

Abstract
Chronic inflammatory diseases are the most important causes of mortality in the world today and are on the rise. We now know that immune-driven inflammation is critical in the etiology of these diseases, though the environmental triggers and cellular mechanisms that lead to their development are still mysterious. Many chronic inflammatory diseases are associated with significant shifts in the microbiota towards inflammatory configurations, which can affect the host both by inducing local and systemic inflammation and by alterations in microbiota-derived metabolites. This review discusses recent findings suggesting that shifts in the microbiota may contribute to chronic disease via effects on the immune system.
Keywords: Microbiota, dysbiosis, metabolic syndrome, inflammasome
IMMUNITY, THE MICROBIOTA AND CHRONIC INFLAMMATORY DISEASE
In the past decade our knowledge of chronic inflammatory disease (CID) has been transformed by a new understanding of the central role of the immune system in driving pathology. The immune system functions as a ‘rheostat for the entire body; seeking out physiological disturbances, (infectious or otherwise), and rectifying them via both inflammatory immune responses, but also critical repair/regulatory processes such as the clearance of dead cells and the restoration of the physical barrier [1, 2]. Thus, many of the most chronic and debilitating diseases afflicting humankind can be seen as a shift in the immune response away from repair/regulation, and towards immune-driven inflammatory responses. Some of these diseases are also associated with a significant shift in the composition of the microbiota [3–7]. The microbiota is a significant source of both nutritional metabolites and inflammatory innate immune signals [8]. Therefore, compositional shifts in the microbiota are of potential importance as modifiers and triggers of disease, in the context of genetic predisposition and environmental factors.
CID can encompass both autoimmune disease, such as systemic lupus erythematosus or rheumatoid arthritis, that are obviously associated with defects in the immune system and responses against self antigens in addition to auto-inflammatory disorders such as metabolic syndrome and cardiovascular disease, that have more recently been associated with immune-driven inflammation and are not dependent upon autoantigen recognition. In this review. we will focus more on links between the microbiota, immunity and the non-autoimmune inflammatory diseases; though a link between the microbiota and many autoimmune diseases is undeniable and reviewed elsewhere [9, 10].
The incidence of many CID is on the rise worldwide, without a clearly defined explanation. It has been hypothesized that this rise in chronic inflammatory and metabolic diseases is due, at least in part, to a significant narrowing of bacterial diversity associated with the evolutionarily recent phenomena of large scale urban living [11]. Antibiotic usage, dietary modifications and infectious diseases all play important roles in causing alterations in the host’s associated microbiota. In this review we will discuss recent evidence linking CID with shifts in both local and systemic immune responses and disease promoting configurations of the microbiota.
IMMUNE/MICROBIOTA INTERACTION
Humans exist as metaorganisms consisting of host cells in addition to a vast consortium of microbial organisms that live on all of our barrier tissues. The highest density of organisms lives in the intestine, and these organisms provide a tremendous benefit to the host via the enzymatic processing of complex dietary constituents, such as fiber, into metabolites digestible by the host, in addition to many other enzymatic functions. Often these enzymatic pathways can involve multiple genes present in separate bacterial clades [12] and it is perhaps more appropriate to measure those genes that are broadly conserved and critical for function, known as the core metagenome, rather than individual strains of microbes whose maintenance is much more volatile [13]. As a result, a narrowing of organismal diversity may also lead to a loss of components of the metagenome, inducing possible deficits in functionality. Since there is great variation in the composition of the microbiota at lower phylogenetic levels, and significant gaps in our knowledge of the specific function of many strains, ‘dysbiosis’ of the microbiota is sometimes difficult to define and is contextual to the individual and their current state of health. Indeed it has been shown that the glycemic response to any particular nutritional input is unique to the individual and dependent upon the microbiota [14]. This study shows that although we have begun to understand how the host, their diet and their associated microbiota work together to influence health, the details appear to be quite complex and individualized. A microbiota that is ‘dysbiotic’ in one individual may be perfectly healthy in the context of different behavior and genetic predisposition. Without a more fundamental understanding of the core metagenome and how it affects health ubiquitously and comprehensively, describing ‘dysbiosis’ is a difficult task. Therefore, while it is undeniable that the microbiota can shift into states that promote inflammation, (thereby meeting the definition of dysbiosis) whether these states are universal is not clear, and thus we will forgo use of the term dysbiosis, in favor of describing those configurations of the microbiota associated to disease.
The microbiome impacts multiple organ systems, including host immunity. The immune system is responsible for maintaining tissue homeostasis in the intestines as it must remain vigilant against invasive microbes while limiting overt inflammatory responses against the vast array of benign organisms that make up the microbiota [15]. To maintain this perilous relationship with the microbiota, the host has developed a series of immune mechanisms that severely limit and regulate the interaction between intestinal epithelia and the microorganisms that reside within the lumen [16]. This series of ‘checks and balances’ is absolutely critical to host health, because a breakdown of the intestine’s barrier function and immune homeostasis can contribute to inflammatory bowel disease (IBD) in addition to many other chronic diseases [6, 7, 17–19]. The host has also evolved multiple products that foster a healthy and diverse microbiota, including the secretion of IgA and bile acids and the fucosylation of the intestinal epithelium [20–22]. The exact mechanisms of these host factors in shaping the microbiota are not clear but are complex in that they both limit growth of some bacteria while benefiting others and are therefore part of a larger effort to maintain health in the microbiome [23].
There are multiple ways in which the immune system surveys the microbiota to detect alterations. One of the primary ways is through tonic sensation via innate immune pattern recognition receptors. Indeed, steady-state innate immune signaling via specific components of Bacteroides spp. have been shown to assist the development of a healthy immune system and antagonize the development of autoinflammatory disorders, such as asthma [24–26]. However, we now understand that the immune system also measures ‘keystone’ metabolites to determine the presence of a functional microbiota. This idea has been best demonstrated by several studies showing that Short-Chain Fatty Acids (SCFAs), which are metabolites derived from the microbiota-dependent breakdown of dietary fiber and are a key energy source for enterocytes, support immune homeostasis in the gastrointestinal tract. Specifically, SCFAs promote the induction and maintenance of regulatory Foxp3+ T cells (Tregs) in the colon and can limit experimentally induced colitis [27–29]. Critically, SCFA can also act systemically, leading to the diminishment of innate cell responses and pathology in a model of asthma [30, 31]. Similarly, a vitamin A metabolite, retinoic acid, whose production has been shown to be partly controlled by the microbiota, also supports immune function in a multitude of ways [32]. Microbiota-derived metabolites have also been shown to modulate the immune response through activation of the inflammasome and the production of IL-18 and anti-microbial peptides from enteric cells [33].
In addition to sensing of microbe-associated molecular patterns (MAMPs) and keystone metabolites, immune cells themselves rely on different metabolites as energy sources for their growth and survival. The most pertinent example of this idea is the preference of adipose tissue and fat metabolism for regulatory T cells (Tregs) and M2 anti-inflammatory macrophages that, as will be discussed in detail, may be critical to the etiology of metabolic syndrome [34]. As the microbiota affects the availability and absorption of nutrients via both their own enzymatic capabilities, and the modification of host products such as bile acids; this is an important and often overlooked effect of the microbiota on the development of disease [35].
THE MICROBIOTA AND CHRONIC DISEASES OF THE MUCOSA
The intestine is home to the largest and most metabolically active microbial community in the human body. Therefore, it is perhaps unsurprising that the predominant inflammatory diseases of the gut, Crohn’s Disease (CD) and Ulcerative Colitis (UC) are intertwined with shifts in the microbiota. In both diseases, inflammatory immune responses in the intestine against the microbiota induce shifts towards more aggressive members of the population. The most salient example of this effect is the outgrowth of gram negative Proteobacteria, in particular the family Enterobacteriaceae in a subset of patients with IBD [3]. CD specifically has been associated with the outgrowth of opportunistic ‘Adherent-Invasive’ E. coli that can complicate the disease [36]. Additionally, IBD is also associated with a loss of benign gram positive Clostridia, such as Faecalbacterium prausnitzii, which are associated with health and the provision of metabolites such as SCFA from food [37]. Multiple animal models now show that a transmissible microbiota can predispose to IBD susceptibility [38, 39]. However, given the inconsistent efficacy of fecal transplant and antibiotic treatment for IBD it would be a significant overstatement to suggest that the microbiota is sufficient to induce the disease [19, 40–42]. One view on the etiology of these diseases is that barrier and immune dysfunction leads to an outgrowth of those organisms capable of withstanding the intestinal inflammatory response [43]. Indeed, blooms of E. coli are enabled by their ability to use inflammatory nitrogenous compounds as metabolites [44]. Unfortunately, the modules associated with survival in inflamed environments tend to be found in organisms with invasive capabilities and the outgrowth of these organisms lead to a further exacerbation of immune activation and inflammation [45–47]. For example, Clostridium difficile infection is a common complication of patients with IBD [48]. It stands to reason that the most inflammatory organisms are more likely to induce an IgA antibody response in the intestine [49]. Thus the fact that the IgA bound fraction of intestinal bacteria from IBD patients is sufficient to predispose to disease in a mouse model is excellent evidence for the hypothesis that a shift to an inflammatory configuration contributes to development of the disease [50]. However, interestingly none of the organisms discussed above was enriched in the IgA bound fraction of bacteria from IBD patients, and there was no ‘core’ IgA-bound microbiome, perhaps indicating that a loss of diversity in the ‘core’ microbiome in general is more important than the outgrowth of any particular organism.
The model for chronic inflammation at barrier sites, wherein genetic predisposition, immune-driven inflammation and an altered microbiota combine to drive disease, is not unique to the intestine. One clear example is Cystic Fibrosis where defective salt transport leads to thickened mucus, reduced function in lung phagocytes and disrupted mucociliary transport, which allows for the overgrowth of Proteobacteria on all of the patient’s mucosal surfaces, most notably their lungs, leading to chronic inflammation and damage to the alveoli [51]. In addition, Atopic Dermatitis is associated with immune responses against Staphylococcus, explaining perhaps why antibacterial wraps are effective in combating severe cases [52].
THE MICROBIOTA, OBESITY AND METABOLIC SYNDROME
Obesity and associated pathologies, including Type II Diabetes (T2D) and dyslipidemia, are a worldwide epidemic and affect an increasing number of people each year. The mechanism of T2D is now believed to be a block in insulin signaling driven by inflammatory cytokines [53]. In lean individuals, the fat is dominated by M2 macrophages, eosinophils, group 2 innate lymphoid cells and Tregs, all of which contribute in their own way to adipose tissue homeostasis [34]. In obese individuals, the adipose tissue is profoundly altered as it becomes invaded by inflammatory M1 macrophages that secrete large amounts of TNFα, IL-1β and IL-6, all of which contribute to an inhibition of PI3K/Akt signaling downstream of the insulin receptor [54]. Blockade of these inflammatory cytokines has shown modest efficacy in restoring insulin sensitivity in these patients, providing further support for this hypothesis [53]. Thus T2D is a metabolic disease whose symptoms are driven by alterations to the immune response, and identifying the underlying stressor would be of tremendous benefit to the development of novel therapeutics.
It is undeniable that in most cases, the primary cause of these diseases is increased access to high calorie foods, in particular foods high in fat and simple sugars. Gordon and colleagues have now established that obesity is also associated with a configuration of the microbiota that has an increased fraction of Firmicutes, reduced Bacteroidetes, and is capable of providing additional calories to the host for a given caloric intake [5] (Figure 1). Interestingly, the obese microbiota is both a symptom and a contributor to disease, as transfer of this microbiota to germ-free mice leads to significant increases in adiposity in recipients [55]. Building from these studies, it has been demonstrated that the microbiota of obese patients actually produces significantly less SCFA than that of lean identical twins [56]. Whether these SCFAs are contributing to regulation of inflammation via dampening of the immune response through Tregs and innate immune cells, or via regulating other aspects of metabolism or satiety is not entirely clear, but will be an important area for future research. The microbiota can also block adiposity. Indeed, an additional finding of these studies is that gnotobiotic mice transplanted with the microbiota of obese patients can be invaded by a healthy “lean microbiota”, if the mice are placed on a standard diet low in fat [56]. As well, the presence of mucinophilic bacteria, Akkermansia muciniphila, has been associated with a reduction of inflammation and protection from T2D, in mouse models [57, 58] (Figure 1). Therefore, while the microbiota of patients with obesity resists weight loss, it is amenable to change and as such may represent a potential target for treatment.
Figure 1. The microbiota affects metabolic syndrome via the immune system.

a) Obesity is associated with an increase in Firmicutes and a decrease in Bacteroidetes. Firmicutes provide an increased amount of calories to the host by increased harvest of energy from the diet. b) Outgrowth of Proteobacteria is associated with metabolic syndrome and has been shown to increase the frequency of IFNγ-producing T cells in the host, which in turn is associated with increased concentrations of serum LPS. Serum LPS and IFNγ may then drive the development of pro-inflammatory M1 macrophages in the adipose tissue. M1 macrophages express significantly higher amounts of TNFα and IL-1β than the resident M2 macrophages of the gut, and both cytokines contribute to insulin resistance. Beige adipose cells further contribute to health by metabolizing lipids to heat instead of storing them and are supported directly and indirectly by the cytokines IL-4, IL-13, IL-25 and IL-33. c) Mucophilic bacteria Akkermansia combat many of the effects of Proteobacteria outgrowth, including IFNγ production, and can alleviate symptoms associated with metabolic syndrome in animal models.
Beyond nutrient provision, the microbiota can also directly contribute to inflammatory cytokine expression at the core of T2D, via activation of the immune system. In mouse models, this has been clearly shown by the phenotype of TLR5 knockout mice. TLR5 knockout mice show a strikingly increased adiposity, insulin resistance and inflammatory cytokine production, while raised on standard mouse chow [59]. TLR5 is the innate immune receptor for bacterial flagellin, and mice deficient in this receptor have significantly increased populations of Enterobacteriaceae that can transmit many of the phenotypes in wild-type mice via fecal transfer [60] (Figure 1). Interestingly, the outgrowth of Enterobacteriaceae has also been associated with obesity and T2D in humans, though this finding is confounded by the effects of the diabetes medication, metformin [4, 61]. Much like CD, obesity, though not metabolic syndrome specifically, is correlated with reduced microbial diversity [62, 63]. Therefore, a shift towards a more invasive and inflammatory microbiota that induces changes to the immune milieu of the adipose compartment from M2 to M1 macrophage may contribute to the development of metabolic syndrome [64]. Indeed, a number of studies have linked obesity with a ‘leaky gut’, as measured by an increase in serum LPS, and increased intestinal adherence of Enterobacteriaceae [18, 65, 66]. In animal models, provision of a high fat diet and associated affects on the microbiota are sufficient to induce a significant increase in IFNγ expressing Th1 T cells in the small intestine which may traffic to associated adipose depots and directly affect inflammation [67]. The exact mechanism of Proteobacteria outgrowth in obesity and T2D remains unknown and will be an important area for future research. The effect of the microbiota on metabolic disease has also been demonstrated by experiments on animal models of liver disease and cirrhosis. The liver, via the hepatic portal vein, is directly downstream of the small intestine and acts as a barrier to the further trafficking of microbiota-derived products [68]. Mice deficient in components of the inflammasome harbor an altered microbiota with inflammatory potential [69]. When these animals are placed on specific diets associated with non-alcoholic steatohepatitis (NASH) their disease is pronounced and transmissible via the microbiota to co-housed animals [17]. Increased liver damage is dependent upon TLR signaling and TNFα production, and is associated with heightened presence of TLR ligands in the hepatic portal vein. Similar phenomena involving increased translocation of bacterial products may also contribute to other forms of cirrhosis due to alcohol abuse or as a complication of Cystic Fibrosis [70–72].
The microbiota and the immune system may also contribute to dyslipidemia and the cellular composition of the adipose tissue in a metabolically important way. The fat depots exists in at least two distinct states: (i) ‘white’ which functions to store energy, and (ii) brown/beige fat, which is critical for thermogenesis. Brown fat is present at birth, whereas beige fat develops later in life [73]. Instead of storing energy, beige adipocytes metabolize lipids rapidly to create heat and therefore is typically inversely correlated with adiposity throughout the body [74]. Interestingly, the presence of beige fat is supported directly via the local production of IL-4, IL-13, IL-25 and IL-33 from innate lymphoid cells, eosinophils, macrophages and stroma [75–77]. Since inflammation causes such a switch from from M2 to M1 immune cells downstream effects on adipocytes may explain with it is so detrimental to metabolic health. The microbiota does seem to also play a role in the decision to switch on a transcriptional program associated with brown fat in white adipose tissue, in murine models of hypothermia, where fat ‘beiges’ for heat production. Interestingly, the transfer of the microbiota from hypothermic mice is sufficient to induce beige fat in the transplant recipient [78, 79]. However, in this context, it is unclear whether the mechanism of hypothermic beiging is associated with immune cytokine production and adipose tissue inflammatory state.
THE MICROBIOTA AND CARDIOVASCULAR DISEASE
Cardiovascular disease (CVD) and in particular atherosclerosis is the most common cause of death in high-income countries. The root cause of atherosclerosis is blockages in coronary arteries, which is caused by plaques formed of fat deposits and the development of fat laden macrophages called foam cells. Development of atherosclerosis has long been associated with diets high in animal fats, cholesterol and red meat. Recently, Hazen and colleagues have identified that a chemical derivative of red meat, trimethylamine-N-oxide (TMAO) is a significantly more reliable indicator of risk for CVD than cholesterol, and may drive disease by effects on platelets [80, 81]. TMAO is the product of the bacterial breakdown of meat-derived compounds such as L-carnitine and choline, and the microbiota is absolutely necessary for its production, as germ-free mice are free of TMAO, even when fed a diet high in choline [82]. Plasma levels of TMAO can be correlated with specific taxa of the microbiota, but it will only be through a better understanding of the metagenome of patients suffering CVD that we will be able to understand if there is a particular configuration of the microbiome that contributes to CVD [83]. It is possible that the process of converting compounds to TMAO has some benefit for the microbes that carry it out, and that these organisms would be selected for by a diet high in meat. Indeed, the microbiome of vegans has a significantly reduced ability to convert L-carnitine to TMAO [83].
Chronic HIV infection provides another example of a human disease in which CVD risk is elevated, with a possible contributory role for the gut microbiota. HIV-infected subjects exhibit multiple metabolic symptoms, but critically, a significant increase in the incidence of CVD [84]. HIV infection is associated with an increased abundance of invasive, pro-inflammatory Proteobacteria in the gut microbiota including Enterobacteriaceae, along with a depletion of members of the SCFA-producing Clostridia clade; a microbial profile that superficially resembles that of IBD [85–87]. Possibly related to this shift in the microbiota, HIV-infected subjects also exhibit impaired mucosal barrier function, translocation of microbial products into systemic circulation, and increased markers of innate immune activation associated with risk for the development of CVD [88]. Indeed, the degree to which the HIV-associated gut microbiota profile is observed correlates with markers of circulating LPS as well as inflammatory biomarkers of CVD risk [85, 87, 89–91].
CVD may also be associated with shifts in the oral microbiota. Human periodontitis is characterized by an altered and expanded oral bacterial community [92]. In periodontitis patients, markers of systemic exposure to periodontal bacteria are elevated and correlated with increased risk of CVD across several cohorts [93, 94]. Indeed, mouse models reveal that atherogenesis is accelerated by oral colonization with the periodontitis-associated bacterium, Porphyromonas gingivalis and DNA from oral bacteria has been found in atherosclerotic plaques in mice and humans [95–97].
MODERN LIFE AND CHANGING HOST/MICROBIOTA RELATIONSHIPS
As discussed, a multitude of evidence now suggests that shifts in the microbiota towards inflammatory and low diversity states can contribute to chronic disease. One of the primary ways that modern life has changed our relationship with our resident bacteria is the advent of antibiotics. The invention of antibiotics sparked a revolution that fundamentally changed morbidity and mortality associated with bacterial infections. However, it can be argued that antibiotics have been overused as palliatives instead of therapeutics and in agriculture, where they promote rapid weight gain in livestock [98]. Recently, concerns have been raised that the increased use of antibiotics may be contributing to the rise of metabolic syndrome [11]. Essentially, this hypothesis posits that the same effect that induces increased size in cows and pigs is acting on children via constitutive exposure to trace antibiotics present in meat and milk, and early-life administration to combat infection. In support of this idea, mice fed low dose antibiotics early in life grow 10% larger, and the difference is largely due to increases in adipose tissue [99, 100]. These differences in growth could be transferred with the microbiota and seemed to be associated to shifts in the mucosal immune response [100]. Importantly, after cessation of antibiotic treatment, the microbiota, which had deviated significantly from controls, returned to normal, implying that the negative impact of alteration in microbiota can be subsequently sustained by the immune system [100]. Although not formally shown, one might suspect that these immune effects are penetrating beyond the gastrointestinal compartment and acting upon adipose tissue homeostasis. It is not well understood why the microbiota that is resistant to β lactam antibiotics [100], would be more inflammatory and prone to the induction of metabolic issues. One intriguing possibility is that the most inflammatory members of the microbiota tend to be ‘successful colonizers’ with broad host ranges requiring genetic adaptability and therefore may be more accepting to horizontal gene transfer such as antibiotic resistance. Indeed, it has been shown that Enterobacteriaceae of different genera exchange plasmids at a very high rate in vivo under inflammatory conditions [101].
Another fundamental way that those in high-income countries may have developed altered relationship to the microbiota is through diet and exercise. Our ancestors likely ate diets high in fibrous plant material, and had limited access to the saturated fat and simple sugars that represent today the majority of calories in the ‘Western’ diet. As has already been discussed in this review, the Western diet can lead to metabolic syndrome and obesity and these effects are in part due to shifts in the microbiota that control nutrient uptake and inflammation in the adipose tissue. In addition, the western lifestyle, which tends to less physical activity contributes to shifts in the microbiota as well [102]. Western diets might be also selecting away from those organisms that are more closely adapted to the mammalian gut and dependent upon complex carbohydrates [103]. For example, the breakdown of complex carbohydrates to SCFA requires a cascade of enzymes that are not always present in a single organism and can require the cooperation of an ecological community [12]. The Western diet, taken to extremes, presents a situation where there is no selection for cooperative breakdown of complex molecules and only competition for simple sugars and energy rich fats. One could hypothesize that this situation would benefit the more aggressive and inflammatory members of the microbiota, in particular, Proteobacteria, who can divide in as little as twenty minutes and rely heavily on mono and disaccharides as carbon sources [104], and select against organisms that prefer complex carbohydrates as a carbon source. For example, a diet high in milk fat has been shown to favor the outgrowth of Deltaproteobacteria and predispose to significantly exacerbated disease in the IL-10 knockout mouse model of IBD [20]. Thus, the western diet may not only shift the microbiota towards increased provision of calories, but also may benefit those organisms that are least symbiotic and prone to inflammatory outgrowth.
Worldwide urban life amongst millions of other human beings is a modern construction that we have not evolved to deal with. One of the primary ways that life has changed because of urbanization is infection. As opposed to our ancestors, who were beset by chronic parasitic infections and intermittent outbreaks of zoonoses [105, 106], our current population sizes and global travel patterns supports multiple endemic pathogens. Fortunately, in the last century the combined efficacy of sanitation, public health, antibiotics and vaccines have largely mitigated the lethal consequences of these pathogens but it is undeniable that the frequency and biological effect of infection has been profoundly changed. A primary example of this effect is the worldwide reduction in enteric helminth infection. Intestinal helminths used to be a ubiquitous of human life, but in high-income countries are now quite rare. ‘De-worming’ has obvious benefits, as infections with high burdens of worms can lead to malnutrition and anemia [107]. However, our immune system evolved in the context of exposure to these organisms and as a part of the ‘Hygiene Hypothesis’ their absence is believed to contribute to the rise of CID [108, 109]. Multiple findings in animal models have shown that co-infection with helminthes can affect the immune system both systemically and locally at the mucosal surface [110]. For example, helminth infection can support the production of Tregs at mucosal surfaces, which may prevent the development of food allergy and temper the immune response to viruses via shifting from type I to type II immunity, limiting immunopathology and aiding the healing process [111, 112]. Helminth infection can also promote immunoregulation via induction of increased SCFA production from the microbiota [113]. Animal models of IBD and chronic diarrhea have shown that transient helminth infection may represent an effective treatment [114, 115]. Trials in human patients with UC have been promising, but the identification of the products that drive regulatory immune functions is critical, because infection with live worms present obvious limitations [116]. Additionally, identifying direct host affects from those that act through the microbiota could also assist in developing the most effective therapeutics.
Although improved sanitation has reduced the incidence of enteric infections, they remain a significant burden in high-income countries. For instance, the average child in North America will experience ten diarrheal episodes by their 5th birthday and children in low-income countries are exposed to an even higher number of enteric infections [117, 118] Enteric infection poses an issue for the mucosal immune system, because the microbiota and inciting organisms are not always easily discriminated and it has been hypothesized that infections could be triggering events for CID [119]. Indeed, in many cases bacterial pathogenicity is contextual to the complex relationship between the bacteria, the host and the microbiota. Multiple laboratories have now shown that infection causes a significant shift in the microbiota and in general, these shifts mimic those seen in IBD, characterized by increases in Proteobacteria, specifically Enterobacteriaceae, and a depletion of Firmicutes [45–47, 120]. Given that many enteric infections belong to the same phylogenetic groups as prominent members of the microbiota, a critical question is how the immune system discriminates between opportunistic members of the microbiota and the initiating infectious organism. This is made all the more difficult by the fact that many infections break the epithelial barrier leading to translocation of the microbiota into the host [47, 121, 122]. Recent work indicates that the immune system does not discriminate between the microbiota and the infectious organism and activates microbiota-specific T cells in a comparable manner to those specific to the pathogen [123]. How these microbiota-specific T cells are maintained long-term and possibly contribute to chronic disease is an active area of new research. Infection with enteric pathogens such as Yersinia pseudotuberculosis can also cause damage and induce ‘immunological scarring’, which can chronically affect homeostatic intestinal immune responses by deviating lymphatic traffic away from the lymph nodes and into the fat [124]. Fascinatingly, oral Yersinia infection is also associated to chronic pro-inflammatory shifts in the microbiota, possibly indicating a link with immunological scarring [125]. As well, it is interesting to consider whether this kind of atypical traffic of myeloid cells activated at barrier surfaces into adipose depots is somehow contributing to metabolic disease and IBD. Indeed, CD is characterized by the ‘wrapping’ of the gut with creeping fat, the presence of bacteria within the adipose tissue and the production of inflammatory adipokines [126, 127]. Thus taken together, these three effects of infection; shifts in the microbiota towards more inflammatory organisms, microbiota-specific memory, and immunological scarring, provides a framework within which current or cleared infections can act as the inciting event of a CID [119]. An example in which one or more of these microbiota-dependent factors may be acting, is Environmental Enteropathy (EE) in children. EE is a disease that is prevalent in sub-Saharan Africa and parts of the Indian subcontinent wherein malnutrition and poor sanitation allow for chronic infection leading to a malabsorption syndrome [128]. Recent studies have identified that Enterobacteriaceae and subsequent anti-microbiota immune responses are critical players in the etiology this disease [129, 130]. Fascinatingly, it took two years after restoration of a healthy diet and abatement of chronic enteric infection, for patients with EE to experience full restoration of intestinal absorption, implying that either immune memory against the microbiota and immunological scarring could be maintaining the phenotype [131].
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
It is now clear that the microbiota acts as a genetically distinct organ that is critical for the enzymatic digestion of food, promotion of immune homeostasis and prevention of enteric infection. Just like the host-derived organs of the body, the microbiota can be damaged, or, as commonly described, become dysbiotic, leading to a negative impact on dependent host systems. Alterations to the microbiota can certainly contribute to pathology and in cases where these changes include the outgrowth of opportunistic and innately inflammatory bacteria, complicate many diseases (see ‘Outstanding Questions’ and Figure 2). Many studies on the microbiota describe associations to a disease state, and lack clear causative mechanisms. Thus, in order to achieve experimental control, many microbiota studies are carried out in inbred mouse models, which lack both genetic diversity and life history that would fundamentally shape the interaction between the immune system and the microbiota. Additionally, experiments done in germ-free and antibiotic treated mice can be complicated by defects in immune development. This is important because a patient’s history of infection, pathology and treatment may also be critical to understanding their current disease state. Going forward, it is only through a more holistic understanding of the individualized genetic (host and microbial) and environmental triggers of complex diseases that we will be able to cure them.
Figure 2. Potential linkages between the environment, microbiota, immune system and chronic inflammatory disease.
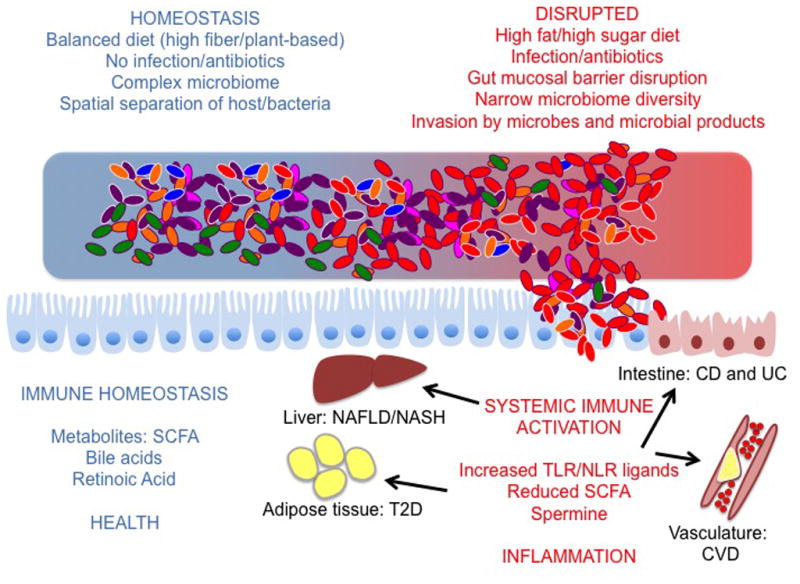
At homeostasis, the microbiota assists the host in converting the diet into metabolites that foster a healthy host/microbiota relationship. These metabolites bolster the barrier between the host and the microbiota, preventing systemic immunity and inflammation. Disruption of the microbiota due to environmental factors (such as diet, infection, antibiotics etc.) or host factors can lead to a narrowing of microbial diversity and a shift in the metabolites derived from the microbiota. Shifts in this relationship can also lead to increased invasion of host tissue by bacteria and bacterial products. Together, a shift in metabolites and increased translocation of microbial products is believed to contribute to immune activation at the core of chronic inflammatory disease (IBD = Inflammatory Bowel Disease; CVD = Cardiovascular Disease; T2D = Type II Diabetes; NASH/NAFLD = Non-alcoholic Steatohepatitis/Non-alcoholic Fatty Liver Disease).
OUTSTANDING QUESTIONS.
Can we define a core metagenome that is associated with health and conversely deficient metagenomes that are dysbiotic in all hosts? Alternatively, is a healthy metagenome also contextual to host genetics and environment and shifting temporally?
Can we define those aspects of modern living that are contributing to the increase in chronic inflammatory disease via induction of shifts in the microbiota?
Is infection a trigger for chronic inflammatory disease via the modification of the microbiota/host relationship?
If the microbiota is a critical contributor to disease, can we modify the microbiota with pre and probiotics to alleviate symptoms exacerbated by inflammatory organisms? If the immune response is critical, can we modulate the immune response downstream of the microbiota to modulate disease phenotypes?
Can we catalog the microbiota-derived metabolites that affect the immune response and understand how the balance of these metabolites may allow for assessment of the health of the host/microbiome relationship.
Shifts in the microbiota, such as those induced by early life antibiotic use, seem to have a systemic immune memory. Can these ‘memory’ factors be defined and more importantly, can we intervene, to prevent long-term health impacts
TRENDS BOX.
Chronic inflammatory disease is associated with shifts in the microbiota; often towards reduced diversity and increased inflammatory character
The immune system can identify shifts in the microbiota both by direct interaction with bacteria in addition to ‘sensing’ of microbiota-derived metabolites
Immune activation associated with shifts in the microbiota can have far-reaching systemic effects and contribute to disease
The incidence in chronic disease is on the rise and has been linked to a number of environmental factors, including antibiotic use, diet and infection. There is evidence that these factors act on the host via shifting the microbiota
Acknowledgments
The authors would like to apologize that due to length requirements, not all work in this burgeoning field could be discussed and properly cited. This work was supported by NIAID K22 AI108719 (T.W.H) and the NIH intramural program (V.K.R, I.V-C and Y.B.). The authors would like to thank K. Gopalakrishna and J. Tometich for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54:281–288. doi: 10.1016/j.molcel.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank DN, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin J, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 5.Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 6.Berni Canani R, et al. The role of the commensal microbiota in the regulation of tolerance to dietary allergens. Curr Opin Allergy Clin Immunol. 2015;15:243–249. doi: 10.1097/ACI.0000000000000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. 2012;10:655–666. doi: 10.1038/nrmicro2848. [DOI] [PubMed] [Google Scholar]
- 8.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Longman RS, et al. Microbiota: host interactions in mucosal homeostasis and systemic autoimmunity. Cold Spring Harb Symp Quant Biol. 2013;78:193–201. doi: 10.1101/sqb.2013.78.020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yurkovetskiy LA, et al. Microbiota and autoimmunity: exploring new avenues. Cell Host Microbe. 2015;17:548–552. doi: 10.1016/j.chom.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flint HJ, et al. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6:121–131. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
- 13.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeevi D, et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell. 2015;163:1079–1094. doi: 10.1016/j.cell.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Belkaid Y, et al. Effector and memory T cell responses to commensal bacteria. Trends Immunol. 2013;34:299–306. doi: 10.1016/j.it.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hooper LV, et al. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henao-Mejia J, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amar J, et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med. 2011;3:559–572. doi: 10.1002/emmm.201100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaser A, et al. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devkota S, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawamoto S, et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity. 2014;41:152–165. doi: 10.1016/j.immuni.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 22.Pickard JM, et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature. 2014;514:638–641. doi: 10.1038/nature13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wahlstrom A, et al. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Mazmanian SK, et al. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 25.An D, et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell. 2014;156:123–133. doi: 10.1016/j.cell.2013.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olszak T, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arpaia N, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furusawa Y, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 29.Smith PM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macia L, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. 2015;6:6734. doi: 10.1038/ncomms7734. [DOI] [PubMed] [Google Scholar]
- 31.Maslowski KM, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall JA, et al. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35:13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levy M, et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell. 2015;163:1428–1443. doi: 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathis D. Immunological goings-on in visceral adipose tissue. Cell Metab. 2013;17:851–859. doi: 10.1016/j.cmet.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buffie CG, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rolhion N, Darfeuille-Michaud A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1277–1283. doi: 10.1002/ibd.20176. [DOI] [PubMed] [Google Scholar]
- 37.Sokol H, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15:1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 38.Garrett WS, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elinav E, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moayyedi P, et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology. 2015;149:102–109. e106. doi: 10.1053/j.gastro.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Rossen NG, et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients With Ulcerative Colitis. Gastroenterology. 2015;149:110–118. e114. doi: 10.1053/j.gastro.2015.03.045. [DOI] [PubMed] [Google Scholar]
- 42.Sokol H. Probiotics and antibiotics in IBD. Dig Dis. 2014;32(Suppl 1):10–17. doi: 10.1159/000367820. [DOI] [PubMed] [Google Scholar]
- 43.Winter SE, Baumler AJ. Dysbiosis in the inflamed intestine: chance favors the prepared microbe. Gut Microbes. 2014;5:71–73. doi: 10.4161/gmic.27129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winter SE, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339:708–711. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Craven M, et al. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn’s disease. PloS one. 2012;7:e41594. doi: 10.1371/journal.pone.0041594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heimesaat MM, et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- 47.Molloy MJ, et al. Intraluminal containment of commensal outgrowth in the gut during infection-induced dysbiosis. Cell Host Microbe. 2013;14:318–328. doi: 10.1016/j.chom.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berg AM, et al. Clostridium difficile infection in the inflammatory bowel disease patient. Inflamm Bowel Dis. 2013;19:194–204. doi: 10.1002/ibd.22964. [DOI] [PubMed] [Google Scholar]
- 49.Cullender TC, et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe. 2013;14:571–581. doi: 10.1016/j.chom.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palm NW, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158:1000–1010. doi: 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang YJ, LiPuma JJ. The Microbiome in Cystic Fibrosis. Clin Chest Med. 2016;37:59–67. doi: 10.1016/j.ccm.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233–246. doi: 10.1111/j.1600-065X.2011.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 54.McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48. doi: 10.1016/j.immuni.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 55.Turnbaugh PJ, et al. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ridaura VK, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Everard A, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneeberger M, et al. Akkermansia muciniphila inversely correlates with the onset of inflammation, altered adipose tissue metabolism and metabolic disorders during obesity in mice. Sci Rep. 2015;5:16643. doi: 10.1038/srep16643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vijay-Kumar M, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carvalho FA, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12:139–152. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Forslund K, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528:262–266. doi: 10.1038/nature15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cotillard A, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585–588. doi: 10.1038/nature12480. [DOI] [PubMed] [Google Scholar]
- 63.Le Chatelier E, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 64.Burcelin R, et al. Immuno-microbiota cross and talk: the new paradigm of metabolic diseases. Semin Immunol. 2012;24:67–74. doi: 10.1016/j.smim.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 65.Martinez-Medina M, et al. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut. 2014;63:116–124. doi: 10.1136/gutjnl-2012-304119. [DOI] [PubMed] [Google Scholar]
- 66.Cani PD, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 67.Garidou L, et al. The Gut Microbiota Regulates Intestinal CD4 T Cells Expressing RORgammat and Controls Metabolic Disease. Cell Metab. 2015;22:100–112. doi: 10.1016/j.cmet.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 68.Balmer ML, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med. 2014;6:237ra266. doi: 10.1126/scitranslmed.3008618. [DOI] [PubMed] [Google Scholar]
- 69.Elinav E, et al. Analysis of microbiota alterations in inflammasome-deficient mice. Methods Mol Biol. 2013;1040:185–194. doi: 10.1007/978-1-62703-523-1_14. [DOI] [PubMed] [Google Scholar]
- 70.Fiorotto R, et al. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology. 2011;141:1498–1508. 1508e1491–1495. doi: 10.1053/j.gastro.2011.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brenner DA, et al. Role of Gut Microbiota in Liver Disease. J Clin Gastroenterol. 2015;49(Suppl 1):S25–27. doi: 10.1097/MCG.0000000000000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang L, et al. Intestinal REG3 Lectins Protect against Alcoholic Steatohepatitis by Reducing Mucosa-Associated Microbiota and Preventing Bacterial Translocation. Cell Host Microbe. 2016;19:227–239. doi: 10.1016/j.chom.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu J, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cohen P, Spiegelman BM. Brown and Beige Fat: Molecular Parts of a Thermogenic Machine. Diabetes. 2015;64:2346–2351. doi: 10.2337/db15-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brestoff JR, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519:242–246. doi: 10.1038/nature14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee MW, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. 2015;160:74–87. doi: 10.1016/j.cell.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qiu Y, et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell. 2014;157:1292–1308. doi: 10.1016/j.cell.2014.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suarez-Zamorano N, et al. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med. 2015;21:1497–1501. doi: 10.1038/nm.3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chevalier C, et al. Gut Microbiota Orchestrates Energy Homeostasis during Cold. Cell. 2015;163:1360–1374. doi: 10.1016/j.cell.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 80.Zhu W, et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell. 2016;165:111–124. doi: 10.1016/j.cell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang WH, et al. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol. 2014;64:1908–1914. doi: 10.1016/j.jacc.2014.02.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koeth RA, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stein JH, Hsue PY. Inflammation, immune activation, and CVD risk in individuals with HIV infection. JAMA. 2012;308:405–406. doi: 10.1001/jama.2012.8488. [DOI] [PubMed] [Google Scholar]
- 85.Dillon SM, et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014;7:983–994. doi: 10.1038/mi.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lozupone CA, et al. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe. 2013;14:329–339. doi: 10.1016/j.chom.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vujkovic-Cvijin I, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. 2013;5:193ra191. doi: 10.1126/scitranslmed.3006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 89.Dinh DM, et al. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis. 2015;211:19–27. doi: 10.1093/infdis/jiu409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mutlu EA, et al. A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog. 2014;10:e1003829. doi: 10.1371/journal.ppat.1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vazquez-Castellanos JF, et al. Altered metabolism of gut microbiota contributes to chronic immune activation in HIV-infected individuals. Mucosal Immunol. 2015;8:760–772. doi: 10.1038/mi.2014.107. [DOI] [PubMed] [Google Scholar]
- 92.Costalonga M, Herzberg MC. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol Lett. 2014;162:22–38. doi: 10.1016/j.imlet.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gmur R, et al. Double-blind analysis of the relation between adult periodontitis and systemic host response to suspected periodontal pathogens. Infect Immun. 1986;52:768–776. doi: 10.1128/iai.52.3.768-776.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mustapha IZ, et al. Markers of systemic bacterial exposure in periodontal disease and cardiovascular disease risk: a systematic review and meta-analysis. J Periodontol. 2007;78:2289–2302. doi: 10.1902/jop.2007.070140. [DOI] [PubMed] [Google Scholar]
- 95.Gibson FC, 3rd, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:2801–2806. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- 96.Koren O, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4592–4598. doi: 10.1073/pnas.1011383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lalla E, et al. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2003;23:1405–1411. doi: 10.1161/01.ATV.0000082462.26258.FE. [DOI] [PubMed] [Google Scholar]
- 98.Silbergeld EK, et al. Industrial food animal production, antimicrobial resistance, and human health. Annu Rev Public Health. 2008;29:151–169. doi: 10.1146/annurev.publhealth.29.020907.090904. [DOI] [PubMed] [Google Scholar]
- 99.Cho I, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488:621–626. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cox LM, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705–721. doi: 10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stecher B, et al. Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc Natl Acad Sci U S A. 2012;109:1269–1274. doi: 10.1073/pnas.1113246109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Clarke SF, et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut. 2014;63:1913–1920. doi: 10.1136/gutjnl-2013-306541. [DOI] [PubMed] [Google Scholar]
- 103.Sonnenburg ED, Sonnenburg JL. Starving our microbial self: the deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 2014;20:779–786. doi: 10.1016/j.cmet.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kamada N, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McNeill WH. Plagues and peoples. Anchor Press; 1976. [Google Scholar]
- 106.Trueba G, Dunthorn M. Many neglected tropical diseases may have originated in the Paleolithic or before: new insights from genetics. PLoS Negl Trop Dis. 2012;6:e1393. doi: 10.1371/journal.pntd.0001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hotez PJ, et al. The global burden of disease study 2010: interpretation and implications for the neglected tropical diseases. PLoS Negl Trop Dis. 2014;8:e2865. doi: 10.1371/journal.pntd.0002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weinstock JV. Do We Need Worms to Promote Immune Health? Clin Rev Allergy Immunol. 2015;49:227–231. doi: 10.1007/s12016-014-8458-3. [DOI] [PubMed] [Google Scholar]
- 109.Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol. 2011;11:375–388. doi: 10.1038/nri2992. [DOI] [PubMed] [Google Scholar]
- 110.Maizels RM, et al. Immune modulation and modulators in Heligmosomoides polygyrus infection. Exp Parasitol. 2012;132:76–89. doi: 10.1016/j.exppara.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Grainger JR, et al. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J Exp Med. 2010;207:2331–2341. doi: 10.1084/jem.20101074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Osborne LC, et al. Coinfection. Virus-helminth coinfection reveals a microbiota-independent mechanism of immunomodulation. Science. 2014;345:578–582. doi: 10.1126/science.1256942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zaiss MM, et al. The Intestinal Microbiota Contributes to the Ability of Helminths to Modulate Allergic Inflammation. Immunity. 2015;43:998–1010. doi: 10.1016/j.immuni.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leung J, et al. Heligmosomoides polygyrus abrogates antigen-specific gut injury in a murine model of inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:1447–1455. doi: 10.1002/ibd.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Broadhurst MJ, et al. Therapeutic helminth infection of macaques with idiopathic chronic diarrhea alters the inflammatory signature and mucosal microbiota of the colon. PLoS Pathog. 2012;8:e1003000. doi: 10.1371/journal.ppat.1003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Helmby H. Human helminth therapy to treat inflammatory disorders -where do we stand? BMC Immunol. 2015;16:12. doi: 10.1186/s12865-015-0074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vernacchio L, et al. Diarrhea in American infants and young children in the community setting: incidence, clinical presentation and microbiology. Pediatr Infect Dis J. 2006;25:2–7. doi: 10.1097/01.inf.0000195623.57945.87. [DOI] [PubMed] [Google Scholar]
- 118.Kosek M, et al. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull World Health Organ. 2003;81:197–204. [PMC free article] [PubMed] [Google Scholar]
- 119.Cadwell K, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lupp C, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 121.Meinzer U, et al. Yersinia pseudotuberculosis effector YopJ subverts the Nod2/RICK/TAK1 pathway and activates caspase-1 to induce intestinal barrier dysfunction. Cell Host Microbe. 2012;11:337–351. doi: 10.1016/j.chom.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 122.Hasegawa M, et al. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity. 2014;41:620–632. doi: 10.1016/j.immuni.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hand TW, et al. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science. 2012;337:1553–1556. doi: 10.1126/science.1220961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fonseca DM, et al. Microbiota-Dependent Sequelae of Acute Infection Compromise Tissue-Specific Immunity. Cell. 2015;163:354–366. doi: 10.1016/j.cell.2015.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kamdar K, et al. Genetic and Metabolic Signals during Acute Enteric Bacterial Infection Alter the Microbiota and Drive Progression to Chronic Inflammatory Disease. Cell Host Microbe. 2016;19:21–31. doi: 10.1016/j.chom.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Peyrin-Biroulet L, et al. Mesenteric fat as a source of C reactive protein and as a target for bacterial translocation in Crohn’s disease. Gut. 2012;61:78–85. doi: 10.1136/gutjnl-2011-300370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zulian A, et al. Differences in visceral fat and fat bacterial colonization between ulcerative colitis and Crohn’s disease. An in vivo and in vitro study. PLoS One. 2013;8:e78495. doi: 10.1371/journal.pone.0078495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Korpe PS, Petri WA., Jr Environmental enteropathy: critical implications of a poorly understood condition. Trends Mol Med. 2012;18:328–336. doi: 10.1016/j.molmed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brown EM, et al. Diet and specific microbial exposure trigger features of environmental enteropathy in a novel murine model. Nat Commun. 2015;6:7806. doi: 10.1038/ncomms8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kau AL, et al. Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci Transl Med. 2015;7:276ra224. doi: 10.1126/scitranslmed.aaa4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lindenbaum J, et al. Subclinical malabsorption in developing countries. Am J Clin Nutr. 1972;25:1056–1061. doi: 10.1093/ajcn/25.10.1056. [DOI] [PubMed] [Google Scholar]