

Abstract
This study demonstrated that glycyrrhizate (GAS) could protect HEPG2 cells against damage and apoptosis induced by H2O2 (1600 μM, 4 h). Cell viability assay revealed that GAS was noncytotoxity at concentration 125 µg/mL, and GAS (5 μg/mL, 25 μg/mL, and 125 μg/mL) protected HepG2 cells against H2O2-induced cytotoxicity. H2O2 induced the HepG2 cells apoptosis, obvious morphologic changes were observed after Hochest 33258 staining, and more apoptotic cells were counted in flow cytometry assay compared to that of the natural group. Pretreatment GAS (5 μg/mL, 25 μg/mL, and 125 μg/mL) prior to H2O2 reverses the morphologic changes and reduced the apoptotic cells in HepG2 cells. GAS reduced the release of MDA, increased the activities of superoxide dismutase, and diminished the release of ALT and AST during oxidative stress in HepG2 cells. After Elisa kit detecting, GAS inhibited the caspase activity induced by H2O2, GAS decreased the level of caspase-3 and caspase-9 from mitochondria in dose-dependent manner. Western blot results showed that pretreatment GAS upregulated the expression of Bcl-2 and decreased the expression of Bax. These results reveal that GAS has the cytoprotection in HepG2 cells during ROS exposure by inhibiting the caspase activity in the mitochondria and influencing apoptogenic factors of the expression of Bax and Bcl-2.
1. Introduction
Oxidative stress is a crucial factor that contributes to aging and multiple degenerative diseases, because it can alter biological molecules such as DNA, proteins, and lipids [1]. Chronic oxidative stress may also promote the onset or progression of chronic liver diseases including nonalcoholic fatty liver disease (NAFLD), cirrhosis, hepatitis, and hepatic carcinoma, which if not treated properly are likely to advance to end-stage liver diseases requiring surgical intervention [2]. Major sources of cellular ROS include the mitochondrial respiratory chain and enzymatic reactions mediated by enzyme systems such as xanthine oxidoreductase, nitric oxide (NO) synthase (NOS), and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases [3]. The physiological and pathological relevance of these sources is different depending on the specific disease and tissue/organ. Superoxide (O2 −) and hydrogen peroxide (H2O2), primary ROS products, are indeed involved in a broad range disease progress. Previous studies indicated that extracellular H2O2 increased intracellular ROS levels via multiple mechanisms including loss of intracellular ROS antioxidants such as glutathione (GSH) [4] or decreased mitochondrial membrane permeability followed by mitochondrial ROS release [5]. Many researches have confirmed that the mechanisms underlying various types of hepatic injuries are induced by robust generation of intracellular reactive oxygen species (ROS) [6, 7]. Extracellular H2O2 has been used to induce oxidative injury in hepatocytes at different concentration [8, 9]. Therefore, H2O2 was used to induce oxidative injury in HepG2 cells in our study.
Licorice (Glycyrrhiza glabra Linn; family: Leguminosae) has been widely used for several centuries in China, especially in Chinese herbal compound. The Licorice has been reported for antipyretic, antimicrobial, hepatoprotective, antioxidant, anxiolytic, expectorant, laxative, and diuretic properties [10–13]. Glycyrrhizate (GAS) was one of the main active ingredients of Licorice. Pharmacology studies in recent years show that GAS has multiple pharmacological effects, including adrenal cortical hormone-like effect, anti-inflammatory antihypertensive effect [14], antiaging and enhancing immune function, the enhancement of body's physiological function, and the inhibitory cancer cell growth. But, few researches on GAS can provide enough data particularly with regard to their mechanism to protect cells against oxidative damage. Here we investigated the effects of GAS on oxidative HepG2 cells damage induced by H2O2 and elucidated the potential mechanism involved in the cytoprotective effect of GAS.
2. Materials and Methods
2.1. Chemicals
GAS was obtained from China Institute for Food and Drug control (Beijing, China); the purity of GAS is 98%. Caspase-3 and caspase-9 Elisa kits were purchased from Wuhan Xinqidi Biological Technology Co., Ltd. (Wuhan, china). Hoechst 33258 stains were purchased from Sigma-Aldrich, USA. ALT, AST, MDA, and SOD kits were obtained from the Nan Jing Jian Cheng Bioengineering Institute (Nanjing, China). The tubulin antibody was purchased from Beyotime (Shanghai, china). Annexin V-FITC/propidium iodide (PI) apoptosis detection kit was purchased from BD company (American). Anti-Bax antibody and anti-Bcl-2 antibody were purchased from Abcam company (UK). HepG2 cells were purchased from Beijing Concord Cell Resource Center (Beijing, China).
2.2. Cell Culture
HepG2 cells were cultured in a humidified atmosphere of 95% air plus 5% CO2 in a 37°C incubator in DMEM supplemented with 10% FBS, 100 μM streptomycin, and 100 U/mL penicillin.
2.3. Determination of Cytotoxicity by MTT Assay
Cell toxicity was induced by exposure to H2O2; cells were challenged with H2O2 (1600 μM) for 4 hours. The MTT assay is based on the principle that viable cells convert MTT into an formazan crystals, whose absorbance at 490 nm was read in a microplate reader. It has been divided into 5 groups, natural group, H2O2 group, and GAS (5 μg/mL, 25 μg/mL, and 125 μg/mL). Briefly, HepG2 cells were cultured in 96-well microtiter plates in a final volume of 100 μL culture medium per well. After incubation for 24 hours at 37°C and 5% CO2, the GAS groups were pretreated with GAS (5 μg/mL, 25 μg/mL, and 125 μg/mL) for 12 h. After that, the other four groups, except from the nature group, were treated with H2O2 (1600 μM) for 4 hours. Then, the supernatant was licked off and washed once, 20 μL of MTT (5 mg/mL) in PBS solution was added to each well, and the plate was further incubated for 4 hours. Most of the medium was removed and 100 μL of DMSO was added into the wells to solubilize the crystals. Finally the optical density (OD) was measured by microplate reader at wave length of 490 nm. All viability assays were performed in duplicate; its percentage growth inhibition was calculated using the following formula:
| (1) |
2.4. Hoechst 33258 Staining
Briefly, preparations of fixed cells were rinsed three times with PBS, permeabilised with 70% ethanol (30 s), and incubated with a solution of Hoechst 33258 (2 μg/mL) for 30 min at room temperature (RT). Then, the cells were observed by fluorescence microscope (Leica, Germany).
2.5. The Detection of ALT, AST, MDA, and SOD
ALT, AST, MDA, and SOD in HepG2 cells were measured with commercial kits according to the manufacturer's recommendations. ALT, AST, and MDA levels and SOD activity were expressed as U/g.prot.
2.6. Flow Cytometry
Flow cytometry was performed as described before [15]. Briefly, the HepG2 cells were washed with cold PBS 2 times and then were resuspended in binding buffer (10 mM Hepes/NaOH (pH 7.4), 0.14 M NaCl, and 2.5 mM CaCl2), and FITC Annexin V and PI were added. After incubation at room temperature for 15 min in the dark, flow cytometry was analyzed. Annexin V-FITC and PI double staining were regarded as late apoptotic or necrotic cells.
2.7. Caspase-3 and Caspase-9 Activity Elisa Kit Assays
Briefly, HepG2 cells were washed twice by PBS, digested with trypsin (0.25%), and collected after centrifugation for 5 mins. Then, the HepG2 cells were lysed on ice (lysate 1 mL : PMSF 100 μL : 11 μL protease inhibitor), the protein was collected, and caspase-3 and caspase-9 were measured with commercial kits according to the manufacturer's recommendations.
2.8. Western Blotting Analysis
The expressions of Bcl-2 and Bax were detected by Western blotting. HepG2 cells were exposed to 1600 μM H2O2 with or without GAS. Cytoplasmic extracts were prepared with 150 μL cell lysis buffer (lysis buffer : phenylmethylsulfonyl fluoride (PMSF) : protease inhibitor cocktail = 100 : 1 : 1) on ice for 30 min, then the centrifuged supernatant was collected, and the protein concentration was quantified using the detergent-compatible (DC) protein assay kit (Bio-Rad, Richmond, CA, USA). Proteins were mixed with 2x sodium dodecyl sulphate (SDS) sample buffer. A total of 40 μg of proteins were separated in a 10% (w/v) polyacrylamide gel and blotted on a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). The blots were blocked for 2 h and incubated with polyantibodies Bcl-2, Bax, and tubulin at 1 : 2000 dilution for 12 h. Subsequently, the membranes were washed in buffer (PBS with 0.1% (v/v) Tween-20) and then incubated with horseradish peroxidase link-coupled rat anti-rabbit-antibody at 1 : 1000 in blocking buffer. In all experiments, Ponceau staining was carried out to control equal loading and the bands were visualized by electrogenerated chemiluminescence (ECL) Western blotting system (GE Healthcare Biosciences, USA). Protein levels were also analyzed by ImageJ software. The data shown are representative of at least three experiments.
2.9. Statistical Analysis
All data are expressed as mean ± standard deviation (SD) and were analyzed using SPSS 13.0 software (SPSS, Chicago, IL). p < 0.05 was considered statistically significant.
3. Result
3.1. GAS Inhibits H2O2-Induced Cell Death of HepG2 Cells
To determine the proper working concentrations of H2O2, MTT assay was applied to detect the viability that HEPG2 cells were exposed to different concentrations of H2O2 with 4 h (data not shown). With the concentration of H2O2 increasing, cell survival rate was gradually decreased, and we found that 1600 μM H2O2 caused cell viability to decrease by about 50%; its survival rate has been reduced to (52.20 ± 1.13)% compared to natural group. Therefore we exposed HepG2 cells to concentration of 1600 μM H2O2 for 4 hours to establish an oxidative stress injury model. However, GAS pretreatment groups (5 μg/mL, 25 μg/mL, and 125 μg/mL) effectively protected HepG2 cells from H2O2-induced cell death (n = 6, p < 0.01), and GAS alone has no effect on the cell viability of HepG2 cells (Figure 1). Moreover, the antioxidant Vitamin C pretreatment also can reverse the cell death induced by H2O2, and there is no significant difference between the GAS group (125 μg/mL) and Vitamin C group. The data indicates that GAS, similar to Vitamin C does, effectively protects HepG2 cells from H2O2-induced cell death.
Figure 1.
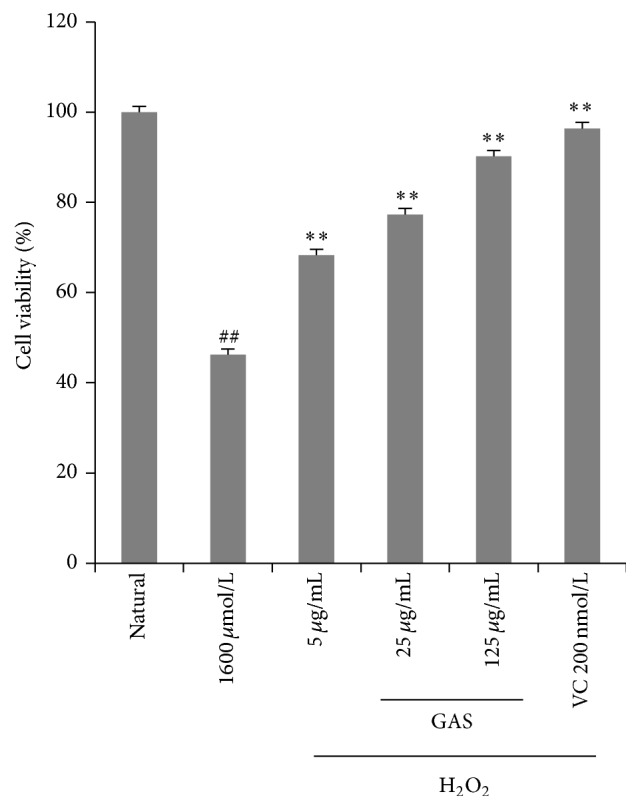
Cell viability of HepG2 cells following different concentrations of GAS pretreatment prior to H2O2 exposure (1600 μM, 4 h) was measured by MTT assay. With increase of the concentration of H2O2, cell survival rate was gradually decreased, and with H2O2 (1600 μM) for 4 hours, its survival rate has been reduced to 52.20 ± 1.13% compared to that of natural group. GAS groups (5 μg/mL, 25 μg/mL, and 125 μg/mL) alleviated the cytotoxicity of HepG2 cells induced by H2O2, and increased cell viability. The annotation ## indicates a p value < 0.05 versus natural group; the annotation ∗∗ indicates a p value < 0.01 versus H2O2 group.
3.2. GAS Inhibits H2O2-Induced HepG2 Cells Apoptosis
The Hoechst 33258 staining and Annexin V-FITC/PI double-staining were applied to further evaluate the protective effect of GAS on H2O2-induced HepG2 cells apoptosis. The results of flow cytometry demonstrated that more apoptotic cells were countered in H2O2-treated group compared with natural group. The apoptotic percentage in the natural group was 5.30 ± 1.20% (Figure 2(a)), which was significantly reduced than that in the H2O2 treated group (42.66 ± 1.54%) (p < 0.01) (Figure 2(b)). On the contrary, pretreatment of HepG2 cells with GAS (25 μg/mL, 125 μg/mL) prior to H2O2 showed the significant inhibition on cell apoptosis induced by H2O2 (p < 0.01). The apoptotic percentage in the GAS groups is 19.52 ± 1.19% (GAS 125 μg/mL), 29.31 ± 1.32% (GAS 25 μg/mL), and 34.11 ± 1.44% (GAS 5 μg/mL), respectively, and the apoptotic percentage in the Vitamin C groups is 11.31 ± 2.10%.
Figure 2.

Cells were stained with Annexin V-FITC/PI for verifying the apoptotic or necrotic cell ratio. GAS inhibited H2O2-induced HEPG2 cells apoptosis. (a) Natural group; (b) H2O2 group; (c) H2O2 + Vitamin C (200 nM) group; (d) H2O2 + GAS (125 μg/mL) group; (e) H2O2 + GAS (25 μg/mL) group; (f) H2O2 + GAS (5 μg/mL group).
After Hoechst 33258 staining, the HepG2 cells in the natural group showed normal shape with round intact nuclei (Figure 3(a)), whereas the H2O2-treated cells became more scarce and showed reduced nuclear size, extensive blebbing, strong fluorescent spot, and pyknotic nuclei (Figure 3(b)); these indicate condensed chromatin and apoptotic bodies. The GAS pretreatment groups (25 μg/mL, 125 μg/mL) show protective effect; it altered the morphologic changes in HepG2 induced by H2O2 (Figures 3(d), 3(e), and 3(f)).
Figure 3.

Hoechst 33258 staining indicated that GAS inhibits H2O2-induced HepG2 cells apoptosis. Morphologic changes in nuclei observed with Hoechst 33258 staining under fluorescence microscopy. (a) Natural group. The HepG2 cells showed normal shape with round intact nuclei; (b) H2O2 group, HepG2 cells treated with 1600 μM H2O2 for 4 hours, and the obvious morphologic changes were observed; (c) H2O2 + Vitamin C (200 nM) group; (d) H2O2 + GAS (125 μg/mL) group; (e) H2O2 + GAS (25 μg/mL) group; (f) H2O2 + GAS (5 μg/mL) group. The arrows indicate apoptotic cells. Original magnification is 400x.
3.3. GAS Reduces the Increase of Lipid Peroxidation Induced by H2O2 in HepG2 Cells
Since lipids in cell membrane were prone to oxidation, the effects of GAS in protecting against lipid peroxidation were also investigated. In the study, H2O2 was used as the source of ROS, which led to oxidative damage in HepG2 cells, and the ALT, AST, MDA, and activity of SOD were measured in the study. The results showed that natural HepG2 cells had little basal intracellular ALT, AST, MDA, and high SOD activity. However, after H2O2 (1600 μM) exposure to 4 hours, cells had significantly increased intracellular ALT, AST, and MDA accumulation (p < 0.01) and significantly reduced intracellular SOD activity (p < 0.01). The results confirmed that H2O2 could induce ROS and led to cell damage in HepG2 cells. GAS pretreatment groups protected HepG2 cells damaged by H2O2 (1600 μM) (Figure 4) and attenuated the H2O2-induced changes in ALT, AST, MDA, and SOD activity. HepG2 cells treated with H2O2 alone evoked a significant increase in the MDA level (Figure 4(a)), approximately 50% higher than the natural group. HepG2 cells treated with GAS showed significant reduction (p < 0.05) in MDA levels compared to H2O2 treated group (Figure 4(a)). In the GAS pretreatment groups (5 μg/mL, 25 μg/mL, and 125 μg/mL), SOD activity was dramatically increased compared with H2O2 treated group with a dose-effect relationship (p < 0.01) (Figure 4(b)). Because of the inhibition of GAS on cell damage induced by H2O2, we detected less AST and ALT in GAS treated group compared to that of H2O2 treated group (p < 0.01) (Figures 4(c) and 4(d)).
Figure 4.

Biochemical assay kits were used to determine content of ALT, AST, MDA, and SOD. GAS protected the cells against H2O2-induced lipid peroxidation in HepG2 cells and reduced H2O2-induced reactive protein production. ALT, AST, and MDA content and SOD activity were measured in HepG2 cells. (a) The MDA level in HepG2 cells; (b) the SOD activity in HepG2 cells; (c) the content of AST; (d) the content of ALT. Error bars represent SD (n = 6). Experiments were performed at least three times. The annotation ## indicates a p value < 0.01 versus natural group. The annotation ∗∗ indicates a p value < 0.01 versus H2O2 group.
3.4. GAS Inhibits Activation of Caspase and Expression of Bcl-2 and Bax Induced by H2O2 in HepG2 Cells
As an important modulator of cell death, caspase cascade activation was evaluated in our study. We measured the activities of caspase-9 and caspase-3 by ELISA Kit. As shown in Figures 5(a) and 5(b), caspase-9 and caspase-3 were activated by H2O2-induced, and GAS effectively inhibited caspase-9 and caspase-3 activation in a dose-dependent manner. These results demonstrated that GAS prevented H2O2-induced apoptosis through inhibition of mitochondrial-dependent cell death pathways.
Figure 5.

GAS inhibit the apoptosis induced by H2O2 in HepG2 cells: (a) the activity of caspase-9; (b) the activity of caspase-3; (c) the expression of Bcl-2; (d) the expression of Bax. The annotation ## indicates a p value < 0.01 versus natural group. The annotation ∗∗ indicates a p value < 0.01 versus H2O2 group.
The potential role of ROS in Bax and Bcl-2 activation has been indicated in multiple cell death signaling [16, 17]. Bax activation in inducing the cell death by ROS is the critical initiators. Bcl-2 family proteins acts as the gate keepers of cell death at mitochondria, which plays a critical role in regulating Bax induced mitochondrial permeability. As shown in Figures 5(c) and 5(d), Bcl-2 was downexpressed, and the expression of Bax was upregulated after the H2O2 treated in HepG2 cells. GAS pretreatment group upregulated the expression of Bcl-2 and suppressed the Bax expression with a dose-effect relationship. These results demonstrated that GAS inhibited H2O2-induced apoptosis through inhibition of Bcl-2 dependent cell death pathways.
4. Discussion
In our study, HepG2 cells were used as a cellular model to investigate the effects of GAS on the antioxidant defense systems. We had shown that H2O2 markedly decreased the viability of HEPG2 cells, whereas pretreatment with GAS significantly inhibited cell injury, as demonstrated by MTT assay. We also detected content of AST, ALT, and MDA, with and without H2O2; our results indicated that H2O2 could induce HEPG2 cells damage, which led to the increase of AST, ALT, and MDA and reduction of SOD activity. Oxidative stress caused by ROS is responsible for a wide variety of cellular damage and is the most validated mechanism of secondary injury [18]. Following oxidative stress, the overproduction of ROS and subsequently the depletion of antioxidants resulted in the total breakdown of the endogenous antioxidant defense mechanisms, culminating in failure to protect cells from oxidative damage. Among biomarkers of oxidative stress, MDA and SOD are known as two sensitive indicators [19]. MDA is the end product of lipid peroxidation [20] and MDA levels reflect the extent of cell damage due to oxidative stress. H2O2 may induce the generation of ROS at mitochondria which has been widely used as a model exogenous oxidative stress mediated experiment in hepatocellular apoptosis [21]. GAS pretreatment effectively protected HEPG2 cells from H2O2-induced damage. GAS increased the activity of SOD, decreased the level of ALT, AST, and MDA, and inhibited the apoptosis induced by H2O2.
Apoptosis may be activated by the intrinsic or by the extrinsic pathway [22]. Caspases are a group of aspartate specific cysteine protease, which plays a key role in regulating the apoptosis induced by different kind of stimuli including oxidative stress [23]. Functionally, caspase-3 is an important effector in the apoptotic process, and caspase-9 is an initiator of caspase-3 in the mitochondria-dependent pathway [24]. Treatment of H2O2 to the HEPG2 cells increased the caspase-9 expression in cells, which indicated that the mitochondrial pathway plays an important function in H2O2 induced apoptosis in cells. In our study, H2O2 upregulated the expression of caspase-3 and caspase-9 in HepG2 cells treated with H2O2; it indicated that the apoptosis induced by H2O2 through activation of caspases cascade. Bcl-2 proteins are major regulators of mitochondrial cytochrome c and caspases activation. It plays an important role in the regulation of cell apoptosis [25]. This family contains both proapoptotic and antiapoptotic proteins (Bcl-2 and Bcl-XL). Bcl-2 is an important cellular component which can protect against apoptotic cell death. Bax proteins were confirmed that could promote apoptosis. Our study proved that GAS reversed Bax upexpression and Bcl-2 downexpression and suppressed the activity of caspase-9 and caspase-3 in HepG2 cells after exposure to H2O2. It could be concluded that cell death evoked by H2O2 is regulated by Bcl-2 family proteins; Bcl-2 downexpression leads to the release of cytochrome c from the damaged mitochondria, which then binds to the adaptor molecule APAF-1 and an inactive “initiator” caspase, procaspase-9, within a multiprotein complex called the apoptosome. This leads to the activation of caspase-9, which then triggers a cascade of caspases activation (caspase-3 and caspase-7) resulting in the morphological and biochemical changes associated with apoptosis. The Bcl-2 upexpression of GAS may be the key mechanism for antiapoptosis induced by ROS in HepG2 cells.
In summary, the present study shows that H2O2 can induce HepG2 cells injury and induce cells apoptosis. GAS protects human HepG2 cells against H2O2-induced oxidative stress, cells apoptosis, ROS activity, and activities of caspase-9 and caspase-3. GAS also can regulate the expression of Bcl2 and Bax. GAS-mediated protection can be conferred by one or more of the following mechanisms: GAS could reduce the oxidative stress injury. Second, GAS could attenuate apoptosis through inhibiting the subsequent biochemical changes in the Bcl-2 apoptotic pathway. These data help explain the protective action of GAS against cell injuries involving the mitochondrial pathway.
Acknowledgments
This work was funded by National Natural Science Foundation of China (nos. 81260650, 81560685, and 81260615), Natural Science Foundation of Inner Mongolian (no. KJT14BS805), Youth Science and Technology Fellowship Program of Inner Mongolia Educational Department (no. JYT14GNYC03), and Inner Mongolia Department of Education Science Projects (no. JYT14133).
Disclosure
The authors are responsible for the content and writing of this article.
Competing Interests
The authors report no conflict of interests.
References
- 1.Kong K. W., Mat-Junit S., Aminudin N., et al. Protective effects of the extracts of barringtonia racemosa shoots against oxidative damage in hepg2 cells. PeerJ. 2016;4 doi: 10.7717/peerj.1628.e1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y., Yuan D., Yao W., et al. Hyperglycemia aggravates hepatic ischemia reperfusion injury by inducing chronic oxidative stress and inflammation. Oxidative Medicine and Cellular Longevity. 2016;2016:16. doi: 10.1155/2016/3919627.3919627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voltan R., Secchiero P., Casciano F., Milani D., Zauli G., Tisato V. Redox signaling and oxidative stress: cross talk with TNF-related apoptosis inducing ligand activity. The International Journal of Biochemistry & Cell Biology. 2016 doi: 10.1016/j.biocel.2016.09.019. [DOI] [PubMed] [Google Scholar]
- 4.Martín D., Salinas M., Fujita N., Tsuruo T., Cuadrado A. Ceramide and reactive oxygen species generated by H2O2 induce caspase-3-independent degradation of Akt/protein kinase B. Journal of Biological Chemistry. 2002;277(45):42943–42952. doi: 10.1074/jbc.m201070200. [DOI] [PubMed] [Google Scholar]
- 5.Dumont A., Hehner S. P., Hofmann T. G., Ueffing M., Dröge W., Schmitz M. L. Hydrogen peroxide-induced apoptosis is CD95-independent, requires the release of mitochondria-derived reactive oxygen species and the activation of NF-κB. Oncogene. 1999;18(3):747–757. doi: 10.1038/sj.onc.1202325. [DOI] [PubMed] [Google Scholar]
- 6.Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury. Journal of Gastroenterology and Hepatology. 2000;15(7):718–724. doi: 10.1046/j.1440-1746.2000.02207.x. [DOI] [PubMed] [Google Scholar]
- 7.Tribble D. L., Aw T. Y., Jones D. P. The pathophysiological significance of lipid peroxidation in oxidative cell injury. Hepatology. 1987;7(2):377–386. doi: 10.1002/hep.1840070227. [DOI] [PubMed] [Google Scholar]
- 8.Cole K. K., Perez-Polo J. R. Poly(ADP-ribose) polymerase inhibition prevents both apoptotic-like delayed neuronal death and necrosis after H2O2 injury. Journal of Neurochemistry. 2002;82(1):19–29. doi: 10.1046/j.1471-4159.2002.00935.x. [DOI] [PubMed] [Google Scholar]
- 9.Englert R. P., Shacter E. Distinct modes of cell death induced by different reactive oxygen species: amino acyl chloramines mediate hypochlorous acid-induced apoptosis. The Journal of Biological Chemistry. 2002;277(23):20518–20526. doi: 10.1074/jbc.m200212200. [DOI] [PubMed] [Google Scholar]
- 10.Szabo G., Wands J. R., Eken A., et al. Alcohol and hepatitis C virus-interactions in immune dysfunctions and liver damage. Alcoholism: Clinical and Experimental Research. 2010;34(10):1675–1686. doi: 10.1111/j.1530-0277.2010.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X., Lemastersn J. J. Translocation of iron from lysosomes to mitochondria during ischemia predisposes to injury after reperfusion in rat hepatocytes. Free Radical Biology and Medicine. 2013;63:243–253. doi: 10.1016/j.freeradbiomed.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhogal R. H., Weston C. J., Curbishley S. M., Adams D. H., Afford S. C. Autophagy: a cyto-protective mechanism which prevents primary human hepatocyte apoptosis during oxidative stress. Autophagy. 2012;8(4):545–558. doi: 10.4161/auto.19012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hafez M. M., Al-Shabanah O. A., Al-Harbi N. O., et al. Association between paraoxonases gene expression and oxidative stress in hepatotoxicity induced by CCl4 . Oxidative Medicine and Cellular Longevity. 2014;2014:12. doi: 10.1155/2014/893212.893212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeda K. Glycyrrhizin injection therapy prevents hepatocellular carcinogenesis in patients with interferon-resistant active chronic hepatitis C. Hepatology Research. 2007;37(2):S287–S293. doi: 10.1111/j.1872-034x.2007.00199.x. [DOI] [PubMed] [Google Scholar]
- 15.Park J.-J., Lim K.-H., Baek K.-H. Annexin-1 regulated by HAUSP is essential for UV-induced damage response. Cell Death & Disease. 2015;6(2) doi: 10.1038/cddis.2015.32.e1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang S., Liu Q., Liu Y., Qiao H., Liu Y. Zerumbone, a Southeast Asian ginger sesquiterpene, induced apoptosis of pancreatic carcinoma cells through p53 signaling pathway. Evidence-Based Complementary and Alternative Medicine. 2012;2012:8. doi: 10.1155/2012/936030.936030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yodkeeree S., Sung B., Limtrakul P., Aggarwal B. B. Zerumbone enhances TRAIL-induced apoptosis through the induction of death receptors in human colon cancer cells: evidence for an essential role of reactive oxygen species. Cancer Research. 2009;69(16):6581–6589. doi: 10.1158/0008-5472.can-09-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu J.-J., Wang Y.-L. Propofol attenuation of hydrogen peroxide-mediated oxidative stress and apoptosis in cultured cardiomyocytes involves haeme oxygenase-1. European Journal of Anaesthesiology. 2008;25(5):395–402. doi: 10.1017/S0265021508003542. [DOI] [PubMed] [Google Scholar]
- 19.Zhu S., Stavrovskaya I. G., Drozda M., et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417(6884):74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 20.Torun A. N., Kulaksizoglu S., Kulaksizoglu M., Pamuk B. O., Isbilen E., Tutuncu N. B. Serum total antioxidant status and lipid peroxidation marker malondialdehyde levels in overt and subclinical hypothyroidism. Clinical Endocrinology. 2009;70(3):469–474. doi: 10.1111/j.1365-2265.2008.03348.x. [DOI] [PubMed] [Google Scholar]
- 21.Zorov D. B., Filburn C. R., Klotz L. O., et al. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. Journal of Experimental Medicine. 2000;192(7):1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryter S. W., Hong P. K., Hoetzel A., et al. Mechanisms of cell death in oxidative stress. Antioxidants and Redox Signaling. 2007;9(1):49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- 23.Earnshaw W. C., Martins L. M., Kaufmann S. H. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annual Review of Biochemistry. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 24.Jiang B., Xiao W., Shi Y., Liu M., Xiao X. Heat shock pretreatment inhibited the release of Smac/DIABLO from mitochondria and apoptosis induced by hydrogen peroxide in cardiomyocytes and C2C12 myogenic cells. Cell Stress and Chaperones. 2005;10(3):252–262. doi: 10.1379/csc-124r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugioka R., Shimizu S., Funatsu T., et al. BH4-domain peptide from Bcl-xL exerts anti-apoptotic activity in vivo . Oncogene. 2003;22(52):8432–8440. doi: 10.1038/sj.onc.1207180. [DOI] [PubMed] [Google Scholar]