

Abstract
Background & Aims
Neonatal sclerosing cholangitis (NSC) is a severe neonatal-onset cholangiopathy commonly leading to liver transplantation (LT) for end-stage liver disease in childhood. Liver-biopsy findings histopathologically resemble those in biliary atresia (BA); however, in NSC extrahepatic bile ducts are patent, whilst in BA their lumina are obliterated. NSC is commonly seen in consanguineous kindreds, suggesting autosomal recessive inheritance.
Methods
From 29 NSC patients (24 families) identified, DNA was available in 24 (21 families). Thirteen (7 male) patients (12 families) of consanguineous parentage were selected for whole exome sequencing. Sequence variants were filtered for homozygosity, pathogenicity, minor allele frequency, quality score, and encoded-protein expression pattern.
Results
Four of 13 patients were homozygous and two were compound heterozygous for mutations in DCDC2, encoding doublecortin domain containing 2 (DCDC2), expressed in cholangiocyte cilia. Another 11 patients were sequenced: one (with one sibling pair) was compound heterozygous for DCDC2 mutations. All mutations were protein-truncating. In available liver tissue from patients with DCDC2 mutations, immunostaining for human DCDC2 and the ciliary protein acetylated alpha-tubulin (ACALT) showed no expression (n=6) and transmission electron microscopy found that cholangiocytes lacked primary cilia (n=5). DCDC2 and ACALT were expressed in NSC patients without DCDC2 mutations (n=22). Of the DCDC2, one patient died awaiting LT; five came to LT, of whom one died 2 years later. The other 4 are well.
Conclusion
Among 24 NSC patients with available DNA, 7 had mutations in DCDC2 (6 of 19 families). NSC patients in substantial proportion harbour mutations in DCDC2. Their disease represents a novel liver-based ciliopathy.
Keywords: neonate, cholangiopathy, doublecortin domain-containing protein 2, ciliopathy, acetylated alpha tubulin
Graphical Abstract

Lay summary
Neonatal sclerosing cholangitis (NSC) is a rare genetic form of liver disease presenting in infancy. Through Next Generation Sequencing we identified mutations in the gene encoding for doublecortin domain containing 2 (DCDC2) protein in a group of NSC children. DCDC2 is a signalling and structural protein found in primary cilia of cholangiocytes. Cholangiocytes are the cells forming the biliary system which is the draining system of the liver.
Introduction
Neonatal sclerosing cholangitis (NSC) is a rare form of severe liver disease first reported in 8 children presenting in early infancy with jaundice, hepatosplenomegaly, pale stools, and high serum γ-glutamyltransferase activity (GGT) [1]. Ductular proliferation, moderate portal-tract inflammation, and fibrosis were found at liver biopsy. Percutaneous cholangiography confirmed intrahepatic cholangiopathy in all; 2 had earlier undergone laparotomy to exclude biliary atresia (BA). Most developed biliary cirrhosis. Three patients were born to consanguine parents, suggesting recessive inheritance. The term NSC was first used for high-GGT neonatal-onset cholangiopathy in another consanguine sibling pair. Biliary cirrhosis required liver transplantation (LT) for survival [2]. A distinct hepatorenal disorder was later suggested in 2 siblings with renal disease, high GGT activity, and, on endoscopic retrograde cholangiopancreatography (ERCP) and liver biopsy, early-onset changes like those of primary sclerosing cholangitis [3]. Cholangiopathy in children has been attributed to immune dysregulation (autoimmune sclerosing cholangitis, immunodeficiency or Langerhans cell histiocytosis [4]) and to single-gene disorders (deficiency of multidrug associated protein 3 (MDR3), encoded by ABCB4 [5], claudin-1 deficiency [6] or Kabuki syndrome [7, 8]); as with BA, it also may have multiple different causes [9].
The aim of this study was to identify genes mutated in NSC patients seen at King’s College Hospital [10]. We describe the clinical and laboratory features, presentation, and disease progression of NSC in these patients; the process and results of whole exome sequencing (WES) in a subgroup of these patients, with Sanger-sequencing confirmation of candidate-gene mutations and selective sequencing of candidate genes, when possible, in the remaining patients; and the findings within liver and biliary tract on immunohistochemical assessment of encoded-protein and comparison-protein expression as well as on ultrastructural study.
Patients and Methods
Patients
The diagnosis of NSC was assigned to 29 patients (24 families) whose disorder clinically presented during infancy; who had cholestasis with elevated GGT; and in whom cholangiopathy was demonstrated on histopathologic study or imaging. Exclusion criteria were evidence of ichthyosis-like skin lesions, extrahepatic abnormalities suggesting Alagille syndrome, mutations in ABCB4, or immune dysregulation. Patients had normal serum immunoglobulin values (IgM, IgG, IgA), lacked demonstrable autoantibodies (anti-nuclear, -smooth muscle, -liver – kidney microsome, -mitochondrial, -gastric parietal cell), and had normal complement levels (C3 / C4).
Histologic features of cholangiopathy and cholestasis, present in all available specimens (28 patients), included porto-septal bridging fibrosis, ductular reaction, hepatocellular metallothionein deposits, and intralobular bile-pigment accumulations (Figure 1A). MDR3 expression was demonstrated immunohistochemically (Figure 1F, inset) in all specimens. Radiological features included irregular dilatation and strictures in intrahepatic or extrahepatic bile ducts, consistent with a cholangiopathy.
Fig. 1. Liver biopsy histology and immunohistochemistry in NSC patients with and without mutations in DCDC2.
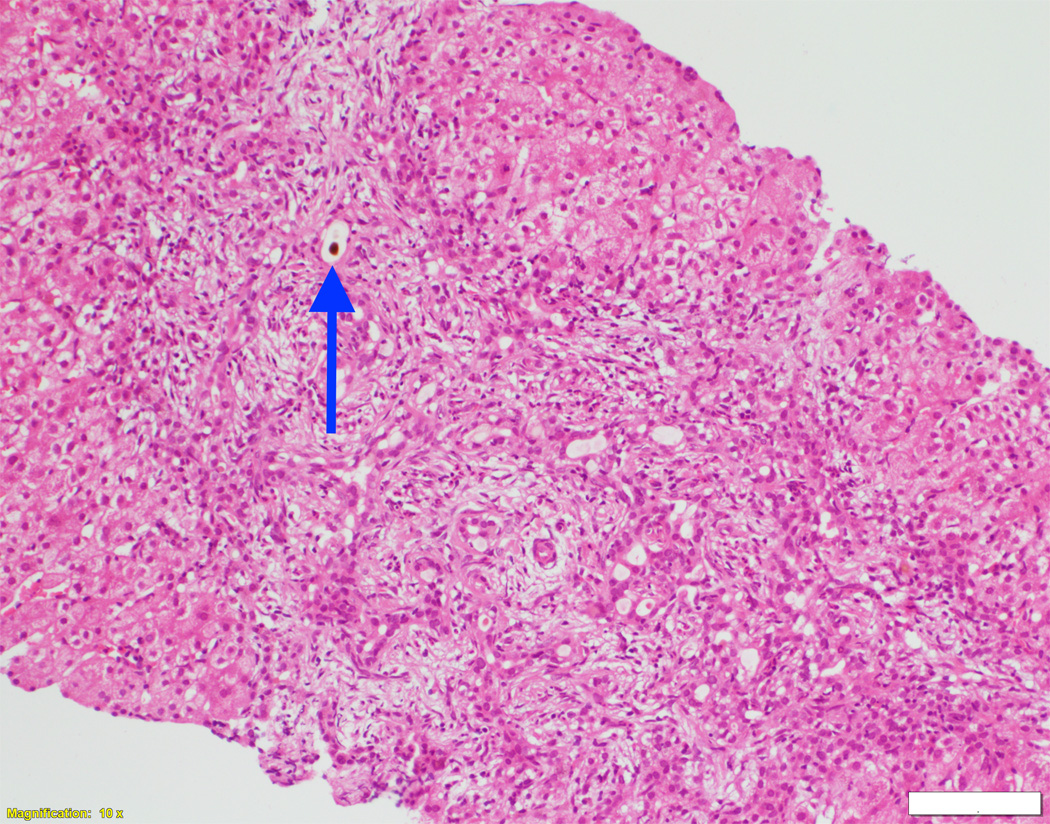
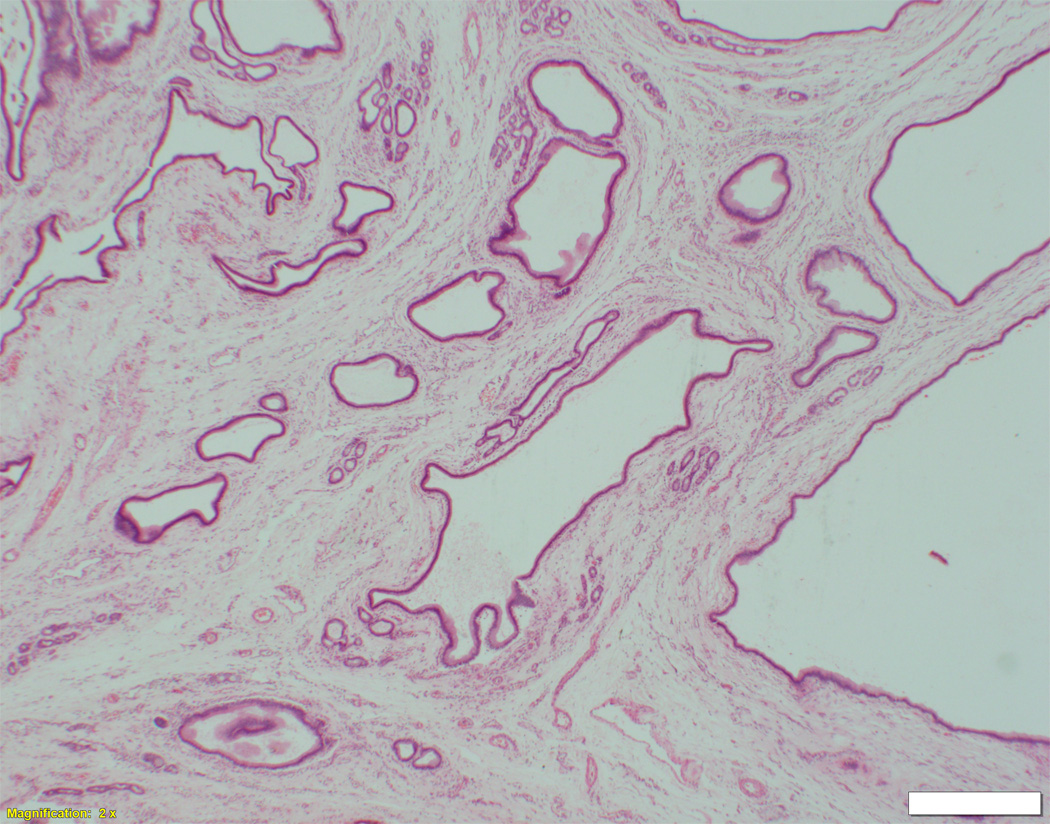
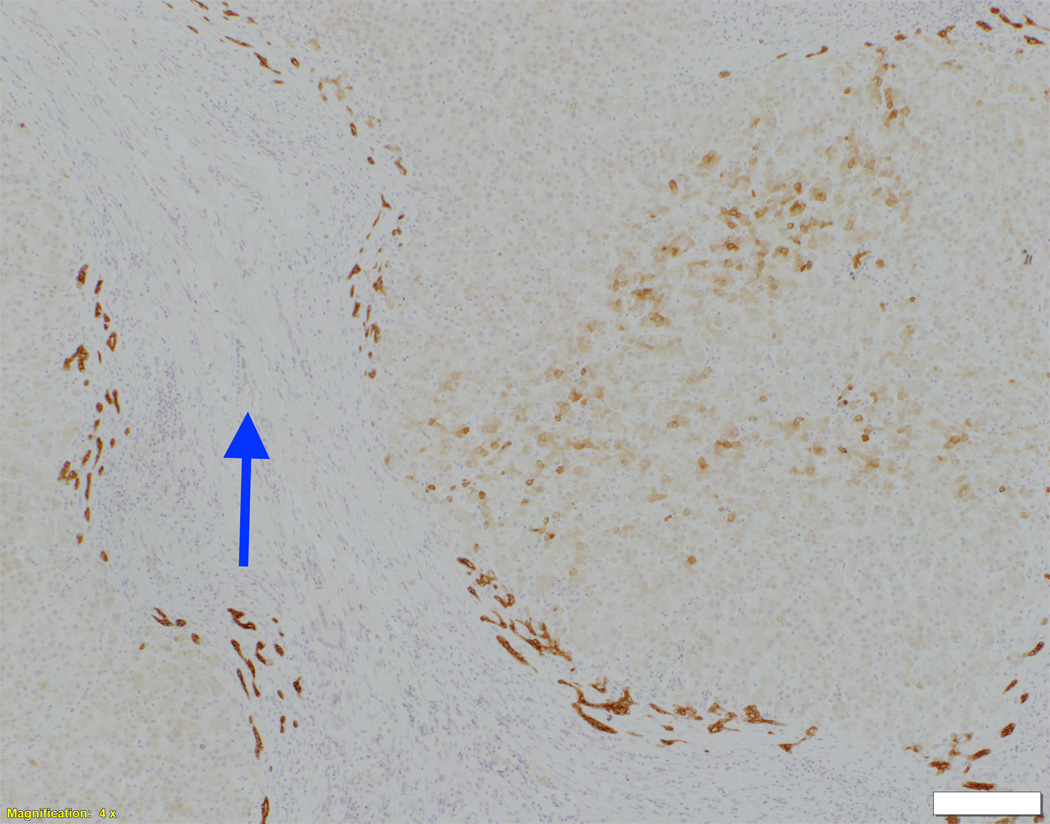
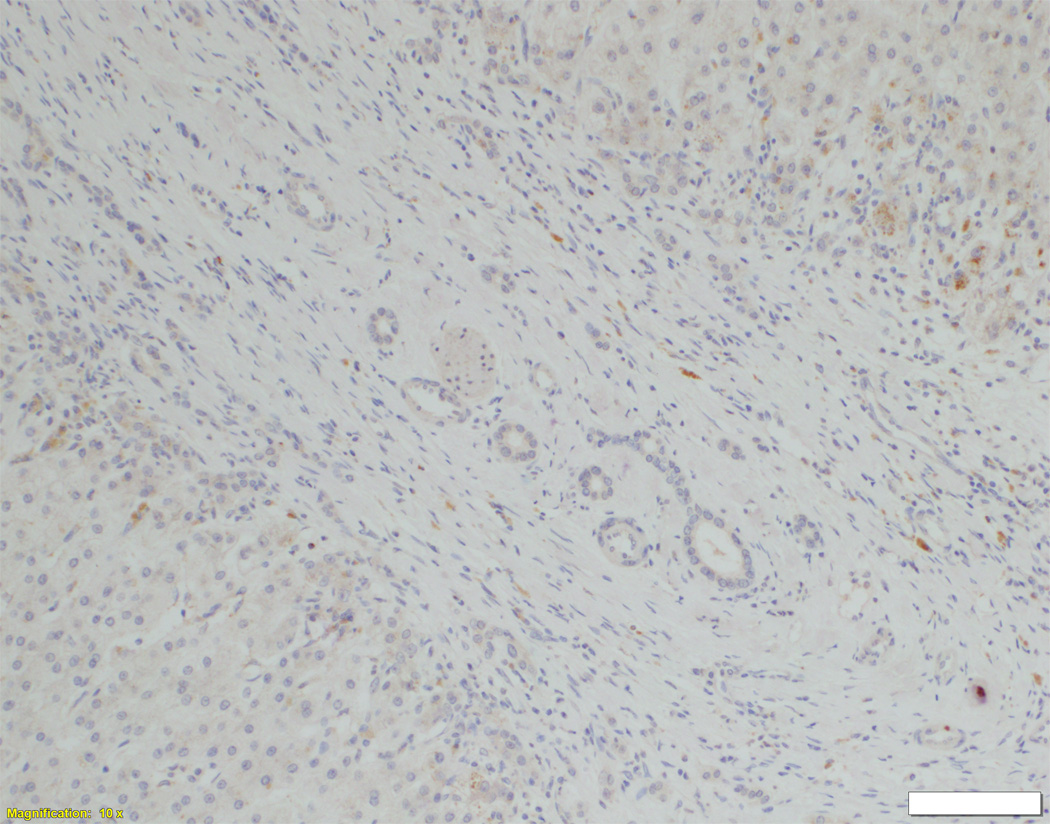
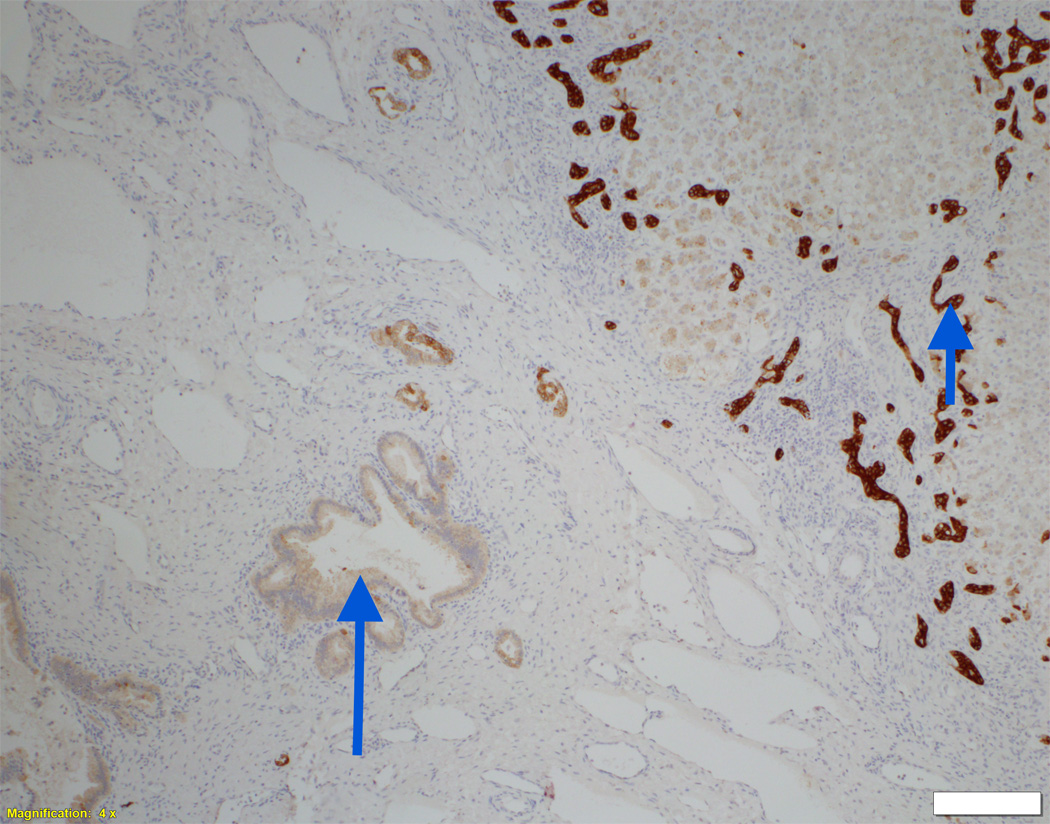
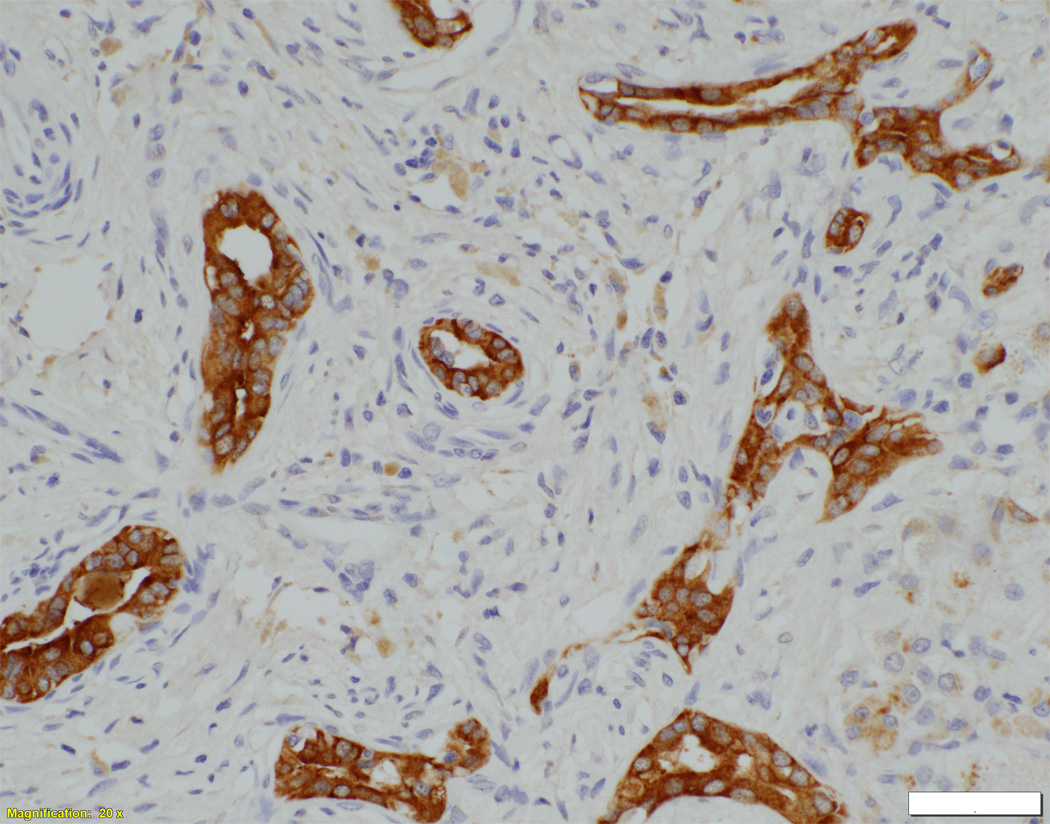
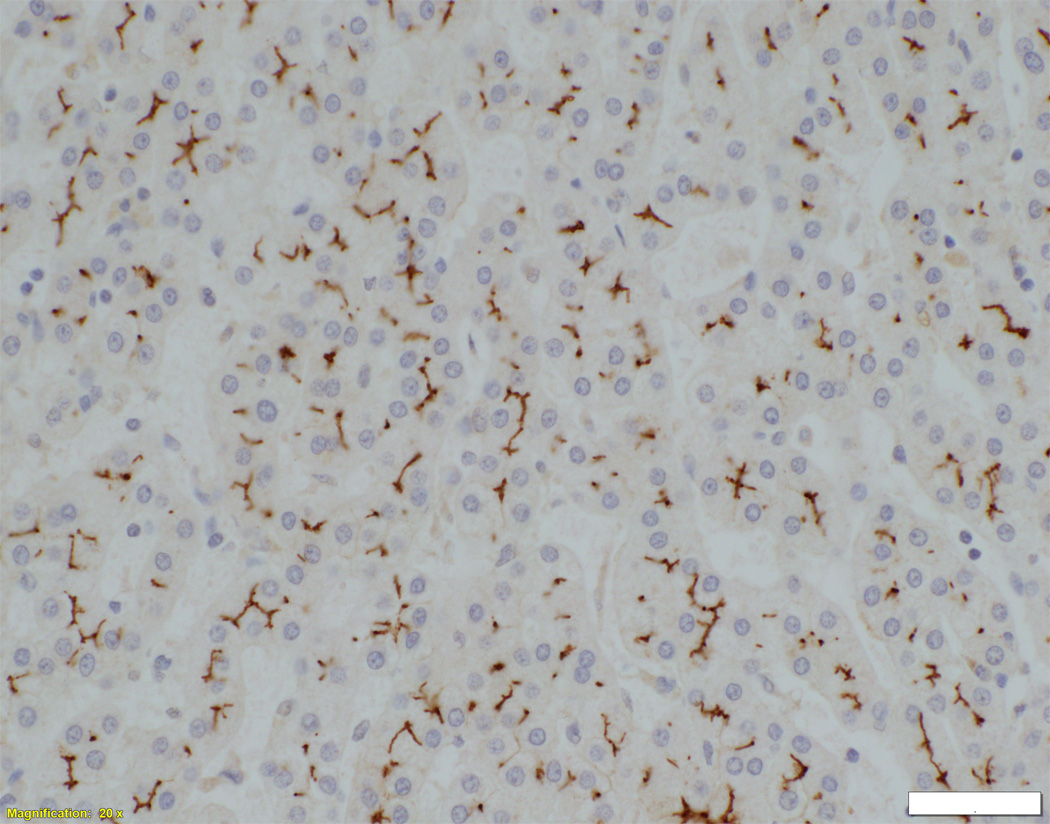
Liver biopsy at 4 months from an NSC patient with DCDC2 mutations (Patient 6) showing expansion of portal areas with ductal bile plugs (arrow) and ductular reaction ((A), H&E, magnification 100×. Calibration bar = 100 micrometres). Variable ectasia of peri-hilar bile ducts in a hepatectomy specimen from an NSC patient with DCDC2 mutations ((B), Patient 4, H&E, magnification 20×. Calibration bar = 500 micrometres). In the periphery of a hepatectomy specimen from an NSC patient with DCDC2 mutations, cytokeratin 7 (CK7) immunostaining demonstrates a ductular reaction, but no bile ducts are detected in portal areas broadened by fibrosis (arrow). Aberrant expres sion of CK7 is seen within the lobule, indicating chronic cholestasis ((C), Patient 4, magnification 40×. Calibration bar = 200 micrometres). DCDC2 immunostaining in NSC patients with DCDC2 mutations demonstrated lack of expression in large peri-hilar bile ducts as well as small interlobular bile ducts ((D), main image from Patient 4, absence of DCDC2 immunostaining in peri-hilar bile ducts, magnification 20×. Calibration bar = 500 micrometres. Inset from Patient 5, liver biopsy at 9 weeks, absence of DCDC2 immunostaining in interlobular bile ducts, magnification 100×. Calibration bar = 100 micrometres). A hepatectomy specimen from an NSC patient with no DCDC2 mutations shows weak and focal DCDC2 staining in large peri-hilar bile ducts (long arrow) and strong diffuse staining in neoductules (short arrow) ((E), Patient 12, magnification 40×. Calibration bar = 200 micrometres). Strong cytoplasmic and apical biliary epithelial expression of DCDC2 is seen within interlobular bile ducts from an NSC patient without DCDC2 mutations ((F), main image, Patient 17, liver biopsy at 35 weeks, DCDC2 immunostaining, magnification 200×. Calibration bar = 50 micrometres). Multidrug resistance protein 3 immunostaining demonstrated canalicular expression in NSC patients with DCDC2 mutations (inset, Patient 3, liver biopsy at 8 weeks, magnification 200×. Calibration bar = 50 micrometres).
Stored blood or DNA was available in 24 patients (19 families). Blood was retrieved for WES from the Paediatric Liver Centre biobank for 13 children (12 families) chosen for parental consanguinity and availability of DNA suitable for next generation sequencing (supplementary information, Table S1). The remaining 11 patients subsequently underwent Sanger sequencing of DCDC2. Parental or patient consent had previously been obtained for research investigation in accordance with institutional guidelines. Ethical-review committee approval for this specific study was obtained, with samples anonymised before use.
Whole Exome Sequencing
WES was undertaken using the Roche Nimblegen SeqCap EZ Human Exome Library v2.0, as per manufacturer’s protocol. Initial analysis focused on finding variants distributed in a pattern consistent with autosomal-recessive disease inheritance. WES to permit cataloguing of genetic variation in patients followed published protocols [11]. Variants were annotated with Variant Effect Predictor and loaded into Gemini software [12]. Variants with minor-allele frequency > 1% in the 1000 Genome or the Exome Sequencing Project data were excluded, as were intergenic variants and variants that were flagged as low quality or potential false-positives (quality scores ≤ 30; long homopolymer runs > 5; low quality by depth < 5; occurrence within a cluster of single-nucleotide polymorphisms). Variants of interest (see above) were prioritised for biological relevance.
Sanger sequencing
Sanger sequencing confirmed variants identified by WES in the first set of patients. Forward and reverse primers were designed and annealing temperatures were set for genes of interest (Supplementary information, Table S2). PCR amplification and sequencing reactions were performed using standard protocols [13, 14].
Histopathologic and ultrastructural studies
Archival formalin-fixed, paraffin-embedded liver-biopsy or hepatectomy materials obtained for clinical diagnosis were available from 12 of the 13 patients in whom WES was conducted. For each patient, tissue sections were cut at 4 µm and stained with haematoxylin-eosin and with orcein. To exclude MDR3 deficiency, parallel sections were immunostained with P311-26, a monoclonal antibody against MDR3 (Alexis Biochemicals ALX-801-028, Nottingham, UK); as a control, parallel sections also were immunostained with M2-lll-6, a monoclonal antibody against a homologous bile-canaliculus transporter, human multidrug resistance-associated protein 2 (Alexis Biochemicals ALX-801-016-C250). Sections also were immunostained using a mouse anti-human DCDC2 monoclonal antibody (Santa Cruz / Insight Biotechnology, Wembley, UK; DCDC2 [C4], sc-166051, recognizing C-terminus amino-acid residues 331–476; 1:50 dilution, 10 minutes pre-treatment at pH9) with BondMax reagents and automated equipment (Leica Microsystems, Milton Keynes, UK). For comparison purposes, hepatobiliary marking for DCDC2 expression in control patients with cholestasis (BA, alpha-1-antitrypsin storage disorder, primary sclerosing cholangitis, primary biliary cirrhosis, Wilson disease) and in tissue from patients without cholestasis also was assessed. To identify primary cilia, parallel sections were immunostained using a mouse monoclonal antibody against acetylated alpha tubulin (ACALT; Sigma-Aldrich (Gillingham, Dorset,UK); clone 6-11B-1; 1:6,000 dilution, epitope unmasked by heating at 100°C for 20 minutes in citrate buffer at pH6).
Liver material from 5 probands, primarily fixed in paraformaldehyde / glutaraldehyde, either at bedside on sampling or on retrieval from −80°C storage, was post-fixed (OsO4) and embedded in resin. Ultrathin sections stained with uranyl acetate / lead citrate were evaluated by transmission electron microscopy (TEM), with particular attention to cholangiocytes. NSC cases with no DCDC2 mutations, and other cholestatic disorders as described above, were used as controls for ultrastructural studies.
Results
Demographics
Twenty-nine children (15 male) from 24 families met inclusion criteria (Table 1 & supplementary Table S1). Parental consanguinity was identified in 16 patients from 12 families. Ethnic background was Arab (8 cases), European (11), and South Asian (9), with mixed ancestry in 1 child. Five patients underwent no genetic testing due to lack of DNA.
Table 1.
Demographical, biochemical, radiological and histological data on NSC patients with DCDC2 mutations. MDR3/MRP2 immunostaining was present in liver tissue, where available. M, male; F, female; ERCP, Endoscopic retrograde cholangiopancreatography; MRCP, Magnetic resonance cholangiopancreatography; GI, gastrointestinal; GGT, γ-Glutamyltransferase; GR, Greek; MDR3, multi drug resistance protein 3; MRP2, multidrug resistance associated protein 2; LT, liver transplantation; NSC, neonatal sclerosing cholangitis; LFTs, liver function tests.
| Patient | Gender/Origin/ Consanguinity |
Age at presentation |
Presenting symptoms |
GGT (IU/L) |
ERCP/MRCP (intrahepatic cholangiopathy) |
Liver histology | LT/age at LT |
Follow up |
|---|---|---|---|---|---|---|---|---|
| 1 | F/Asian/Yes | 20 weeks | Jaundice, pale stools, abnormal LFTs |
247 | Yes/No (+) | N/a | No/listed | Died at 16 years |
| 2 | F/Caucasian (GR)/No |
21 weeks | Jaundice, GI bleeding, ascites, splenomegaly |
447 | Yes | Liver biopsy at 8 months. Porto- portal bridging fibrosis. Ductular reaction with ductal bile plugs. Giant cell change of hepatocytes is not a significant feature. Hepatectomy specimen at 10 years: Biliary pattern cirrhosis. Peripheral ductopaenia. Cholestasis. Copper binding protein deposition. |
Yes 10 years |
12 years |
| 3 | M/Arabic/Yes | 6 weeks | Jaundice, GI bleeding |
711 | N/a | Liver biopsy at 8 weeks. Ductal plate malformation. Small calibre portal vein radicles. Ductal bile plugs. Hepatocellular cholestasis. Giant cell change of hepatocytes is not a significant feature. Hepatectomy specimen at 14 years: Biliary pattern cirrhosis. Peripheral ductopaenia. Cholestasis. Copper binding protein deposition. |
Yes 14 years |
16 years |
| 4 | F/Caucasian (GR)/No |
4 weeks | Jaundice, splenomegaly |
210 | Yes | Hepatectomy at 15 years. Porto- portal bridging fibrosis and partial nodularity. Peripheral ductopaenia. Ectasia and cystic dilatation of perihilar bile ducts. |
Yes 15 years |
Died at 17 years |
| 5 | M/Caucasian (GR)/No |
6 weeks | Jaundice, splenomegaly |
962 | Yes | Liver biopsy at 9 weeks: Porto- portal bridging fibrosis. Ductular proliferation with cholangiopathic features. Canalicular and hepatocellular cholestasis. Giant cell change of hepatocytes is not a significant feature. Liver biopsy at 6 years. Mild fibrosis of portal tracts. Focal interlobular bile duct loss and cholangiopathic features in remaining bile ducts. Features of chronic cholestasis. |
No | 6 years |
| 6 | M/Caucasian (GR)/No |
7 weeks | Jaundice, pale stools, hepatomegaly, splenomegaly |
365 | Yes/No (+) | Liver biopsy at 4 months. Porto- portal bridging fibrosis. Ductular proliferation and ductal bile plugs. Canalicular cholestasis. Giant cell change of hepatocytes is not a significant feature. Hepatectomy: Biliary pattern cirrhosis. Peripheral ductopaenia. Ectasia and cystic dilatation of perihilar bile ducts. Cholestasis. Abundant copper binding protein deposition. |
Yes 15 years |
18 years |
|
7 (sibling of 4) |
F/Caucasian (GR)/No |
1 week | Jaundice | 196 | Yes | Liver biopsy at 10 weeks. Ductal plate malformation. Ductal bile plugs. Giant cell change of hepatocytes is not seen. Liver biopsy at 9 years of age. Mild portal fibrosis. Interlobular portal tract ductopaenia. Features of chronic cholestasis with rosette formation in lobules. Hepatectomy at 14 years. Biliary pattern cirrhosis with peripheral ductopaenia. Ectasia and cystic dilatation of perihilar bile ducts. Cholestasis. Abundant deposition of copper binding protein. |
Yes 14 years |
24 years |
Clinical features, disease course, and investigations
Most patients presented in their first year of life with a median age of 6 weeks [range, 1–57]. Patient 19, who was asymptomatic, was identified (raised liver enzymes) aged 4 years through family screening. Patient 28 had been ill for an undefined period when, aged 5 years, medical attention was sought due to jaundice and pale stools (Table 1 & supplementary Table S1).
Presenting features were jaundice (27), pale stools (10), hepatomegaly (1), coagulopathy (3), gastrointestinal bleeding (3), ascites (2), and splenomegaly (9); some patients manifested disease in more than one way. Median values [and ranges] for serum bilirubin were 111 µmol/l [5–250], GGT 435 IU/l [148–1,120], alkaline phosphatase 694 IU/l [215–1,200], aspartate aminotransferase 179 IU/l [35–482], and albumin 36 g/l [25–48]. ERCP was performed in 15 patients and magnetic resonance cholangiopancreatography (MRCP) in one. Bile duct changes varied. Attenuation and irregular strictures of the common bile duct were usual, with clubbed and irregular smaller ducts, segmental dilatation and stricturing anywhere from common hepatic duct to third-order ducts (Figure 2). Gallbladders were normal. No patient had cholelithiasis.
Fig. 2. Magnetic resonance cholangiography image from patient 25, aged 10 months, demonstrating intrahepatic cholangiopathy with bile-duct irregularity and atypical smooth extrahepatic ductal dilatation.

Liver biopsy was performed at KCH in 25 patients. Liver-biopsy material obtained elsewhere from 3 patients was reviewed at KCH. Patient 29 underwent cholocystojejunostomy aged 18 years for persistent cholestasis. Percutaneous transhepatic cholangiography at age 21 years showed no obstruction; however, biliary emptying was delayed. ERCP a year later showed papillary stenosis and sphincterotomy was performed, without relief. In view of persistent cholestasis, she underwent LT aged 23 years.
Extrahepatic disease was recorded in 5 patients. Three patients (1, 6, and 12) developed renal disease. Patient 1 suffered subarachnoid haemorrhage from a posterior cerebral artery aneurysm, which was clipped at age 12 years. She had previously required splenic embolization twice for thrombocytopenia ascribed to portal hypertension and underwent splenectomy aged 13 years. End-stage liver disease developed, as did end-stage renal disease requiring dialysis. She died aged 16 years from a catastrophic oesophageal variceal haemorrhage. Patient 6 developed hepatorenal syndrome type 2 before LT; this resolved, with restoration of normal renal function, after LT. Patient 12 developed end-stage renal failure and 2 years after LT underwent a living-related-donor renal transplant aged 4 years. Her explanted kidney showed thrombotic microangiopathic changes possibly related to her immunosuppression with no cystic changes or features suggestive of nephronophthisis. Patient 11 had Prader-Willi syndrome. Patient 2 developed hepatopulmonary syndrome, fully reversed after LT. No history of intellectual impairment, neurologic dysfunction, sensorineural hearing loss, dysmorphism, osteochondrodysplasia, or other renal disease was recorded.
In all, 16 patients (55%) underwent LT and 2 died of end-stage liver disease whilst awaiting LT. Two patients died after LT, one of unknown causes while in her native country and the other from complications of end-stage renal disease. Liver graft function was unremarkable in both. Overall mortality was 14%. Patients had a median follow-up of 12 years [range, 2–34].
DNA sequencing
WES in 13 patients identified a total of 998 homozygous or compound-heterozygous variants in 310 genes inherited in an autosomal recessive mode. Genes were categorized by number of patients in whom homozygous variations were identified. In 64 genes 2 or more patients had homozygous mutations. Genes with homozygous variants were filtered further based on predicted impact of mutation, prioritising any protein-truncating mutations through the introduction of a stop codon directly or downstream via frameshift changes. Such mutations were found in 24 genes. These were investigated individually, with priority based on liver tissue expression recorded in GeneCards (http://www.genecards.org) and Online Mendelian Inheritance in Man (OMIM; http://www.omim.org). Eight genes exhibited homozygous variation in 3-patient groups and copy number variant analysis was undertaken. Compound heterozygous changes were also investigated in these candidate genes.
Six (of 13) patients had mutations in DCDC2 (OMIM #605755), encoding doublecortin domain containing protein 2 (DCDC2). Four patients had homozygous changes (2 frameshift, 2 stop codon) and 2 patients had compound heterozygous changes (2 frameshift, 1 stop codon). All mutations were therefore predicted to be protein truncating (Table 2). Sanger sequencing of DCDC2 in patient 7, an affected sibling of patient 4, found the same mutations in exons 4 and 7 as those identified by WES in her sister. Sanger sequencing of DCDC2 in the remaining 10 NSC patients (8 families), numbered 15–24 in the supplementary material table, found no mutations. In the parents of patient 5, Sanger sequencing confirmed heterozygous status in the father (c.123_124delGT) and mother (c.890T>A). DNA was not available from any other parents. WES data (not shown) however, showed quite convincingly that no patient was hemizygous rather than homozygous for a given DCDC2 mutation.
Table 2.
Mutations in DCDC2 identified by WES and confirmed by Sanger sequencing. Mutations are described based on NM_001195610
| Patient number |
Exon Number | Zygosity | Nucleotide change |
Amino acid change |
|---|---|---|---|---|
| 1 | Exon 5 | Homozygous | c.649A>T | p.(Lys217*) |
| 2 | Exon 7 | Homozygous | c.890T>A | p.(Leu297*) |
| 3 | Exon 6 | Homozygous | c.757insG | p.(Ser253Argfs*4) |
| 4 and 7 | Exon 4 | Heterozygous | c.529dupA | p.(Ile177Asnfs*20) |
| Exon 7 | Heterozygous | c.890T>A | p.(Leu297*) | |
| 5 | Exon 1 | Heterozygous | c.123_124delGT | p.(Ser42Glnfs*72) |
| Exon 7 | Heterozygous | c.890T>A | p.(Leu297*) | |
| 6 | Exon 1 | Homozygous | c.123_124delGT | p.(Ser42Glnfs*72) |
Histopathologic and ultrastructural studies
On biopsy at presentation in patients aged eight months or less, varying degrees of portal-tract fibrosis without oedema were found. Numbers of bile-duct profiles were increased in interlobular portal tracts, with occasional intraductal bile plugs (Figure 1A). Persistence of the ductal plate was identified in two patients (Patients 3 and 7). Two patients were biopsied at later time points (Patient 7 at nine years and Patient 5 at six years). Their specimens demonstrated paucity of interlobular bile ducts and mild portal tract fibrosis. At hepatectomy, however, portal tracts deficient in portal-venule radicles were broadened by fibrosis. Biliary-pattern cirrhosis with parenchymal extinction was found in the periphery. Small septal bile ducts and interlobular bile ducts often were lacking (Figure 1C). Concentric periductal lamellar fibrosis was seen focally, as were disarray and atrophy of ductal epithelium. Hepatocytes, Kupffer cells, and canaliculi contained accumulations of bile pigment, particularly in subcapsular regions. Juxtaseptal hepatocytes contained metallothionein deposits. Large septal bile ducts and hilar bile ducts were preserved, with varying dilatation (Figure 1B). In no specimens were biliary hamartomata (von Meyenburg complexes) identified; however, ectasia of large ducts in some patients suggested Caroli-disease–like changes. MDR3 was normally expressed in all patients (Figure 1F, inset).
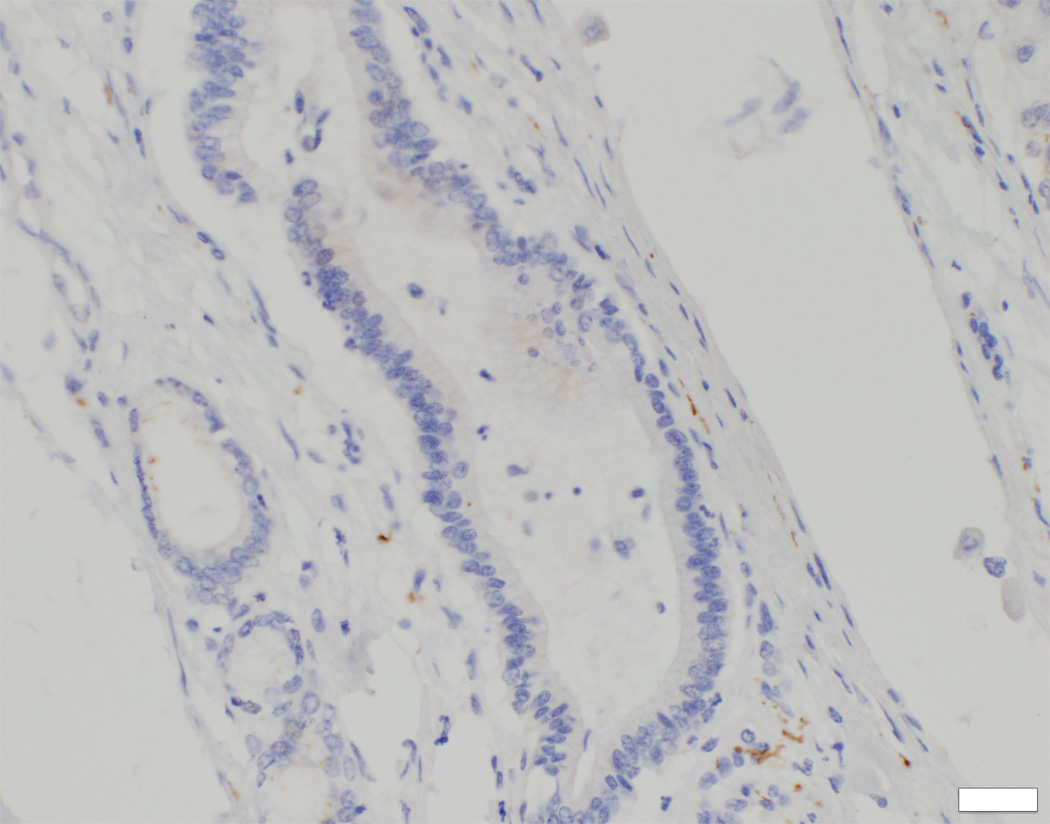
In control material, DCDC2 was generally expressed by cuboidal cholangiocytes (neocholangioles, interlobular bile ducts, small septal bile ducts), but only faint focal marking was seen in columnar cholangiocytes (large septal bile ducts, hilar bile ducts, extrahepatic biliary tract with gallbladder). Similar findings were observed in liver material from the NSC patients with no DCDC2 mutations (Figure 1E). However, in patients with proven DCDC2 mutation, no expression of DCDC2 was found at any site (Figure 1D), consistent with absence of the protein.
Immunostaining for the primary-cilium protein ACALT, conducted to assess presence or absence of primary cilia at cholangiocytes, found good expression in small interlobular ducts as well as in larger septal and perihilar bile ducts (Figure 3C, inset). In DCDC2-mutated probands, however, ACALT expression was entirely absent in septal and perihilar bile ducts, with only very focal and irregular expression in interlobular bile ducts (Figures 3C, 3D). The findings suggested absence of normally constituted primary cilia in association with DCDC2 mutation.
Fig. 3. Ultrastructural studies and immunohistochemistry for ACALT in liver tissue in NSC patients with and without mutations in DCDC2.

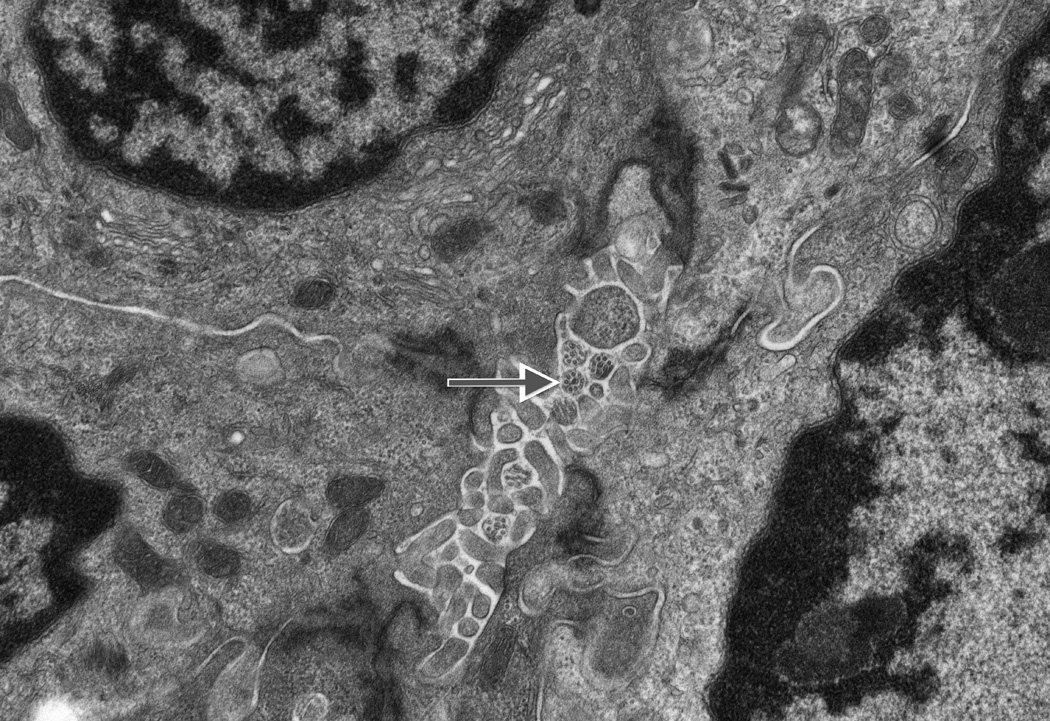
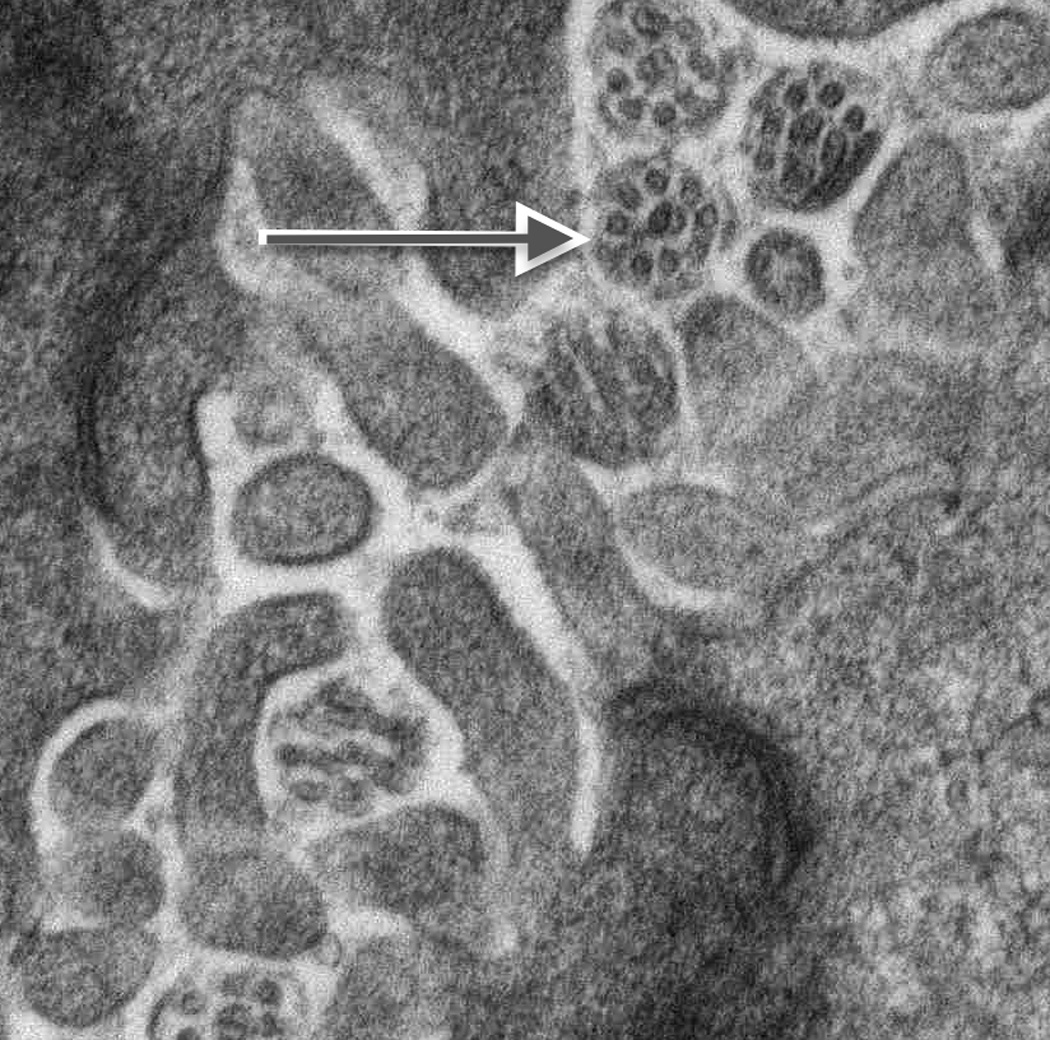
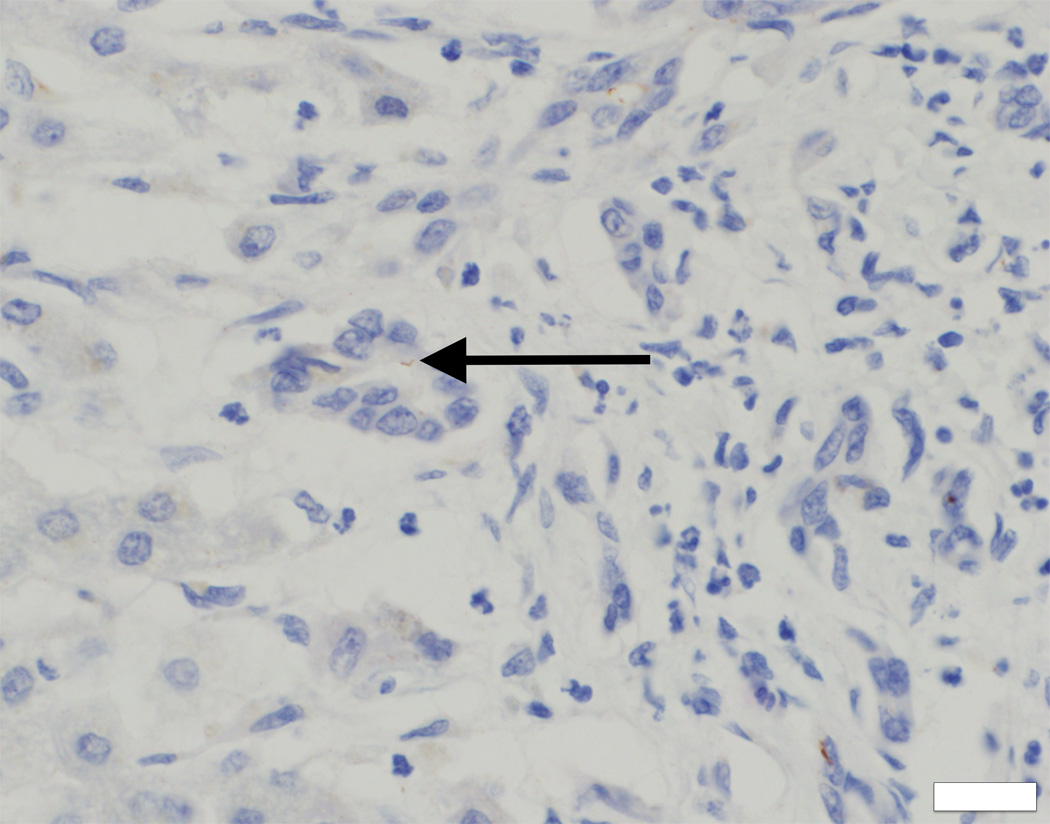
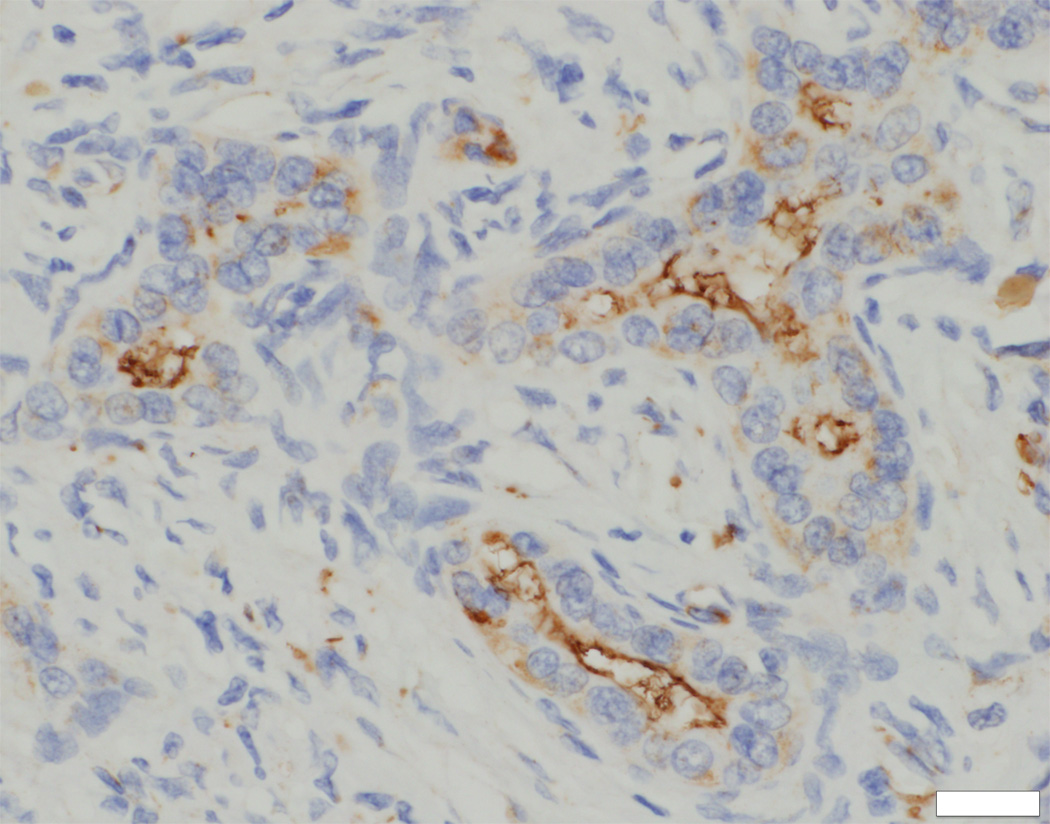
On ultrastructural study, cholangiocyte injury accompanies lack of identifiable primary cilia in patients with NSC and DCDC2 mutation (A and B). Acetylated alpha-tubulin (ACALT) expression at cholangiocytes is lacking in such patients and is present in NSC patients without DCDC2 mutation (C and D). (A). Transmission electron micrograph, interlobular bile duct, with luminal dilatation, loss of microvilli, intraluminal debris, and apical blebbing of cholangiocyte cytoplasm. No primary cilium is apparent. Liver biopsy, age 8 months, Patient 2. Osmium tetroxide / uranyl acetate / lead citrate, original magnification 2,600×; enlarged to facilitate comparison with principal image B (note similar dimensions of mitochondria in both). (B). Transmission electron micrograph, interlobular bile duct, with compact lumen, usual complement of microvilli, unremarkable cholangiocyte cytoplasm, and transversely sectioned primary cilia, with usual internal architecture (arrow; enlarged, also with indicating arrow, inset). Hepatectomy specimen, age 12 years, Wilson-disease patient without known cholangiopathy. Osmium tetroxide / uranyl acetate / lead citrate, original magnification, principal image, 9,200×. (C) ACALT immunostaining in DCDC2 patients demonstrated absent or very focal expression (arrow, main image) in interlobular bile ducts (Patient 4, hepatectomy, magnification 400×, calibration bar = 20 micrometres). By contrast, NSC patients without DCDC2 mutations demonstrated strong apical ACALT expression by cholangiocyte epithelium in bile ducts of all sizes (inset image C, interlobular bile duct in hepatectomy specimen, Patient 14, magnification 400×, calibration bar = 20 micrometres). (D) ACALT expression was not demonstrable in septal or peri-hilar bile ducts in NSC patients with DCDC2 mutations (large septal bile duct from hepatectomy specimen, Patient 4, magnification 200×, calibration bar = 20 micrometres).
In control tissue examined by TEM, primary ciliary structures were identified within interlobular bile ducts and neocholangioles (Figure 3B; green arrow). In the 5 probands with tissue available for TEM, lobular cytoplasmic necrosis, dilatation of canalicular lumina with amorphous bile, blunting of microvilli, and cytoplasmic blebbing into the canalicular lumen were seen. Coarsely granular “Byler bile”, tight-junction abnormalities, and cholangiocellular primary cilia were not identified (Figure 3A).
Discussion
Neonatal sclerosing cholangitis is a rare and severe form of cholangiopathy. Among our 29 patients, 4 died in childhood (14%). More than half (18) developed end-stage liver disease as children, with LT performed in 16. Liver disease like that of NSC has not recurred after LT.
Utilising WES, we identified mutations in DCDC2 (OMIM #605755) in a subgroup of NSC patients. The encoded protein, DCDC2, is part of the microtubule structure involved in ciliary function. DCDC2 belongs to the doublecortin gene family, the first identified member of which is DCX (OMIM #300067). Mutations in DCX, located at Xq23, are associated with subcortical band heterotopia in females and lissencephaly in males [15]. In humans and mouse 11 paralogs in the DCX-repeat gene family have been described [16]. Dcdc2 knockout mice exhibit multiply disrupted development in memory capacity and phonological processing as well as bile duct proliferation and liver fibrosis [17, 18].
Five different mutations in DCDC2 (3 frameshift, 2 introducing premature stop codons; all protein-truncating) were identified in 7 NSC patients, with variants found in exons 1, 4, 5, 6, and 7. The DCDC2 locus contains an antisense transcript, KAAG1 (or RU2AS), on the opposite DNA strand. Six of 7 mutations are predicted to have no effect on this transcript. Only 1 of the 7 mutations was present in the antisense transcript. That our patients’ phenotype is the consequence of mutational effects on this other transcript, rather than on that of DCDC2, thus seems highly unlikely.
Patients 2 – 5 and 7 underwent LT (with living-related donation in patient 3) for cholestasis and severe pruritus at mean age 14 years [range, 10–15]. Patient 4 died suddenly, of unknown causes, 2 years after LT. Patients 2, 3, and 6 are well, with good graft function. Median follow-up from presentation in these 5 patients is 16 years [range, 6–24]. Patient 1 died awaiting LT. How disease has evolved in patient 5, not followed at KCH, is not known.
In 2 of the 6 families with NSC and DCDC2 mutation, the parents acknowledged consanguinity. The other 5 patients were all Greek; however, the parents in their 4 families came from different parts of Greece, and all sets of Greek parents denied consanguinity.
Among the patients with DCDC2 mutation and NSC, only patient 7 had liver cysts; two, described as “small”, were found in preparation for LT. Renal ultrasonography during LT assessment in 6 patients (1 – 4, 6, 7) found only one cyst, 18 mm in diameter, in the right kidney of patient 4. Renal function was normal at listing for LT in all 6 patients. Patient 2 was assessed by cystatin C (0.73 mg/l, n.v. < 1.00 mg/l) and patients 1, 3, 4, 6, and 7 had normal age corrected glomerular filtration rate with a median of 103 ml/min/1.73m2 [range, 92–111). Whilst awaiting LT, patient 1 subsequently developed end-stage renal disease that required dialysis. To date no further renal disease has appeared in the DCDC2-mutated patients.
In our patients with DCDC2-associated NSC, immunohistochemical study found a complete lack of expression of DCDC2. Features at hepatectomy resembled those of congenital hepatic fibrosis centrally and those of sclerosing cholangitis peripherally. Of interest is that in our patients, interlobular and small septal bile ducts (normally lined by cuboidal cholangiocytes, which express DCDC2 uniformly and well) first proliferated and then were lost, whilst bile ducts in larger septa and at the hilum (normally lined by columnar cholangiocytes, which express DCDC2 only weakly and focally) were preserved. Furthermore, some patients, at hepatectomy, exhibited changes that suggested the ductal plate malformation. Models of DCDC2 function that accommodate these observations remain to be developed. Studies of ACALT expression identified parallel loss of both ACALT and DCDC2 expression in DCDC2-mutated patients but not in other NSC patients. On ultrastructural study, cholangiocytes lacked primary cilia, unlike cholangiocytes in other cholestatic disorders with patent biliary-tract lumina[19, 20]. These findings indicate that primary cilia fail to develop normally when biallelic protein-truncating mutation in DCDC2 is present. How loss of ciliary integrity predisposes to inflammation and cholestasis, with the phenotype of neonatalonset cholangiopathy, is at present unclear [21]. We suggest that the absence of DCDC2 may be implicated either in the formation of “cytotoxic” bile or in dysregulation of the cholangiocyte’s homeostatic mechanisms, perhaps via Wnt signalling [17].
Cilia are structures enclosed by the plasma membrane of eukaryotic cells. They are divided into motile cilia such as those of respiratory and genital-tract mucosa and primary cilia (also known as sensory cilia) such as those of cholangiocytes [22]. Specialised cilia also exist, such as the kinocilia of neuroepithelium (see below). Each primary cilium consists of a microtubule-based axoneme and a basal body – a centriole-based microtubule centre from which the axoneme is derived. Primary cilia, when compared to motile cilia, have the basic structure of the 9+0 microtubule arrangement. However, they lack the inner and outer dynein arms and radial spokes of motile cilia, and consequently are non-motile.
Cholangiocyte cilia were first identified in mice in 1963 [23]. Their main function, through utilization of different sensory molecules, is threefold. Firstly, they can detect alterations in bile flow in the biliary-tract lumen via the receptors polycystin 1 & 2; changes in bile flow cause cilia to bend. This stimulus is transduced by polycystins, which form a functional complex that allows calcium ions to enter the cholangiocyte, affecting its function. Secondly, they sense bile composition via purinoreceptors such as P2Y and via interactions with the small extracellular vesicles known as exosomes. P2Y receptors, which are expressed at the cholangiocyte apical membrane and on cilia, are stimulated by specific nucleotide concentrations in bile. Their activation influences cell proliferation and secretion. Exosomes are involved in intercellular communication. They attach themselves to cholangiocyte cilia; this attachment affects the extracellular signal-regulated kinase signalling pathway and influences cell proliferation [24, 25]. Thirdly, cilia act as osmotic sensors. As bile traverses the biliary tract, its osmolality alters due to absorption of bile acids and glucose, or to secretion of bicarbonate ions and water. Transient receptor potential vallinoid type 4 channels regulate intracellular ionised-calcium concentrations in response to changes in osmolality and biliary secretion, with increased cholangiocyte proliferation [26].
DCDC2 has been identified as a candidate gene for dyslexia [27]. DCDC2 is highly expressed in the central nervous system throughout foetal and adult life. It is also present in other organs, including the liver. DCDC2 contains two doublecortin domains, previously described in DCX. These domains are microtubule-binding modifiers. Microtubules are involved in cytoskeletal structure, cell movement and division, and intracellular transport. They are a key component of the internal structure of cilia. Microtubule-associated proteins (MAPs) have a regulatory role mediated via binding to microtubules in a nucleotide-independent process [28]. DCDC2, a known MAP, has the potential to interfere with tubulin binding and microtubule polymerisation, and accordingly with development of normal ciliary structure [28], as suggested by the lack of ACALT in our DCDC2-mutated patients. Ciliary proteins are synthesized in the cytoplasm and endoplasmic reticulum and transported within the cilium via the intraflagellary transport system, which is important in multiple ciliary functions. This system, composed of 20 proteins, relies on kinesin-2 or dyneine 2/1b. It operates along microtubules. MAPs such as DCDC2 affect its operation [29]. DCDC2, as a MAP, is localised at the ciliary axoneme, where it interacts with both the sonic hedgehog and the Wnt signalling pathways [17, 29–31].
Disorders associated with variants in DCDC2 have been described. A homozygous point mutation in DCDC2 (c.1271A>C, p.Gln424Pro) was reported in a Tunisian family with non-syndromic autosomal recessive hearing loss [10]. Immunofluorescence studies in rat inner ear neuroepithelial tissue found that Dcdc2 localized to the primary cilia of nonsensory supporting cells and the kinocilia of sensory hair cells (a type of cilium on the apex of hair cells located in the sensory epithelium of the vertebrate inner ear), with increased density toward the tip. They also demonstrated that DCDC2 mutation could deregulate kinociliary axoneme length and stability with consequent loss of cell function[10].
Nephronophthisis-related ciliopathies (NPHP-RC) are associated with mutations in a variety of genes [32–39]. A recent study of 100 consanguine patients with NPHP-RC identified a homozygous truncating mutation in DCDC2 (the same as that in our patient 1) in a single patient who also had liver disease. High-throughput exon sequencing of DNA from another 800 NPHP-RC families in the study found compound heterozygosity for two mutations in DCDC2, a frameshift mutation (the same as that in our patient 6) and a splice site mutation (c.349-2A>G), in a single patient with hepatic fibrosis but no renal disease up to age of 9 years. Whilst no specific liver phenotype-genotype correlations were found in the patients studied, hepatic portal fibrosis and bile duct proliferation were documented in Dcdc2 knockout mice; in addition, interaction between DCDC2 and Wnt signaling was confirmed in IMCD3 and NIH3T3 cell culture, as was localisation of DCDC2 in the ciliary axoneme and mitotic spindle fibres [17].
Hepatorenal fibrocystic diseases, commonly known as ciliopathies, include autosomal-dominant and -recessive polycystic kidney diseases [40, 41] and Joubert [42], Jeune [43], Bardet-Biedl [44], Meckel-Gruber [45], and oro-facial-digital syndromes [46]. Liver manifestations have been described in congenital hepatic fibrosis, Caroli disease, Jeune and Caroli syndromes [43, 47, 48], and polycystic liver disease [49], with gene mutations confirmed in a proportion of affected patients [50]. Based on our data, a subset of NSC should be included among the ciliopathies.
In our patients with DCDC2 mutations, only one manifested chronic renal disease, although another had a small renal cyst and another suffered transient renal impairment during an episode of liver failure. Other features of hepatorenal ciliopathies, such as osteochondrodysplasia or multiorgan cystic change, were not observed, and central nervous system dysfunction or hearing loss – potential concerns, given the pattern of DCDC2 expression in early life and observations noted above – were not recognised. Our identification of DCDC2 mutations in NSC patients, mostly without renal involvement, suggests a distinct type of ciliopathy.
Of particular interest in DCDC2 deficiency is that biliary-tract inflammation, with scarring and cholestasis, predominates clinically. This is not the case in other hepatorenal ciliopathies. The predominance of liver disease over kidney disease in patients with mutations in DCDC2 highlights differences in the function of primary cilia between the two organs. The mechanisms underlying these differences remain to be determined. Stimuli generated by alterations of bile composition in the proximal biliary tract, and of urine composition in tubuloglomerular structures, could potentially direct different microtubular responses, with organ-specific, distinct phenotypes, such as that described in our probands. In the liver, we can only speculate on whether impairment of DCDC2-mediated function alters bile composition, making bile more damaging, or lowers cholangiocyte defences against normal bile. However, study of bile-mediated injury pathways could improve our understanding of NSC as well as of other early onset cholangiopathies, such as biliary atresia (in which indeed ciliary abnormalities are described)[19, 20]. We anticipate that study of DCDC2 function and dysfunction will provide insights into cholangiocyte biology and regulation of normal bile flow.
Supplementary Material
Acknowledgments
Funding for this project included NIH R01 DK094828 to L.N.B. and R.J.T., the UCSF-King’s College Health Partners Faculty Fellowship Travel Grant (UCSF Academic Senate) to L.N.B., and NIH U01 DK062500 to P. Rosenthal, as well as a gift of funds from A.S. Knisely. WES was undertaken by the University of Washington Center for Mendelian Genomics (UW CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant 1U54HG006493 to Drs. Debbie Nickerson, Jay Shendure, and Michael Bamshad.
List of abbreviations
- ADPKD
Autosomal dominant polycystic kidney disease
- ARPKD
Autosomal recessive polycystic kidney disease
- ACALT
Acetylated alpha tubulin
- BA
Biliary atresia
- Ca2+
Calcium
- CHF
Congenital hepatic fibrosis
- DCDC2
Doublecortin domain containing protein 2
- DCX
Doublecortin gene family
- ERCP
Endoscopic retrograde cholangiopancreatography
- ERK
Extracellular signal-regulated kinase
- GGT
γ-Glutamyltransferase
- IFT
Intraflagellary transport system
- KCH
King’s College Hospital
- LT
Liver transplantation
- MAP
Microtubule-associated protein
- MDR3
Multidrug resistance protein 3
- MRCP
Magnetic resonance cholangiopancreatography
- NPHP-RC
Nephronophthisis-related ciliopathies
- NSC
Neonatal sclerosing cholangitis
- PSC
Primary sclerosing cholangitis
- TEM
Transmission electron microscopy
- WES
Whole exome sequencing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: none
Author contributions
Tassos Grammatikopoulos: Initiated, designed, and performed study, and drafted manuscript
Melissa Sambrotta: Contributed to genetic analysis and manuscript
Sandra Strautnieks: Contributed to genetic analysis and manuscript
Pierre Foskett: Contributed to genetic analysis and manuscript
AS Knisely: Funded study; initiated and supervised liver histopathologic study and contributed to manuscript
Bart Wagner: Contributed to liver electron microscopic study and manuscript
Maesha Deheragoda: Contributed to liver histopathologic study and manuscript
Chris Starling: Contributed to liver histopathologic study
Giorgina Mieli-Vergani: Initiated and supervised study and contributed to manuscript
Joshua Smith: Contributed to genetic analysis and manuscript
University of Washington Center for Mendelian Genomics: Contributed to genetic analysis
Laura Bull: Obtained funding, designed genetic study, contributed to genetic analysis and manuscript
Richard Thompson: Obtained funding, initiated, designed, and supervised study, and contributed to manuscript
REFERENCES
- 1.Amedee-Manesme O, Bernard O, Brunelle F, Hadchouel M, Polonovski C, Baudon JJ, Beguet P, et al. Sclerosing cholangitis with neonatal onset. J Pediatr. 1987;111:225–229. doi: 10.1016/s0022-3476(87)80072-0. [DOI] [PubMed] [Google Scholar]
- 2.Baker AJ, Portmann B, Westaby D, Wilkinson M, Karani J, Mowat AP. Neonatal sclerosing cholangitis in two siblings: a category of progressive intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 1993;17:317–322. doi: 10.1097/00005176-199310000-00016. [DOI] [PubMed] [Google Scholar]
- 3.Bogert PT, LaRusso NF. Cholangiocyte biology. Curr Opin Gastroenterol. 2007;23:299–305. doi: 10.1097/MOG.0b013e3280b079fb. [DOI] [PubMed] [Google Scholar]
- 4.Mieli-Vergani G, Vergani D. Sclerosing cholangitis in the paediatric patient. Best Pract Res Clin Gastroenterol. 2001;15:681–690. doi: 10.1053/bega.2001.0213. [DOI] [PubMed] [Google Scholar]
- 5.de Vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, Deleuze JF, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A. 1998;95:282–287. doi: 10.1073/pnas.95.1.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hadj-Rabia S, Baala L, Vabres P, Hamel-Teillac D, Jacquemin E, Fabre M, Lyonnet S, et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: a tight junction disease. Gastroenterology. 2004;127:1386–1390. doi: 10.1053/j.gastro.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 7.Ewart-Toland A, Enns GM, Cox VA, Mohan GC, Rosenthal P, Golabi M. Severe congenital anomalies requiring transplantation in children with Kabuki syndrome. Am J Med Genet. 1998;80:362–367. [PubMed] [Google Scholar]
- 8.van Haelst MM, Brooks AS, Hoogeboom J, Wessels MW, Tibboel D, de Jongste JC, den Hollander JC, et al. Unexpected life-threatening complications in Kabuki syndrome. Am J Med Genet. 2000;94:170–173. doi: 10.1002/1096-8628(20000911)94:2<170::aid-ajmg10>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Davenport M, Kerkar N, Mieli-Vergani G, Mowat AP, Howard ER. Biliary atresia: the King's College Hospital experience (1974–1995) J Pediatr Surg. 1997;32:479–485. doi: 10.1016/s0022-3468(97)90611-4. [DOI] [PubMed] [Google Scholar]
- 10.Grati M, Chakchouk I, Ma Q, Bensaid M, Desmidt A, Turki N, Yan D, et al. A missense mutation in DCDC2 causes human recessive deafness DFNB66, likely by interfering with sensory hair cell and supporting cell cilia length regulation. Hum Mol Genet. 2015 doi: 10.1093/hmg/ddv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith JD, Hing AV, Clarke CM, Johnson NM, Perez FA, Park SS, Horst JA, et al. Exome sequencing identifies a recurrent de novo ZSWIM6 mutation associated with acromelic frontonasal dysostosis. Am J Hum Genet. 2014;95:235–240. doi: 10.1016/j.ajhg.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. 2013;9:e1003153. doi: 10.1371/journal.pcbi.1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sambrotta M, Strautnieks S, Papouli E, Rushton P, Clark BE, Parry DA, Logan CV, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. 2014;46:326–328. doi: 10.1038/ng.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 15.des Portes V, Francis F, Pinard JM, Desguerre I, Moutard ML, Snoeck I, Meiners LC, et al. doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH) Hum Mol Genet. 1998;7:1063–1070. doi: 10.1093/hmg/7.7.1063. [DOI] [PubMed] [Google Scholar]
- 16.Reiner O, Coquelle FM, Peter B, Levy T, Kaplan A, Sapir T, Orr I, et al. The evolving doublecortin (DCX) superfamily. BMC Genomics. 2006;7:188. doi: 10.1186/1471-2164-7-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schueler M, Braun DA, Chandrasekar G, Gee HY, Klasson TD, Halbritter J, Bieder A, et al. DCDC2 Mutations Cause a Renal-Hepatic Ciliopathy by Disrupting Wnt Signaling. Am J Hum Genet. 2015;96:81–92. doi: 10.1016/j.ajhg.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Truong DT, Che A, Rendall AR, Szalkowski CE, LoTurco JJ, Galaburda AM, Holly Fitch R. Mutation of Dcdc2 in mice leads to impairments in auditory processing and memory ability. Genes Brain Behav. 2014;13:802–811. doi: 10.1111/gbb.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu AS, Russo PA, Wells RG. Cholangiocyte cilia are abnormal in syndromic and non-syndromic biliary atresia. Mod Pathol. 2012;25:751–757. doi: 10.1038/modpathol.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karjoo S, Hand NJ, Loarca L, Russo PA, Friedman JR, Wells RG. Extrahepatic cholangiocyte cilia are abnormal in biliary atresia. J Pediatr Gastroenterol Nutr. 2013;57:96–101. doi: 10.1097/MPG.0b013e318296e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunay-Aygun M. Liver and kidney disease in ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:296–306. doi: 10.1002/ajmg.c.30225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 23.Grisham JW. Ciliated epithelial cells in normal murine intrahepatic bile ducts. Proc Soc Exp Biol Med. 1963;114:318–320. doi: 10.3181/00379727-114-28663. [DOI] [PubMed] [Google Scholar]
- 24.Larusso NF, Masyuk TV. The role of cilia in the regulation of bile flow. Dig Dis. 2011;29:6–12. doi: 10.1159/000324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masyuk AI, Huang BQ, Ward CJ, Gradilone SA, Banales JM, Masyuk TV, Radtke B, et al. Biliary exosomes influence cholangiocyte regulatory mechanisms and proliferation through interaction with primary cilia. Am J Physiol Gastrointest Liver Physiol. 2010;299:G990–G999. doi: 10.1152/ajpgi.00093.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gradilone SA, Masyuk AI, Splinter PL, Banales JM, Huang BQ, Tietz PS, Masyuk TV, et al. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci U S A. 2007;104:19138–19143. doi: 10.1073/pnas.0705964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng H, Smith SD, Hager K, Held M, Liu J, Olson RK, Pennington BF, et al. DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc Natl Acad Sci U S A. 2005;102:17053–17058. doi: 10.1073/pnas.0508591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim MH, Cierpicki T, Derewenda U, Krowarsch D, Feng Y, Devedjiev Y, Dauter Z, et al. The DCX-domain tandems of doublecortin and doublecortin-like kinase. Nat Struct Biol. 2003;10:324–333. doi: 10.1038/nsb918. [DOI] [PubMed] [Google Scholar]
- 29.Massinen S, Hokkanen ME, Matsson H, Tammimies K, Tapia-Paez I, Dahlstrom-Heuser V, Kuja-Panula J, et al. Increased expression of the dyslexia candidate gene DCDC2 affects length and signaling of primary cilia in neurons. PLoS One. 2011;6:e20580. doi: 10.1371/journal.pone.0020580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng H, Powers NR, Tang L, Cope NA, Zhang PX, Fuleihan R, Gibson C, et al. A dyslexia-associated variant in DCDC2 changes gene expression. Behav Genet. 2011;41:58–66. doi: 10.1007/s10519-010-9408-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scholey JM, Anderson KV. Intraflagellar transport and cilium-based signaling. Cell. 2006;125:439–442. doi: 10.1016/j.cell.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 32.Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 33.Hildebrandt F, Otto E, Rensing C, Nothwang HG, Vollmer M, Adolphs J, Hanusch H, et al. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet. 1997;17:149–153. doi: 10.1038/ng1097-149. [DOI] [PubMed] [Google Scholar]
- 34.Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet. 2003;34:455–459. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- 35.Otto E, Hoefele J, Ruf R, Mueller AM, Hiller KS, Wolf MT, Schuermann MJ, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet. 2002;71:1161–1167. doi: 10.1086/344395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 37.Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol. 2008;19:587–592. doi: 10.1681/ASN.2007040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sayer JA, Otto EA, O'Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 39.Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 40.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerres K, Rudnik-Schoneborn S, Deget F, Holtkamp U, Brodehl J, Geisert J, Scharer K. Autosomal recessive polycystic kidney disease in 115 children: clinical presentation, course and influence of gender. Arbeitsgemeinschaft fur Padiatrische, Nephrologie. Acta Paediatr. 1996;85:437–445. doi: 10.1111/j.1651-2227.1996.tb14056.x. [DOI] [PubMed] [Google Scholar]
- 42.Fraser FC, Lytwyn A. Spectrum of anomalies in the Meckel syndrome, or: "Maybe there is a malformation syndrome with at least one constant anomaly". Am J Med Genet. 1981;9:67–73. doi: 10.1002/ajmg.1320090112. [DOI] [PubMed] [Google Scholar]
- 43.Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet. 2012;160C:165–174. doi: 10.1002/ajmg.c.31336. [DOI] [PubMed] [Google Scholar]
- 44.Hurley RM, Dery P, Norady MB, Drummond KN. The renal lesion of the Laurence-Moon-Biedl syndrome. J Pediatr. 1975;87:206–209. doi: 10.1016/s0022-3476(75)80580-4. [DOI] [PubMed] [Google Scholar]
- 45.Barisic I, Boban L, Loane M, Garne E, Wellesley D, Calzolari E, Dolk H, et al. Meckel-Gruber Syndrome: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toriello HV. Are the oral-facial-digital syndromes ciliopathies? Am J Med Genet A. 2009;149A:1089–1095. doi: 10.1002/ajmg.a.32799. [DOI] [PubMed] [Google Scholar]
- 47.Caroli J. Diseases of intrahepatic bile ducts. Isr J Med Sci. 1968;4:21–35. [PubMed] [Google Scholar]
- 48.Caroli J. Intrahepatic bile duct diseases. Rev Med Chir Mal Foie. 1968a;43:211–230. [PubMed] [Google Scholar]
- 49.Tahvanainen E, Tahvanainen P, Kaariainen H, Hockerstedt K. Polycystic liver and kidney diseases. Ann Med. 2005;37:546–555. doi: 10.1080/07853890500389181. [DOI] [PubMed] [Google Scholar]
- 50.Perugorria MJ, Masyuk TV, Marin JJ, Marzioni M, Bujanda L, LaRusso NF, Banales JM. Polycystic liver diseases: advanced insights into the molecular mechanisms. Nat Rev Gastroenterol Hepatol. 2014;11:750–761. doi: 10.1038/nrgastro.2014.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.