraveling a common mechanism for all LRRK2 mutations is critical for understanding LRRK2 role in PD pathogenesis.
Increasing experimental evidence underscores the involvement of LRRK2 in the inflammatory response, supported also by the robust LRRK2 expression in immune cells, including peripheral monocytes and macrophages, and in primary microglia (reviewed in [5]). The link to the innate immune response is further reinforced by the genetic association of LRRK2 with susceptibility to inflammatory bowel disorder [11] and leprosy [12, 13]. Moreover, the expression of LRRK2 is modulated by immune cell-specific signals, like IFNγ and toll-like receptor (TLR) agonists [14–16].
In order to examine inflammatory responses in a disease-relevant context, we extended our previous work on patients’ fibroblasts harboring LRRK2G2019S and LRRK2R1441G mutations by reprogramming the cells and using neurons derived from the induced pluripotent stem cells (iPSCs). In this cellular model, which preserves the endogenous (and regulated) expression of LRRK2, we examined the effect of the mutations on α-synuclein and TNFα-induced NF-κB activation, with the hypothesis that inflammatory stimuli can modulate α-synuclein proteostasis.
Methods
Reprogramming and generation of iPSC lines
The experimental protocol was approved by the Ethical Committee at Hospital Donostia (San Sebastian, Spain), and all procedures adhered to the internal and EU guidelines for research involving derivation of pluripotent cell lines. All subjects gave informed consent for the study using forms approved by the Ethical Committee on the Use of Human Subjects in Research at Hospital Donostia and Onkologikoa Hospital, both in San Sebastian, Spain. Generation of iPSC lines was approved by the Advisory Committee for Human Tissue and Cell Donation and Use, Instituto Carlos III (ISCIII), Madrid, Spain. All procedures were done in accordance with institutional guidelines, and the cell lines have been deposited at the Banco Nacional de Lineas Celulares (BNLC, ISCIII) following the Spanish legislation.
Skin fibroblast cultures from PD patients with mutations in LRRK2 and matched healthy subjects have been previously characterized in our laboratory [6]. We included samples from four men and two women, with a median age of 62.5 ± 13.9 years. Dermal fibroblasts were cultivated as described previously [6].
For this study, we reprogrammed fibroblasts from two LRRK2G2019S and two LRRK2R1441G different patients. We used lentiviral vectors containing c-Myc, Oct-4, Sox-2, and Klf-4 as previously reported [17], and iPS cell lines were characterized following standard procedures defined by the BNLC in accordance with international guidelines. Results for the two LRRK2R1441G (that have not been previously reported in the literature) are shown in Fig. 1. Controls included a human embryonic stem cell line (H9) to control for a possible effect of the lentiviral reprogramming procedure and iPSC lines from healthy individuals, previously generated in our laboratory [17].
Fig. 1.
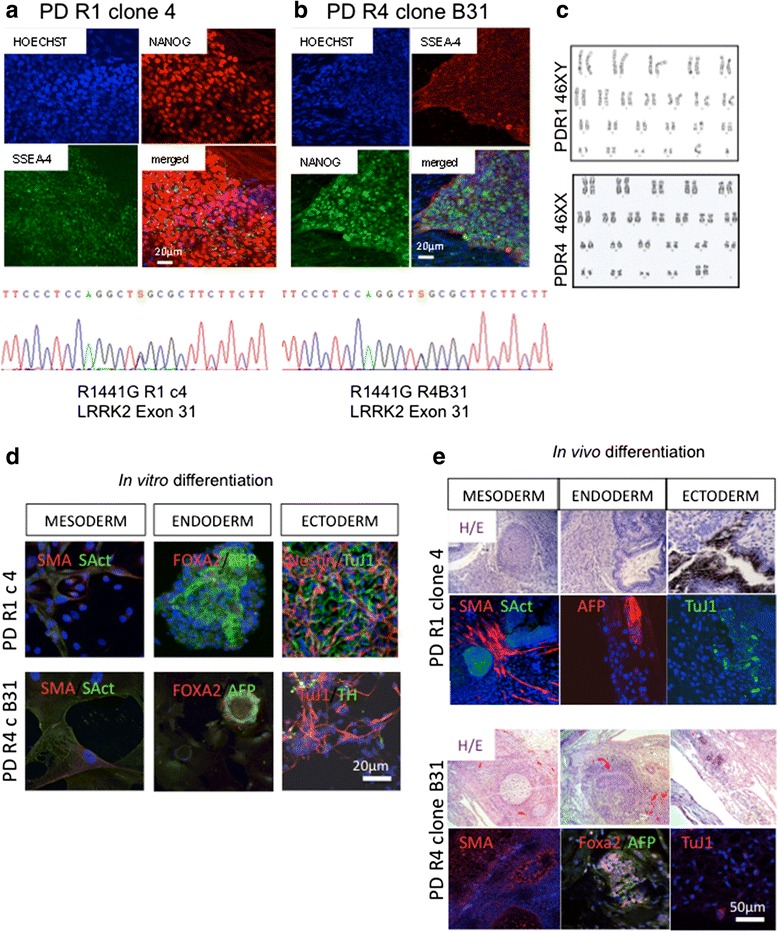
Characterization of pluripotency of the LRRK2R1441G mutant iPSC lines (PD-R1 and PD-R4). a, b Reprogrammed cells formed compact uniform colonies that showed robust and uniform expression of typical pluripotent markers such as NANOG and embryonic stage-specific antigen 4 (SSEA-4) by immunofluorescence. Sequence analysis of exon 31 confirmed the presence of the point mutation in the clones selected for this study. c G-band karyotypes for the selected clones. Pluripotency was confirmed d in vitro by embryoid body formation and trilineage differentiation, analyzed by immunofluorescence and e in vivo by formation of teratomas in NOD-SCID mice that showed cells corresponding to the three germ layers. SMA smooth muscle actin, SAct sarcomeric actin, AFP alpha fetoprotein, TUJ1 βIII-tubulin, TH tyrosine hydroxylase, H/E hematoxylin/eosin. Scale bars are indicated in each panel
Maintenance of iPSCs and differentiation to DA neurons
iPSCs were cultured and differentiated as described in [17] with minor modifications. iPSCs were maintained on irradiated human foreskin fibroblasts (HFF-1, SCRC-1041, ATCC) in hES cell medium made of knockout-DMEM (10829-018, Invitrogen), supplemented with 2 mM Glutamax (#3505003, Invitrogen), 50 nM β-mercaptoethanol (31350-010, Invitrogen), 1× non-essential amino acids (M7145, Sigma), 20% knockout serum (KSR10828028, Invitrogen), 50 U/ml penicillin, 50 μg/ml streptomycin, and 10 ng/ml FGF2 (100-18B, PeproTech). iPS cell colonies were passaged manually once a week.
The protocol used for DA induction and differentiation is shown schematically in Fig. 2a. Undifferentiated cells were plated on Matrigel in hES cell medium (D0), and 1 day later (D1), half of the hES cell medium was replaced with DMEM-F12 supplemented with 1× N2 supplement (#17502048, Invitrogen) and 10 ng/ml FGF2. Neural induction was started at D2 by adding 10 μM SB431542 (#1614, Tocris) and 100 nM LDN-193189 (130-096-226, Miltenyi Biotech). For the induction of DA phenotype, 500 nM SAG (smoothen agonist, #566660, Millipore), and 0.5 nM CHIR 99021 (#13122, Cayman Chemical) were added to the medium. On D12, neural rosettes were mechanically passaged onto 0.1% gelatine (G1393; Sigma), 15 μg/ml polyornithine, 1 μg/ml fibronectin, and 1 μg/ml laminin-coated plates and expanded at high cell densities. At this time, LDN and CHIR 99021 were removed from the medium; SAG was reduced to 20 nM, and 100 ng/ml FGF8 was added. At D14, the medium was also supplemented with 20 ng/ml BDNF and 200 μM ascorbic acid (BASF medium). Subsequent passages of neural progenitors were done using Accutase® (Sigma-Aldrich®). On ∼D20, BASF medium was replaced with BCT-GA medium, composed of neurobasal medium with 1× N2 supplement, 1× B27 supplement (17504-044, Invitrogen), 2 mM Glutamax, 20 ng/ml BDNF, 10 ng/ml GDNF, 200 μM ascorbic acid, 0.5 mM dibutyryl-cAMP, 1 ng/ml TGF-β III, and 1 μg/ml laminin. As shown in Fig. 2a, we defined three stages: induction stage, weeks 1–3 (D0–D20); expansion stage, weeks 4–5 (D28-D35); and maturation stage, week 6–onwards (>D40). All the experiments were carried out in the maturation stage.
Fig. 2.
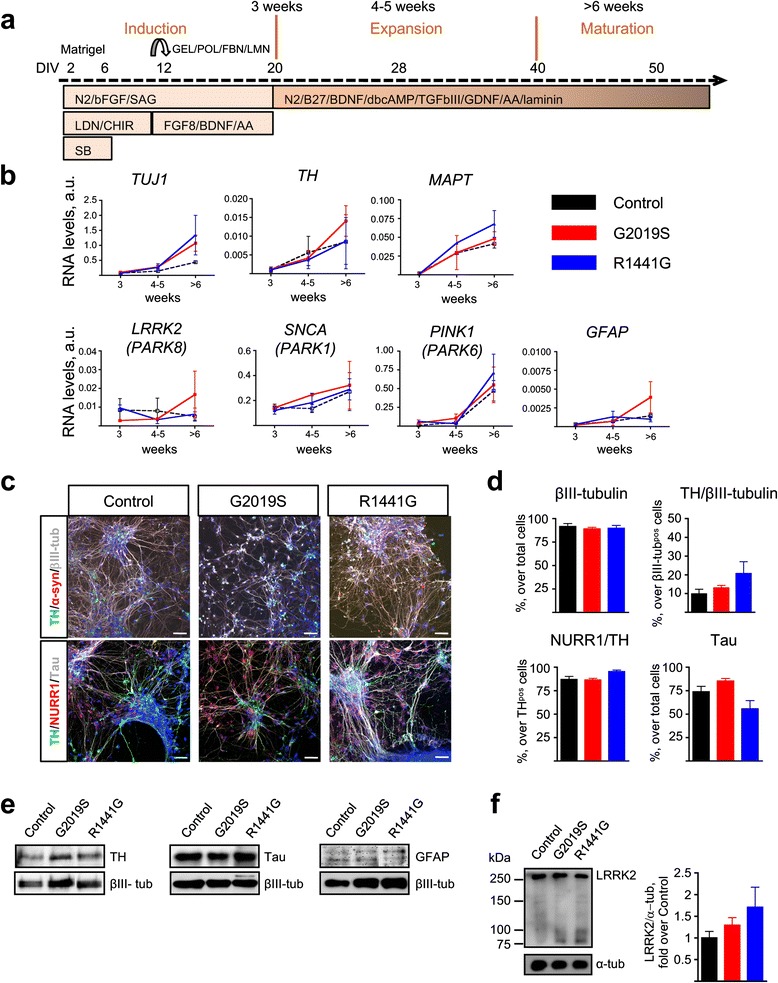
Characterization of iPSC-derived DA neurons with LRRK2 mutations. a Diagram showing the DA differentiation protocol used for neural induction of human iPSC lines. b Temporal gene expression analyzed by qRT-PCR at three time points: induction (3 weeks), expansion (4–5 weeks), and maturation (>6 weeks). Each point represents the mean ± SEM of at least two independent differentiation experiments. c Representative images of mature neuronal cultures showing expression of neuronal (βIII-tubulin, Tau, and α-synuclein) and dopaminergic (TH, NURR1) markers. Nuclei were counterstained with Hoechst. Scale bars: 50 μm. d Quantification of immunostainings. Data are represented as mean ± SEM of counts from at least two different lines for each genotype. e Representative western blot analyses of TH, Tau, and GFAP with βIII-tubulin as loading control in iPSC-derived mature neurons. f Representative immunoblots and quantification of LRRK2 expression in mature neuronal cultures. α-tubulin was the loading control and data were normalized to control WT neurons. Bars represent the mean ± SEM of at least two different lines per genotype. DIV days in vitro, GEL gelatin, POL poly-ornithine, FBN fibronectin, LMN laminin, N2 N2 supplement, bFGF basic fibroblast growth factor, SAG smoothened agonist, LDN LDN-193189, CHIR CHIR99021, SB SB431542, BDNF brain-derived neurotrophic factor, AA ascorbic acid, B27 B27 supplement, dbcAMP dibutyryl cyclic adenosine monophosphate, TGFβIII transforming growth factor βIII, GDNF glial derived neurotrophic factor. See Additional file 2 for uncropped blots
To evaluate the NF-κB activity, cells were stimulated by the addition of TNFα (#210-TA-005, R&D Systems), with 10 and 15 ng/ml as indicated in the “Results” section.
LRRK2 gene knockdown
Endogenous LRRK2 expression was silenced as previously described [6] using the MISSION® shLRRK2 vector (TRCN0000358257, SIGMA) with a multiplicity of infection (MOI) of 8. As a control, cells were transduced with the empty vector (mock control). The viral supernatant was removed 18 h later and replaced with fresh growth medium. The following experiments were carried out 5 days after transduction.
Western blotting
For whole-cell lysate preparation, neurons were harvested using Accutase®, washed in PBS and lysed in RIPA lysis buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.25% sodium deoxycholate) with 1 mM sodium orthovanadate, 1 mM NaF, and a protease inhibitor cocktail (Roche). For probing α-synuclein, the cells were washed with PBS and lysed in a modified RIPA buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% SDS, 0.5% sodium deoxycholate, 5% glycerol). Lysates were briefly sonicated, clarified by centrifugation at 15,000 rpm for 15 min and finally resolved by SDS-PAGE. Immunoprecipitation was performed using Protein-G cross-linked with the anti-p62 antibody (P0067, Sigma). For the kinetic experiments, the cells were stimulated with TNFα, washed with PBS 1×, and lysed in SDS-PAGE loading buffer (2% SDS, 5% 2-mercaptoethanol, 10% glycerol, 0,1% bromophenol blue, 62.5 mM Tris-HCl, pH 6.8). After SDS-PAGE, the proteins were transferred to polyvinylidene difluoride (PVDF) membrane (Immobilon-P, Merck-Millipore). The following antibodies were used at a 1:1000 dilution: LRRK2 (NB110-58771, Novus Biologicals), p62 (P0067, Sigma), Sam68 (C-20, #sc-333, Santa Cruz Biotechnology), IκBα (L35A5, #4814, Cell Signalling), α-synuclein (MA1-12874, Thermo Scientific), Tau (T46, #136400, Invitrogen), βIII-tubulin (mms-435p, Covance), and TH (P60101-0, Pel Freez Biologicals); α-tubulin (DM1A, #3873, Cell Signalling Technology®) was used at 1:5000 dilution. A horseradish peroxidase-conjugated IgG (GE Healthcare) was employed as a secondary antibody. Visualization of HRP-labeled proteins was performed using enzyme-linked chemiluminescence (ThermoFisher Scientific) either detected on X-ray films or directly with a digital CCD camera (Versadoc Imager, Bio-Rad). Bands were quantified by densitometry relative to the corresponding loading control using ImageJ (NIH).
Immunofluorescence
Cells cultured on sterile glass cover slips were fixed with 4% paraformaldehyde for 10 min at room temperature. The cells were then permeabilized and blocked with 10% donkey serum in 0.1% Triton X-100/PBS for 45 min. Primary antibody incubation was performed overnight at 4 °C, using the following antibodies diluted in PBS as indicated: Nanog (1:100, R&D), SSEA-4 (1:100, Hybridoma Bank, U.IOWA), SMA (1:400, Sigma), sarcomeric actin (1:400, Sigma), AFP (1:400, DAKO), FOXA2 (1:100, Santa Cruz Biotechnology), Nestin (1:500, Neuromics), p65 (1:100, sc-372, Santa Cruz), TH (1:1000, P60101-0, Pel Freez Biologicals), α-synuclein (1:100, MA1-12874, Thermo Scientific), Tau (1:200, T46, #136400, Invitrogen), βIII-tubulin (1:1000, prb-435p-100, Covance), NURR1 (1:200, E-20, sc-990, Santa Cruz), and GFAP (1:500, Z0334, Dako). Next, the cells were washed with 10% donkey serum/PBS and incubated for 1 h with Alexa fluorochrome-conjugated secondary antibodies diluted in PBS. After final washes with 0.1× PBS, the cover slips were mounted using ProLong® antifade reagent (Molecular Probes®, Life Technologies). Images were acquired with a Zeiss LSM510 confocal microscope and analyzed using ImageJ (1.49, NIH). For quantification of neuronal markers, tile images (1272 × 1272 μm) were acquired at a ×40 magnification and at least 1000 cells were counted for each cell line. For the evaluation of nuclear p65 translocation, images were randomly acquired at ×63 magnification, and between 150 and 300 cells were scored for each condition.
Real-time RT-PCR
Total RNA was extracted using Trizol® total RNA isolation reagent (Gibco®, Life Technologies), followed by the RNeasy Qiaprep (Qiagen) per manufacturer’s protocol. RNA concentration was quantified using a NanoDrop Spectrophotometer (NanoDrop Technologies). cDNA was synthesized from total RNA using random hexamers according to the GeneAmp® RNA PCR Core Kit (Life Technologies) and the High-Capacity cDNA RT kit (Applied Biosystems®, Life Technologies). Real-time PCR was performed using an Applied Biosystems StepOne™ Detection System. Comparative analysis of gene expression levels (ΔΔCt) was carried out using GAPDH as the reference gene. The sequences of the primers are indicated in Additional file 1.
Luciferase assays
We used the Dual-Luciferase® Reporter (DLR™) Assay System (Promega) to measure the activity of firefly and Renilla luciferases sequentially from a single sample. We used the pNF3ConA-Luc plasmid [6] and the pRL-CMV (#E226A, Promega) for normalization of gene expression. Both plasmids were co-transfected into the neurons by electroporation according to the standard protocols (Neon® Transfection System, Invitrogen™, Life Technologies). Briefly, one million cells were resuspended in 10 μl buffer R containing 1 μg pNF3ConA-Luc, and 100 ng pRL-CMV were subjected to two pulses (1000 V, 10 ms), and re-plated on 12-well plates. Forty-eight hours later, neurons were treated with TNFα for 8 h in neurobasal medium without trophic factor supplementation. Normalized data are expressed as the firefly (NF-κB) divided by the Renilla luciferase activity.
Data transformation and analysis
Data were analyzed using Prism 4.0 (GraphPad Software). All experiments were performed in at least two different cell lines (from different individuals) for each genotype and in at least two independent differentiations. Bar graphs represent average and SEM. Values were normalized as specified in the figure legends. Comparison between groups was carried out by one-way ANOVA with Dunn’s post-test (one variable) or two-way ANOVA with Bonferroni post-test (two variables). Values of P < 0.05 were considered significant.
Results
Reprogramming and derivation of LRRK2 iPSC-derived DA neuronal cultures
We generated iPSCs from four PD patients harboring mutations in LRRK2. Two patients carried the G2019S in the kinase domain of the protein and another two patients carried the R1441G mutation in the ROC-GTPase domain (Fig. 1). Several reports have used LRRK2G2019S and LRRK2R1441C iPSC lines, but this is the first study describing iPS cell lines carrying the LRRK2R1441G mutation. There was no effect of the mutations on the reprogramming process, and we obtained pluripotent cell lines that maintained the original mutation, had normal karyotypes, and had potential to generate the three germ layers both in vitro and in vivo (Fig. 1a–e). Cell lines generated for the study have been deposited in the Spanish repository (BNLC) and are available at http://www.isciii.es/ISCIII/es.
We next differentiated iPSCs towards a dopamine phenotype, using a double-SMAD inhibition protocol, illustrated in Fig. 2a. Neuronal markers were first detected by qPCR during the expansion stage and increased gradually with time (Fig. 2b). We analyzed the expression profile of several PARK genes (LRRK2, SNCA, PINK1). While SNCA and PINK1 levels increased steadily, LRRK2 RNA levels were more variable along the differentiation process but were not different between groups. RNA levels of the glial marker GFAP were low at all times examined (Fig. 2b).
Immunofluorescence analysis of neurons during the maturation stage demonstrated that 90.4 ± 4.4% of cells were βIII-tubulin-positive and 13.3 ± 6.4% of βIII-tubulin-positive cells were TH-positive. Of these TH-positive cells, 88.3 ± 5.5% co-expressed NURR1 (Fig. 2c, d). These in vitro generated neurons showed a normal developmental expression of specific proteins like the microtubule-associated protein Tau, which was present in 73.5 ± 13.6% of cells in the cultures (Fig. 2c, d). Immunoblot analysis of protein extracts confirmed no differences in either TH or Tau content across genotypes. Consistent with the transcriptional profile, the glial marker GFAP was barely detectable (Fig. 2e, Additional file 2). Importantly, LRRK2 protein expression showed no differences between genotypes at 6 weeks (Fig. 2f).
Taken all analyses together, we concluded that there were no differences in the efficiency of neuronal specification and markers’ expression in these cultures; the low numbers of DA neurons that we obtained in these experiments were not related to the presence of LRRK2 mutations.
LRRK2 regulates α-synuclein proteostasis
We analyzed the expression of monomeric soluble α-synuclein in our differentiated neurons, since α-synuclein levels have been reported to be higher in LRRK2G2019S neurons [18, 19].We confirmed that in LRRK2G2019S cultures, α-synuclein levels were twofold higher than that in the control wild-type (WT) cultures at the late stage of differentiation (two-way ANOVA, P < 0.01, LRRK2G2019S v. WT LRRK2, late stage) (Fig. 3a, Additional file 3). Interestingly, from the point of view of age-related neurodegeneration, this phenotype was only observed in mature neurons (two-way ANOVA, effect of time, P < 0.05). Also notably, this appears to be quite specific for LRRK2G2019S neurons, since α-synuclein levels were not increased in the LRKK2R1441G neuronal cultures (Fig. 3a). The autophagic mediator p62, implicated in the removal of α-synuclein, has been reported to be increased in LRRK2G2019S neurons [19]. However, we did not find any differences in p62 basal levels in neurons with either LRRK2 mutation at any stage (Fig. 3b).
Fig. 3.
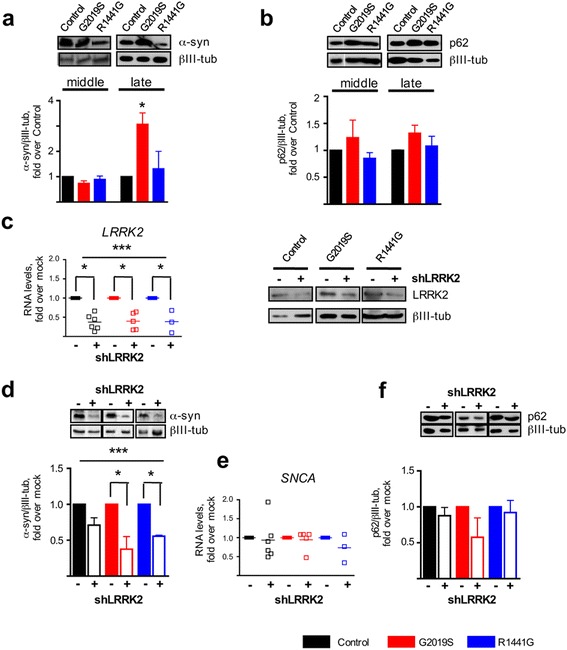
LRRK2 regulation on α-synuclein levels. a Western blot analysis of basal α-synuclein and b p62 levels in neurons during mid and late differentiations. Blots show the α-synuclein soluble monomer of ~15 kDa. Bars represent the mean ± SEM of three to six values, including two different cell lines per group. βIII-tubulin was used as loading control (two-way ANOVA, ** P < 0.01, LRRK2G2019S v. control WT, late stage). c LRRK2 RNA levels 5 days after shRNA lentiviral transduction. The empty vector was used as the mock control. Lines represent the mean ± SEM of three to four independent silencing experiments. A representative immunoblot analysis of LRRK2 protein levels 5 days after transduction is also shown. βIII-tubulin was the loading control. d Immunoblots and corresponding quantification of α-synuclein 5 days after shLRRK2 transduction. Bars represent the mean ± SEM of at least three independent silencing experiments. LRRK2 silencing had a significant effect on α-synuclein protein levels (two-way ANOVA, *** P = 0.0002). Bonferroni post hoc test showed that this effect was limited to LRRK2 mutated mature neurons (**P < 0.05 for both LRRK2G2019S and LRRK2R1441G). e qRT-PCR analysis of SNCA after LRRK2 silencing. f Immunoblots and corresponding quantification of p62 5 days after shLRRK2 transduction. Bars represent the mean ± SEM of at least three independent silencing experiments. See Additional file 3 for uncropped blots
To better define the role of LRRK2 in the regulation of these proteins in neurons, we analyzed α-synuclein after silencing endogenous LRRK2 expression. Analysis 5 days after transduction showed an average decrease in LRRK2 RNA of 60 ± 10%, compared to mock-transduced cells, and a corresponding decrease in LRRK2 protein levels (Fig. 3c). The presence of mutations did not affect silencing efficiency. LRRK2 knockdown significantly reduced α-synuclein protein levels overall, and this effect was driven by the marked decrease observed in the mutant neurons (two-way ANOVA, P < 0.001; Bonferroni post-test, P < 0.05 for both LRRK2G2019S and LRRK2R1441G) (Fig. 3d). This result shows that mutations in LRRK2 affect α-synuclein regulation in neurons. Despite this large effect on protein levels, SNCA RNA did not change in LRRK2 silenced neurons, indicating that the regulation is not taking place at the transcriptional level (Fig. 3d). Therefore, we next examined p62 in LRRK2-silenced neurons, and we did not observe changes in its protein levels, concluding that LRRK2 modulation of α-synuclein is independent of p62, at least in these conditions (Fig. 3e).
Transcriptional effects of LRRK2
We have previously reported a strong effect of LRRK2 silencing on NF-κB target genes in fibroblasts [6]. Therefore, we investigated the role of LRRK2 in the regulation of this pathway in mature neurons. We found that LRRK2 knockdown significantly affected PTGS2 (COX-2), TNFAIP3 (A20), and TNFRSF1A (TNFR1) expression, but not all to the same extent (Fig. 4a, c, e). While shLRRK2 induced a decrease in TNFRSF1A in all genotypes, TNFAIP3 RNA levels were reduced only in LRRK2 mutated neurons. We did not observe significant changes in IL-6, NFKBIA (IκBa), and TNFRSF1B (TNFR2) expression in silenced neurons of any genotype. This indicates that LRRK2 knockdown does not affect all NF-κB target genes equally because these are additionally regulated in each specific cell context. Importantly, LRRK2 silencing did not modify the constitutively expressed genes PTGS1 (COX-1) and βIII-tubulin (TUJ1) (Fig. 4g, h). This confirms the lack of toxicity of LRRK2 silencing in mature human neurons and the specificity of the changes observed. Taken together, these data strengthen the link between LRRK2 and the NF-κB pathway in neurons.
Fig. 4.
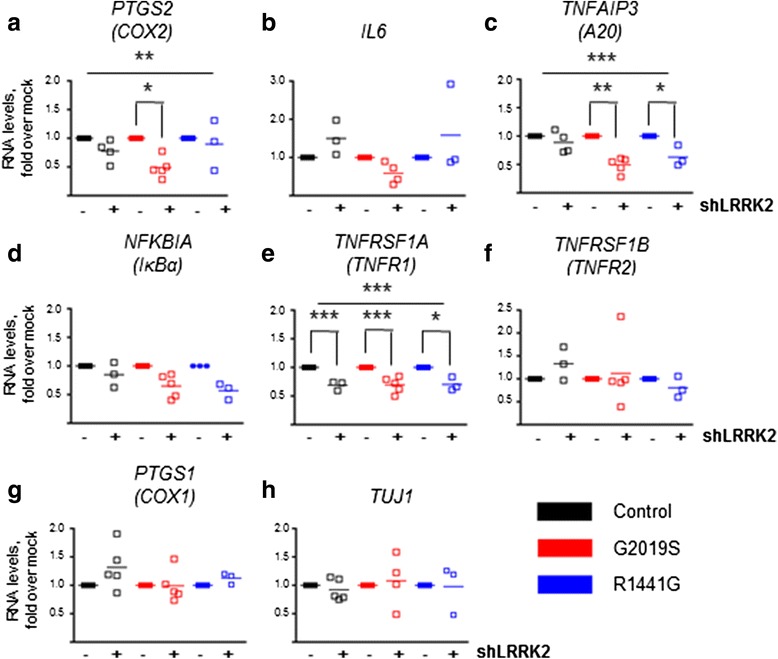
Effect of endogenous LRRK2 silencing on NF-κB target genes. a-h qRT-PCR analyses of inflammatory genes 5 days after LRRK2 silencing. The effect of LRRK2 silencing was evident in the regulation of RNA levels of COX-2, A20, and TNFR1. For COX-2, the effect of LRRK2 silencing was significant (two-way ANOVA, ** P = 0.0021,), and post hoc analyses revealed a significant effect of shLRRK2 on RNA levels specifically in LRRK2G2019S neurons (** P < 0.01). For A20, in addition to the effect of knocking down the expression of LRRK2 (two-way ANOVA, P < 0.0001), the presence of the mutations had also a significant effect (two-way ANOVA, ** P = 0.008). Subsequently, there was an interaction between shLRRK2 and genotype (two-way ANOVA, ** P = 0.008). Indeed, post hoc analyses showed that there was an effect of LRRK2 silencing on A20 levels in LRRK2G2010 (*** P < 0.001) and LRRK2R1441G neurons (** P < 0.01). Finally, for TNFR1, the effect of LRRK2 silencing was significant in all groups (two-way ANOVA *** P < 0.0001; post hoc tests: *** P < 0.001 in Control WT and LRRK2G2019S and ** P < 0.01 in LRRK2R1441G). Mock cells were transduced with the empty vector. Lines represent the mean ± SEM of three to five independent silencing experiments
TNFα effect on α-synuclein protein levels
Our results so far confirmed a regulatory role of LRRK2 on α-synuclein proteostasis, as well as on NF-κB transcriptional activity. Interestingly, there are several NF-κB binding sites in the SNCA promoter, so we next investigated the effect of NF-κB activation with TNFα on α-synuclein levels in this paradigm. We treated mature neurons with TNFα (15 ng/ml) for the times indicated in the figure and observed a modulation of α-synuclein protein levels (Fig. 5a, Additional file 4, RT-ANOVA, effect of time, P < 0.05) with no significant effect of the genotype. A parallel (but not significant) trend was observed for p62 levels (Fig. 5b). The small, transient increases after TNFα challenge in all neurons support our hypothesis that inflammatory stimuli activating the NFĸB pathway can modulate α-synuclein protein levels. We next analyzed the interaction of α-synuclein with the autophagic protein p62 in WT neurons but could not detect any association between the two proteins in differentiated neurons (Fig. 5c). Thus, it is unlikely that modifications in α-synuclein in response to TNFα involve p62 in these conditions.
Fig. 5.
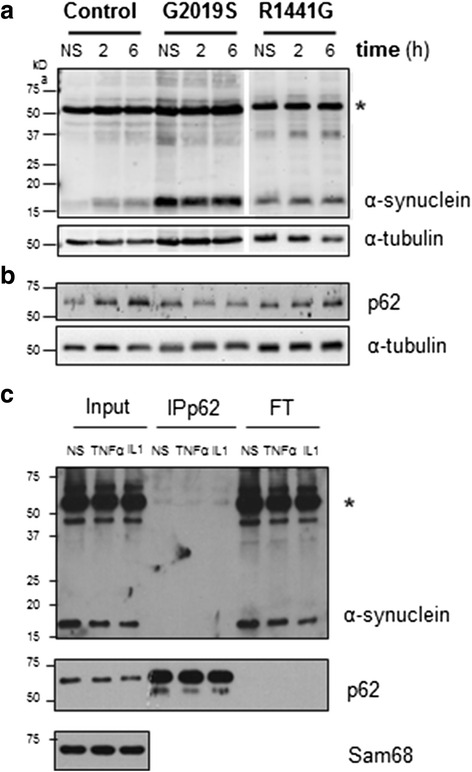
Effect of TNFα on α-synuclein. a Western blot analysis showing α-synuclein and b p62 proteins in mature neurons after treatment with TNFα, at 2 and 6 h. Proteins were resolved in 15% SDS polyacrylamide gels for the visualization of different α-synuclein oligomers. The band at 15 kDa corresponds to the monomer. A band at 50 kDa (asterisk) could correspond to an oligomeric form of α-synuclein. α-tubulin was used as loading control. Statistical analysis showed a significant effect of time on α-synuclein monomer levels in three to six independent experiments, including two different cell lines per genotype (RT-ANOVA, effect of time, P = 0.035). c Control WT neurons were stimulated with TNFα and with IL-1β to evaluate the interaction between p62 and α-synuclein. Co-immunoprecipitation of p62 and α-synuclein using specific antibodies did not show any association under these conditions. Sam68 was used as the loading control. NS non-stimulated, IP immunoprecipitation, FT flow-through. See Additional file 4 for uncropped blots
Mutations in LRRK2 alter NF-κB activation
To examine the impact of LRRK2 mutations on NF-κB function, we treated mature neurons with TNFα (10 ng/ml, 8 h). In all the genotypes, basal NF-κB activity was similar and TNFα induced a significant NF-κB response (two-way ANOVA, P < 0.01). However, while the control WT cells showed a relatively large response (tenfold, P < 0.05, Bonferroni post hoc test), LRRK2G2019S and LRRK2R1441G neurons displayed a more variable activation of smaller amplitude (≤4-fold, n.s., Bonferroni post hoc test) (Fig. 6a).
Fig. 6.
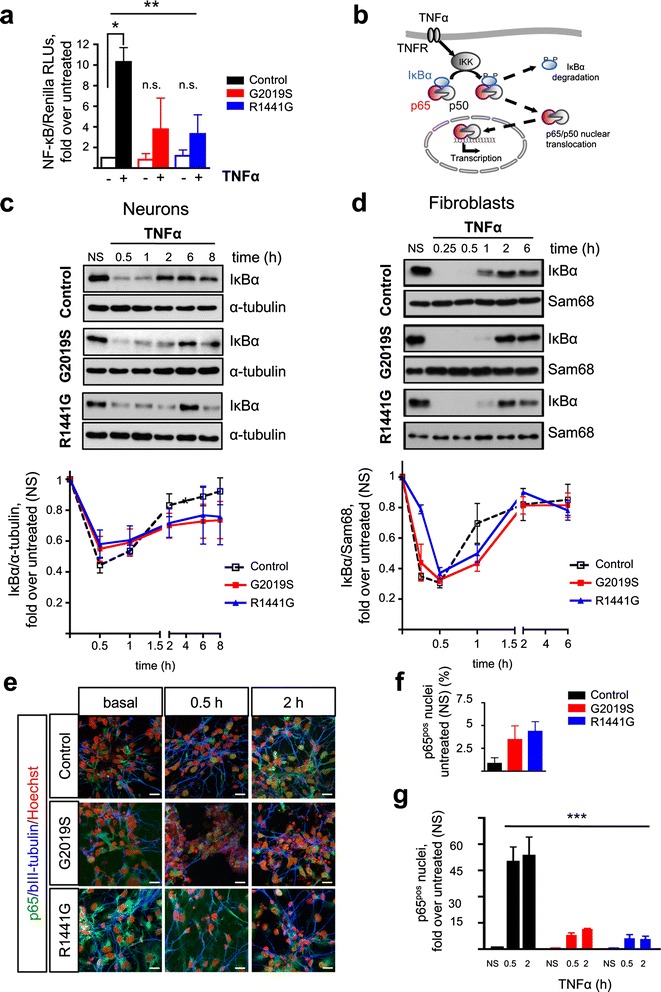
TNFα-induced NF-κB activation in neurons derived from iPSCs with LRRK2 mutations. a TNFα (10 ng/ml, 8 h) induced a significant NF-κB activation (two-way ANOVA, ** P < 0.01) but only in control WT neurons (* P < 0.05, post hoc test) and not in LRRK2G2019S or LRRK2R1441G neurons (n.s.). Bars represent the mean ± SEM of two to four determinations, including two different lines per genotype and expressed as fold values over untreated. In addition, basal NF-κB activities are normalized to the activity in the control WT neurons. b Schematic representation of NF-κB pathway activation. c Time-course of IκBα protein levels after treatment with TNFα (10 ng/ml). Immunoblots were quantified and normalized to non-stimulated (NS) samples. Each point in the curve is the mean ± SEM of four to five independent experiments, including two different cell lines per genotype. α-tubulin was used as the loading control. Statistical analysis showed a significant effect of time on IκBα protein levels (two-way RT-ANOVA, *** P < 0.0001). d For comparison, the same experiment is shown in fibroblasts. Points represent the mean ± SEM of three to six independent experiments, including at least two different cell lines per genotype. Sam68 was used as the loading control. Statistical analysis showed a significant effect of time on IκBα protein levels (two-way ANOVA, *** P < 0.0001). e Representative immunofluorescence staining of p65 (green) and βIII-tubulin (blue) in mature neuronal cultures incubated with TNFα (15 ng/ml, 0.5 and 2 h). Nuclei were counterstained with Hoechst 33342 (red). Scale bar, 20 μm. f Quantification of p65 immunoreactivity at baseline and g after TNFα incubation showing a significant effect of genotype (two-way ANOVA, *** P < 0.001). Bars represent the mean ± SEM of counts from two different lines per group in two to three independent experiments. NS non-stimulated. See Additional file 5 for uncropped blots
Degradation of IκBα, which retains the NF-κB effector dimer p65/p50 in the cytoplasm, is an essential event for the activation of canonical NF-κB pathway following a TNFα challenge (Fig. 6b). Time-course analysis of IκBα levels showed a rapid degradation in response to TNFα in all genotypes corroborating activation of the pathway (two-way RT-ANOVA, effect of time, P < 0.0001). Quantification of the area under the curve (AUC) revealed a delay in the recovery of IκBα in the neurons with LRRK2 mutations (Fig. 6c, Additional file 5). During the degradation phase (0–0.5 h), the AUCs were 0.3620, 0.3874, and 0.3950 for the control WT, LRRK2G2019S, and LRRK2R1441G neurons (C < G < R), respectively. In contrast, during the recovery phase (1–8 h), the AUCs were 5.936, 4.965, and 5.155 for the control WT, LRRK2G2019S, and LRRK2R1441G neurons (C > R > G), respectively. Indeed, 2 h after the TNFα stimulation, IκBα levels were back to 80% of baseline in the control WT neurons while in LRRK2 mutant neurons, the levels reached only ~65% at this time. Moreover, neurons with LRRK2 mutations failed to get back to the initial values at the latest time examined (8 h). Interestingly, fibroblast cultures from these patients displayed a delayed IκBα recovery rate as well. However, in fibroblasts, IκBα levels were back to baseline in all genotypes by 2 h (Fig. 6d), suggesting that the NF-κB transcriptional defect associated with LRRK2 mutations is more pronounced in the neurons.
We next evaluated p65 nuclear translocation in mature neurons by IF at 0.5 and 2 h after TNFα stimulation (15 ng/ml). These time points were chosen to coincide with the IκBα degradation and recovery phases observed by western blot. Notably, already at baseline, LRRK2G2019S and LRRK2R1441G cultures had a higher percentage of cells displaying a clear nuclear p65 signal (Fig. 6e, f) (0.8 ± 0.4, 3.6 ± 1.6, and 4.7 ± 1.8 in the control WT, LRRK2G2019S, and LRRK2R1441G, respectively). TNFα induced an increase in p65 nuclear localization in the control WT cultures (50- and 53-fold over non-stimulated baseline at 0.5 and 2 h, respectively). This response to TNFα was slower and significantly attenuated in all LRRK2 mutated neurons (8 and 11.5 at 0.5 and 2 h in LRRK2G2019S; 6.2 and 5.8 at 0.5 and 2 h in LRRK2R1441G, two-way ANOVA, F = 24.68, P < 0.001) (Fig. 6e, g). These results show that neurons with LRRK2G2019S and LRRK2R1441G mutations have a defect in p65 translocation underlying the protracted NF-κB transcriptional response, similar to the defect that we described before in patients’ fibroblasts [6].
Discussion
Taking advantage of iPSC technology, we investigated NF-κB signaling in patient-specific neurons harboring the G2019S and R1441G mutations in LRRK2. Some LRRK2G2019S, LRRK2R1441C, and LRRK2I2020T iPSC lines have been previously established and characterized [19–27]. For this study, we derived two novel iPSC lines with the R1441G substitution, which is frequent in the Basque Country, in addition to two G2019S-iPSC lines. Reprogramming and differentiation into DA neurons was similar in all mutant lines. The efficiency of DA neuronal specification, rather limited in this study, was nonetheless comparable to that obtained from a hES cell line (H9) and to that reported in other studies [19–27]. With this small percentage of DA neurons, our results are better viewed as representative of a mature heterogeneous neuronal population. Importantly, all experiments were carried out at 6 weeks or later, when cultures mainly contained mature neurons (90% of all cells) with normal Tau expression levels and distribution. We could not detect LRRK2 during the neural induction stage (1–3 weeks), but protein expression increased at later stages following a normal developmental profile [28, 29]. LRRK2 protein levels were similar in all genotypes and, in agreement with previous reports, LRRK2 endogenous baseline expression was not increased in LRRK2G2019S neurons [19, 20]. This is in stark contrast with overexpression studies that have proposed an increased dimerization and protein stability related to the mutation effect on kinase activity [25, 30]. For LRRK2I2020T and LRRK2R1441C, the stability of the protein has been reported to be impaired [27, 31].
There is overwhelming genetic and pathological evidence for the involvement of α-synuclein in PD. Indeed, missense mutations and multiplications in SNCA [32–35] cause familial autosomal dominant forms of PD. α-synuclein is also the main component of the proteinaceous inclusions (Lewy bodies and neurites) considered the pathological hallmark of PD [2, 36]. Furthermore, α-synuclein propagation has been proposed to underlie disease progression in a prion-like spreading manner, although this is debatable [37]. In agreement with previous studies in iPSC-derived neurons, we found elevated levels of α-synuclein in LRRK2G2019S neurons at the mature stage [18, 19, 22]. In contrast, α-synuclein was not increased in the LRRK2R1441G neurons. This difference can be related to the unequal effect of the two mutations on LRRK2 kinase activity because only the G2019S mutation robustly increases it [8]. Indeed, a recent paper further supports the hypothesis of a direct link between the enhanced kinase activity in the LRRK2G2019S neurons and the increase in α-synuclein levels (and subsequent formation of inclusions), as both LRRK2 specific kinase inhibitors and α-synuclein knockdown prevented inclusion formation in mutants, in vitro and in vivo [38]. In our study, LRRK2 silencing decreased α-synuclein expression in human LRRK2 mutated neurons, underscoring the tight connection between these two PARK gene products.
Importantly, we found that TNFα modulates α-synuclein dynamics in iPSC-derived neurons. Other studies have shown that TNFα and TLR activation in neurons increase α-synuclein levels by inhibiting autophagy [39, 40]. In this study, TNFα transiently increased α-synuclein levels, which could eventually favor protein aggregation and pathogenicity [41, 42]. Unfortunately, because of the small magnitude of the changes, inter-individual differences and technical limitations, we cannot discuss here the effect of the LRRK2 mutations on different alpha-synuclein molecular forms, which could be relevant for disease pathogenesis and deserves further work.
Wild-type LRRK2 has been proposed to activate inflammatory signaling. Overexpression of LRRK2 in vitro up-regulated the canonical NF-κB pathway [14, 16, 43, 44], while the effect of PD-associated LRRK2 mutations is less clear [14, 43]. Similarly, there is no consensus on the role of LRRK2 kinase activity on the stimulation of the NF-κB cascade [14–16, 44–46]. Discrepancies may be partly due to the use of different cellular systems and stimulation conditions and, in the case of LRRK2 inhibitors, to off-target effects. On the other hand, knockdown experiments of endogenous LRRK2 expression in primary microglia and immortalized immune cell lines down-regulated inflammatory signaling, even in the absence of a pro-inflammatory stimulus [14, 15, 44, 45], which is also in agreement with our findings in LRRK2-silenced fibroblasts [6]. In neurons, we found a differential regulation of NF-κB transcriptional targets, which may be dependent on cell-specific factors. Importantly, we could identify a significant effect on COX-2, validating our previous findings in fibroblasts and underscoring the preservation of the phenotype regarding the NF-κB pathway in these and other experimental models [6, 47]. From a practical point of view, this is rather convenient as dermal fibroblasts are easily accessible and expandable and can be used to screen for disease modifiers regarding this pathway. Nevertheless, neurons allowed us to explore neuronal specific proteins (such as α-synuclein) and pathways.
The low endogenous expression of LRRK2 in iPSC-derived neurons and the heterogeneous nature of the cultures resulted in relatively small TNFα-induced NF-κB response. Still, it was sufficient to detect the reduction in NF-κB transcriptional activation in response to TNFα in both LRRK2G2019S and LRRK2R1441G neuronal cultures. Furthermore, the recovery of IκBα protein, which is a direct transcriptional product providing feedback pathway regulation, was also impaired in mutant neurons. Given that IκBα degradation in the mutants was normal, our data pointed to a defect downstream p65 release from IκBα. Indeed, p65 nuclear translocation was defective in LRRK2 mutants. In addition, LRRK2 mutated neurons displayed slightly increased levels of nuclear p65 in the absence of any stimulus, supporting a leaky regulation of the system. These defects in iPSC-derived neurons corroborate our previous observations in patients’ fibroblasts, albeit with minor differences.
Mechanistically, our results imply that both the G2019S and R1441G mutations impair canonical NF-κB signaling at the level of p65 nuclear translocation and/or downstream transcriptional activation. Indeed, in LRRK2 mutant neurons primed with TNFα, IκBα degradation was normal like in LRRK2-silenced microglia treated with LPS [44]. p65 nuclear translocation may require LRRK2 scaffold function, perhaps through interaction with 14-3-3 proteins that could be disrupted by LRRK2 mutations [48–50]. On the other hand, mutations could alter this pathway at other levels, like the DNA-binding capacity of the transcription factor. In line with this, recent reports described higher levels of the phosphorylated form of the NF-κB inhibitory subunit p50 in LRRK2-silenced microglia, which correlated with subsequent higher p50 binding to DNA and transcriptional repression [45]. It would be very informative to characterize this response in neurons from sporadic PD patients.
Conclusions
These results validate in neurons our previous findings in patients’ fibroblasts regarding NF-κB signaling modulation by LRRK2 and underscore the usefulness of the iPSC-neuron paradigm to study time-dependent, neuron-specific alterations in a context that retains endogenous expression of pathogenic proteins. iPSC-derived neurons carrying the G2019S and R1441G mutations in LRRK2 showed impaired canonical NF-κB signaling and altered NF-κB target gene transcription regulation upon LRRK2 knockdown. Temporal analysis following a TNFα challenge revealed a protracted recovery of IκBα protein, concomitant with defective p65 nuclear translocation in both LRRK2G2019S and LRRK2R1441G neurons. Although basal α-synuclein protein levels were increased in LRRK2G2019S mature neurons, and not in LRRK2R1441G neurons, LRRK2 silencing down-regulated α-synuclein protein expression in both. This led us to hypothesize that NF-κB and α-synuclein pathways driving PD progression might converge. Indeed, TNFα elevated α-synuclein levels, although we could not detect an effect of LRRK2 mutations. Further studies are needed to understand how long-term neuroinflammation impacts on α-synuclein dynamics and the contribution of LRRK2 mutations to this pathway.
Acknowledgements
We are grateful to all the subjects that participated in the study, to Dr. Angel García Martin for the control samples and technical advice, and to Dr. Cesar Trigueros for the reagents. Some antibodies were obtained from the Developmental Studies Hybridoma Bank (DSHB) developed under the auspices of the NICHD and maintained by the Department of Biology, University of Iowa, Iowa City, IA 52242.
Funding
This study is funded by grants from the Spanish Ministry of Economy and Competitiveness (MINECO), Fondo de Investigaciones Sanitarias PI15/00486, the European Commission FP7 Health -278871, and the Joint Program in Neurodegenerative Diseases AC 14/0041 (DAMNDPATHS) to RSP.
Availability of data and materials
The authors declare that they used standard and commercially available software, databases, and application/tool for the data analysis. In addition, the authors declare that they do not have a link to include for the data and that all data and methods of analysis are included in the manuscript (or additional files). The authors will be able to share the software, databases, and all the relevant raw data described in the manuscript for testing by reviewers. The IPS cell lines supporting the conclusions of this article are available in the Spanish National Bank: http://www.isciii.es/ISCIII/es/contenidos/fd-el-instituto/fd-organizacion/fd-estructura-directiva/fd-subdireccion-general-investigacion-terapia-celular-medicina-regenerativa/fd-centros-unidades/fd-banco-nacional-lineas-celulares/fd-lineas-celulares-disponibles/lineas-de-celulas-iPS.shtml
Authors’ contributions
RLdeM and VL contributed to the acquisition and analysis of the data and the drafting and revision of the manuscript. AZ, AS, NV, AG, and JAB contributed to the acquisition and analysis of the data. ALdeM contributed to the critical revision of the manuscript. MR contributed to the data interpretation and the critical revision of the manuscript. RSP contributed to the study design, the data acquisition, analysis and interpretation, and the drafting and critical revision of the manuscript. All contributed to the revision and approval of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study was approved by the Ethical Committee on the Use of Human Subjects in Research in Euskadi, Spain. All subjects gave informed consent for the study using forms approved by the Ethical Committees on the Use of Human Subjects in Research at Hospital Donostia and Onkologikoa, San Sebastián; Generation of iPSC lines was approved by the Advisory Committee for Human Tissue and Cell Donation and Use, Instituto Carlos III, Ministry of Health, Spain.
Abbreviations
- DA
Dopamine
- iPSC
Induced pluripotent stem cells
- IκBα
Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, alpha
- LRRK2
Leucine-rich repeat kinase 2
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- PD
Parkinson’s disease
- ROC
Ras of complex proteins
- SNCA
Synuclein alpha
- TNF
Tumor necrosis factor
- TNFR
Tumor necrosis factor receptor
- WT
Wild-type
Additional files
Primer sequences used in RT-qPCR analyses.
Uncropped blots related to Fig. 2.
Uncropped blots related to Fig. 3.
Uncropped blots related to Fig. 5.
Uncropped blots related to Fig. 6.
Contributor Information
Rakel López de Maturana, Email: rmaturana@inbiomed.org.
Valérie Lang, Email: vlang@inbiomed.org.
Amaia Zubiarrain, Email: azubiarrain@inbiomed.org.
Amaya Sousa, Email: amayasousa@gmail.com.
Nerea Vázquez, Email: nerea.vazquezeguzkiza@osakidetza.eus.
Ana Gorostidi, Email: ana.gorostidi@biodonostia.org.
Julio Águila, Email: julio.aguila.benitez@ki.se.
Adolfo López de Munain, Email: adolfojose.lopezdemunainarregui@osakidetza.net.
Manuel Rodríguez, Email: manuel.rodriguez@itav.fr.
Rosario Sánchez-Pernaute, Phone: +34 943 309 064, Email: rpernaute@inbiomed.org, Email: rossapernaute@gmail.com.
References
- 1.Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Gomez-Suaga P, Fdez E, Fernandez B, Martinez-Salvador M, Blanca Ramirez M, Madero-Perez J, Rivero-Rios P, Fuentes JM, Hilfiker S. Novel insights into the neurobiology underlying LRRK2-linked Parkinson’s disease. Neuropharmacology. 2014;85:45–56. doi: 10.1016/j.neuropharm.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 4.van der Brug MP, Singleton A, Gasser T, Lewis PA. Parkinson’s disease: from human genetics to clinical trials. Sci Transl Med. 2015;7:205ps220. doi: 10.1126/scitranslmed.aaa8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russo I, Bubacco L, Greggio E. LRRK2 and neuroinflammation: partners in crime in Parkinson’s disease? J Neuroinflammation. 2014;11:52. doi: 10.1186/1742-2094-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez de Maturana R, Aguila JC, Sousa A, Vazquez N, Del Rio P, Aiastui A, Gorostidi A, Lopez de Munain A, Sanchez-Pernaute R. Leucine-rich repeat kinase 2 modulates cyclooxygenase 2 and the inflammatory response in idiopathic and genetic Parkinson’s disease. Neurobiol Aging. 2014;35:1116–1124. doi: 10.1016/j.neurobiolaging.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 7.Greggio E, Cookson MR. Leucine-rich repeat kinase 2 mutations and Parkinson’s disease: three questions. ASN Neuro. 2009;1(1):e00002. doi: 10.1042/AN20090007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheng Z, Zhang S, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, Solanoy HO, Drummond J, Zhang X, Ding X, et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med. 2012;4:164ra161. doi: 10.1126/scitranslmed.3004485. [DOI] [PubMed] [Google Scholar]
- 10.Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016;5:e12813. doi: 10.7554/eLife.12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Umeno J, Asano K, Matsushita T, Matsumoto T, Kiyohara Y, Iida M, Nakamura Y, Kamatani N, Kubo M. Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17:2407–2415. doi: 10.1002/ibd.21651. [DOI] [PubMed] [Google Scholar]
- 12.Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, Cui Y, Yan XX, Yang HT, Yang RD, et al. Genomewide association study of leprosy. N Engl J Med. 2009;361:2609–2618. doi: 10.1056/NEJMoa0903753. [DOI] [PubMed] [Google Scholar]
- 13.Marcinek P, Jha AN, Shinde V, Sundaramoorthy A, Rajkumar R, Suryadevara NC, Neela SK, van Tong H, Balachander V, Valluri VL, et al. LRRK2 and RIPK2 variants in the NOD 2-mediated signaling pathway are associated with susceptibility to Mycobacterium leprae in Indian populations. PLoS One. 2013;8:e73103. doi: 10.1371/journal.pone.0073103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–5585. doi: 10.4049/jimmunol.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. 2012;32:1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol. 2011;12:1063–1070. doi: 10.1038/ni.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aguila JC, Blak A, van Arensbergen J, Sousa A, Vazquez N, Aduriz A, Gayosso M, Lopez Mato MP, de Lopez Maturana R, Hedlund E, et al. Selection based on FOXA2 expression is not sufficient to enrich for dopamine neurons from human pluripotent stem cells. Stem Cells Transl Med. 2014;3:1032–1042. doi: 10.5966/sctm.2014-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, Hoing S, Hargus G, Heck SA, Dhingra A, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 2013;12:354–367. doi: 10.1016/j.stem.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 19.Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med. 2012;4:380–395. doi: 10.1002/emmm.201200215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012;4:141ra190. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16:394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su YC, Qi X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet. 2013;22:4545–4561. doi: 10.1093/hmg/ddt301. [DOI] [PubMed] [Google Scholar]
- 24.Sanders LH, Laganiere J, Cooper O, Mak SK, Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov FD, et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis. 2014;62:381–386. doi: 10.1016/j.nbd.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skibinski G, Nakamura K, Cookson MR, Finkbeiner S. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J Neurosci. 2014;34:418–433. doi: 10.1523/JNEUROSCI.2712-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ho DH, Kim H, Kim J, Sim H, Ahn H, Seo H, Chung KC, Park BJ, Son I, Seol W. Leucine-rich repeat kinase 2 (LRRK2) phosphorylates p53 and induces p21(WAF1/CIP1) expression. Mol Brain. 2015;8:54. doi: 10.1186/s13041-015-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohta E, Nihira T, Uchino A, Imaizumi Y, Okada Y, Akamatsu W, Takahashi K, Hayakawa H, Nagai M, Ohyama M, et al. I2020T mutant LRRK2 iPSC-derived neurons in the Sagamihara family exhibit increased Tau phosphorylation through the AKT/GSK-3beta signaling pathway. Hum Mol Genet. 2015;24:4879–4900. doi: 10.1093/hmg/ddv212. [DOI] [PubMed] [Google Scholar]
- 28.Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, Dawson TM, West AB. Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci. 2007;8:102. doi: 10.1186/1471-2202-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zechel S, Meinhardt A, Unsicker K, von Bohlen Und Halbach O. Expression of leucine-rich-repeat-kinase 2 (LRRK2) during embryonic development. Int J Dev Neurosci. 2010;28:391–399. doi: 10.1016/j.ijdevneu.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20:4209–4223. doi: 10.1093/hmg/ddr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greene ID, Mastaglia F, Meloni BP, West KA, Chieng J, Mitchell CJ, Gai WP, Boulos S. Evidence that the LRRK2 ROC domain Parkinson’s disease-associated mutants A1442P and R1441C exhibit increased intracellular degradation. J Neurosci Res. 2014;92:506–516. doi: 10.1002/jnr.23331. [DOI] [PubMed] [Google Scholar]
- 32.Kara E, Kiely AP, Proukakis C, Giffin N, Love S, Hehir J, Rantell K, Pandraud A, Hernandez DG, Nacheva E, et al. A 6.4 Mb duplication of the alpha-synuclein locus causing frontotemporal dementia and Parkinsonism: phenotype-genotype correlations. JAMA Neurol. 2014;71(9):1162–71. doi: 10.1001/jamaneurol.2014.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 34.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 35.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 36.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamberts JT, Hildebrandt EN, Brundin P. Spreading of alpha-synuclein in the face of axonal transport deficits in Parkinson’s disease: a speculative synthesis. Neurobiol Dis. 2015;77:276–283. doi: 10.1016/j.nbd.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JP, Milnerwood AJ, Unni VK, Hirst WD, Yue Z, Zhao HT, et al. G2019S-LRRK2 expression augments alpha-synuclein sequestration into inclusions in neurons. J Neurosci. 2016;36:7415–7427. doi: 10.1523/JNEUROSCI.3642-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang MX, Cheng XY, Jin M, Cao YL, Yang YP, Wang JD, Li Q, Wang F, Hu LF, Liu CF. TNF compromises lysosome acidification and reduces alpha-synuclein degradation via autophagy in dopaminergic cells. Exp Neurol. 2015;271:112–121. doi: 10.1016/j.expneurol.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 40.Kim C, Rockenstein E, Spencer B, Kim HK, Adame A, Trejo M, Stafa K, Lee HJ, Lee SJ, Masliah E. Antagonizing neuronal Toll-like receptor 2 prevents synucleinopathy by activating autophagy. Cell Rep. 2015;13:771–782. doi: 10.1016/j.celrep.2015.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun. 2015;6:7314. doi: 10.1038/ncomms8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gillardon F, Schmid R, Draheim H. Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience. 2012;208:41–48. doi: 10.1016/j.neuroscience.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Kim B, Yang MS, Choi D, Kim JH, Kim HS, Seol W, Choi S, Jou I, Kim EY, Joe EH. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One. 2012;7:e34693. doi: 10.1371/journal.pone.0034693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russo I, Berti G, Plotegher N, Bernardo G, Filograna R, Bubacco L, Greggio E. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-kappaB p50 signaling in cultured microglia cells. J Neuroinflammation. 2015;12:230. doi: 10.1186/s12974-015-0449-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu G, Aliaga L, Cai H. alpha-synuclein, LRRK2 and their interplay in Parkinson’s disease. Future Neurol. 2012;7:145–153. doi: 10.2217/fnl.12.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchez-Pernaute R, Ferree A, Cooper O, Yu M, Brownell AL, Isacson O. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson’s disease. J Neuroinflammation. 2004;1:6. doi: 10.1186/1742-2094-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muda K, Bertinetti D, Gesellchen F, Hermann JS, von Zweydorf F, Geerlof A, Jacob A, Ueffing M, Gloeckner CJ, Herberg FW. Parkinson-related LRRK2 mutation R1441C/G/H impairs PKA phosphorylation of LRRK2 and disrupts its interaction with 14-3-3. Proc Natl Acad Sci U S A. 2014;111:E34–43. doi: 10.1073/pnas.1312701111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Wang QJ, Pan N, Lee S, Zhao Y, Chait BT, Yue Z. Phosphorylation-dependent 14-3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson’s disease. PLoS One. 2011;6:e17153. doi: 10.1371/journal.pone.0017153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doggett EA, Zhao J, Mork CN, Hu D, Nichols RJ. Phosphorylation of LRRK2 serines 955 and 973 is disrupted by Parkinson’s disease mutations and LRRK2 pharmacological inhibition. J Neurochem. 2012;120:37–45. doi: 10.1111/j.1471-4159.2011.07537.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors declare that they used standard and commercially available software, databases, and application/tool for the data analysis. In addition, the authors declare that they do not have a link to include for the data and that all data and methods of analysis are included in the manuscript (or additional files). The authors will be able to share the software, databases, and all the relevant raw data described in the manuscript for testing by reviewers. The IPS cell lines supporting the conclusions of this article are available in the Spanish National Bank: http://www.isciii.es/ISCIII/es/contenidos/fd-el-instituto/fd-organizacion/fd-estructura-directiva/fd-subdireccion-general-investigacion-terapia-celular-medicina-regenerativa/fd-centros-unidades/fd-banco-nacional-lineas-celulares/fd-lineas-celulares-disponibles/lineas-de-celulas-iPS.shtml