

Abstract
Quantifying confocal images to enable location of specific proteins of interest in three-dimensional (3D) is important for many tissue engineering (TE) applications. Quantification of protein localization is essential for evaluation of specific scaffold constructs for cell growth and differentiation for application in TE and tissue regeneration strategies. Although obtaining information regarding protein expression levels is important, the location of proteins within cells grown on scaffolds is often the key to evaluating scaffold efficacy. Functional epithelial cell monolayers must be organized with apicobasal polarity with proteins specifically localized to the apical or basolateral regions of cells in many organs. In this work, a customized program was developed using the LabVIEW platform to quantify protein positions in Z-stacks of confocal images of epithelial cell monolayers. The program's functionality is demonstrated through salivary gland TE, since functional salivary epithelial cells must correctly orient many proteins on the apical and basolateral membranes. Bio-LabVIEW Image Matrix Evaluation (Bio-LIME) takes 3D information collected from confocal Z-stack images and processes the fluorescence at each pixel to determine cell heights, nuclei heights, nuclei widths, protein localization, and cell count. As a demonstration of its utility, Bio-LIME was used to quantify the 3D location of the Zonula occludens-1 protein contained within tight junctions and its change in 3D position in response to chemical modification of the scaffold with laminin. Additionally, Bio-LIME was used to demonstrate that there is no advantage of sub-100 nm poly lactic-co-glycolic acid nanofibers over 250 nm fibers for epithelial apicobasal polarization. Bio-LIME will be broadly applicable for quantification of proteins in 3D that are grown in many different contexts.
Keywords: : tissue engineering, image processing, nanofibers, LabVIEW, salivary gland development
Introduction
In the field of tissue engineering (TE), many researchers are utilizing the properties of synthetic materials to trigger a response in the regeneration or development of damaged tissues.1–3 Synthetic polymeric scaffolds are used in a bottom-up approach to initiate the formation of a healthy functional tissue in vitro that can later be used for transplantation.4 The creation of nanoscale and microscale environments as seen in vivo is difficult to achieve, and successful tissue formation is challenging to verify without proper characterization methods. Although there are many biological assays that can be used to establish scaffold performance, to be able to quantitatively evaluate cell structure and protein localization in three-dimensional (3D) images simultaneously would be invaluable.
Quantification of the spatial distribution of proteins in 3D requires optical microscopy. Other techniques that are useful for quantification of protein abundance, such as western blot analysis, and mass spectrometry, cannot provide such information. Western blot uses sodium dodecyl sulfate–polyacrylamide gel electrophoresis to separate proteins by charge and labeled antibodies to detect specific proteins of interest.5 Mass spectrometry measures the amount and type of molecules in samples by the production of gas-phase ions with varying charge-to-mass ratios.6 Although informative, both assays only distinguish the presence of selected proteins and not their spatial configuration within the tissues. On the other hand, laser scanning confocal microscopy (LSCM) is frequently used to investigate how well the growing cell population in a tissue construct mimics the in vivo tissue environment. With LSCM, researchers are able to localize proteins within 3D samples using antibodies labeled with fluorescent markers to detect both their presence and their localization. LSCM allows image acquisition of a volume of space amenable to rendering a 3D image, but cannot be used to quantify the actual amount of fluorescence detected and how much the respective protein is localized without other supportive processing platforms.
Image processing is currently the methodology that is rapidly increasing in popularity to quantitatively interpret images obtained from LSCM. In particular, ImageJ and IMARIS are frequently used to analyze the dimensional stacks of images. ImageJ is a public domain image processing platform developed at the National Institute of Health to perform a number of different forms of image processing.7 Such processing parameters used for investigation include cellular counts, histograms of fluorescent intensity, cellular heights/widths, and detection of protein interactions using channel overlays. Fluorescence signal distribution is converted into binary images that are analyzed for specific protein location.7,8 After this conversion is performed, the user is able to count cells in the image frame or manually trace images for protein localization, and cell heights and widths.
Soscia et al. analyzed cell–cell junctions using confocal X-Z projection images and the ImageJ freehand tool. Lines were drawn across cell membranes at both the apical and basal ends; quantified data are produced for fluorescent pixels that fall in the same plane as the line.9 However, this analysis is limited to one-dimensional or two-dimensional (2D) frames and freehand tools facilitate user bias and are low throughput. Additionally, the segmentation of 3D information into only 2D image analysis loses critical information. IMARIS (Bitplane) is a stand-alone software module that takes microscopy datasets and reconstructs them into 3D and four-dimensional (4D) images. Four-dimensional images allow the review of information in moving 3D images with the fourth dimension being time. This platform is beneficial for visualization purposes, but is not ideal for providing detailed protein detection with respect to a synthetic scaffold's topography. A software platform with the capability to perform 3D data analysis of the fluorescence information gained by LSCM in a low bias and high-throughput manner is critical in many TE applications.
One needs to develop a custom-based software to accomplish protein localization in 3D samples. Some methods for morphology quantification have been demonstrated using MATLAB™. Rytlewski et al. established computational quantification of 3D structures in fibrin-based in vitro models.10 In this study, they compiled confocal Z-stacks and assembled the information into 3D models for statistical analysis using MATLAB software. The morphology parameters include volume, vascular network length, number of networks, and degree of network branching. Also, Xavier et al. created a MATLAB-based software toolbox capable of analyzing confocal images of biofilms and determining area profiles of microbial colonization, biovolume, colonization fraction, and average height of microcolonies.11 Kozlowski and Weimer also used MATLAB as a platform to obtain automated image analysis when calculating morphologies to describe the shape of immune cells of the brain, microglia.12
Correlating the structure and functions of cells with various scaffolds to judge how well they perform against one another is a significant area of interest. One tissue of considerable importance in bioengineering applications is salivary glands. Salivary glands contain multiple epithelial populations.13 Saliva is secreted by acinar cells, whereas ductal cells modify it and transport saliva to the mouth.14 Tight junctions are proteins present in these epithelial cell monolayers that aid in barrier functions and the direction of saliva secretion. When making a bioinspired scaffold, it is important that these proteins organize in an apically localized fashion with respect to the formation of the epithelial cell monolayer.15 This would mean that after the cell monolayer is mature, tight junction proteins localize toward the apical membrane, forming barriers against small molecules passing between adjacent cells and participating in creation of a saliva flow gradient.16
Physical material size has been shown to be very important when trying to develop a specific tissue of interest.17 Studies have shown that material feature size heavily influences the morphological and functional development of cells.18 Nanofiber scaffolds have been engineered to resemble the basement membrane for growth of salivary gland cell lines. Jean-Gilles et al. and Sequiera et al. observed a more in vivo-like morphology of submandibular immortalized mouse salivary gland epithelial (SIMS) cells cultured on nano-sized fibers (∼250 nm diameter) rather than micro-sized fiber mats.14,19 Cantara et al. noted that poly lactic-co-glycolic acid (PLGA) nanofibers promote, but are insufficient to support assembly of tight junctions in salivary gland cell lines.16
One known chemical requirement for epithelialization is the basement membrane protein, laminin. This protein is critical in assembly and organization for polarized epithelial morphogenesis.20 Cantara et al.16 noted that laminin-conjugated PLGA nanofiber scaffolds more effectively promoted apical localization of tight junction proteins than did PLGA nanofibers; however, this difference was not quantified. The natural extracellular matrix is composed of fibrous proteins having diameters ranging from 50 to 150 nm.21 Since previous studies did not investigate nanofiber scaffolds having diameters in this range, further investigation of how cellular morphology changes at even smaller fiber diameters that better mimic the structure of basement membrane proteins, such as sub-100 nm fibers, was conducted.
In this study, a unique image processing platform amenable to the investigation of functional scaffolds for research in TE is described. Bio-LabVIEW Image Matrix Evaluation (Bio-LIME) is automated and minimizes user bias. Confocal images of salivary gland cell lines grown on PLGA nanofiber scaffolds that were chemically conjugated to laminin and promote apical localization of tight junctions were compared with unconjugated PLGA nanofiber scaffolds as a proof of concept study for Bio-LIME. Additionally, confocal images of 250 nm versus sub-100 nm diameter PLGA nanofiber scaffolds were analyzed in Bio-LIME to assess whether sub-100 nm fibers provide an additional benefit to salivary epithelial cell apicobasal polarization. The information contained in confocal Z-stack fluorescent images was processed into individual pixel quantities and transformed into quantitative data. Bio-LIME establishes a method for the full characterization of scaffold performance for many research applications in TE, with analysis ranging from cell heights/widths, cell count, and protein localization to scaffold thickness.
Materials and Methods
Materials
PLGA with a lactic-to-glycolic acid ratio of 85:15 and a molecular weight of 95,000 Da was purchased from DURECT LACTEL. Hexafluoroisopropanol (HFIP) and laminin-111 (Cat No. L2020) were obtained from Sigma-Aldrich. Dulbecco's phosphate-buffered saline (PBS), Dulbecco's modified Eagle's medium (DMEM), and fetal bovine serum (FBS) were obtained from Invitrogen. Sulforhodamine B (SRB) for fiber staining (Cat. No. S-1307) was purchased from Life Technologies. VECTABOND Reagent was purchased from Vector Laboratories. Anti-E-cadherin (Cat. No. 610182) was from BD Biosciences. 4′,6-Diamidino-2-phenylindole (DAPI for nuclei staining), and AlexaFluor-488 phalloidin (to stain F-actin) were purchased from Life Technologies. Rabbit anti-Zonula occludens-1 (ZO-1) was purchased from ThermoScientific (Cat No. 402200). 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC; Cat No. 39391), N-hydroxysuccinimide (NHS) (Cat No. 130672), and 2-(N-morpholino) ethanesulfonic acid (MES) solution (Cat No. M1317) were purchased from Sigma-Aldrich. Alexa Fluor-647 AffiniPure F(ab′)2 Fragment donkey anti-rabbit immunoglobulin G secondary antibody (Cat No. 711606152) and donkey serum were from Jackson ImmunoResearch. Fluoro-Gel mounting medium was acquired from EMS, and p-phenylenediamine (PPD; Cat. No. P6001) was from Sigma-Aldrich.
Cell maintenance
SIMS cells22 were cultured and expanded in tissue culture-treated plastic flasks with DMEM containing 10% FBS and 1% penicillin–streptomycin. Cell cultures were incubated with 5% CO2 at 37°C.
Cell seeding
Before cell seeding, all scaffolds were UV sterilized for 1 h. SIMS cells were trypsinized, centrifuged into a pellet, and resuspended in fresh media (DMEM, 10% FBS, 1% penicillin–streptomycin). The cell solution was passed through a 70 μm cell strainer to further break up cell clumps. Cells were seeded on scaffolds at a concentration of 6.0 × 104 cells/mL and cultured for 6 days.
Preparation of nanofibers
The nanofibrous mats were fabricated by means of electrospinning on glass coverslips coated with VECTABOND reagent, as reported previously.19 Two batches of fibers were electrospun using 8% w/w and 2% w/w PLGA in HFIP solvent, to yield 247 ± 39 nm fiber diameters (250 nm fibers) and 58 ± 39 nm fiber diameters (sub-100 nm fibers), respectively (Fig. 1A, B). One percent w/w NaCl was added to each solution to enhance the conductivity of the solution. Eight percent PLGA solution was fed at a rate of 3 μL/min into 25-G needle. A potential of 10 kV was applied between the needle and the grounded collector plate, which was 15 cm below the needle. The electrospinning parameters had to be reduced to electrospin 2% PLGA solution to yield the fibers of lower diameter. The potential was reduced to 6 kV, flow rate to 0.8 μL/min, and distance to 5 cm. Both types of fibers were spun at an ambient temperature and a relative humidity of 30%. After electrospinning, the fiber mats were placed into a desiccating cabinet to remove the remaining solvent and preserve from fluctuating temperature and humidity within the laboratory. All PLGA solutions contained 1% SRB dye as a source of detection using confocal microscopy. Fiber scaffolds were analyzed using a scanning electron microscope, where images were collected and analyzed using ImageJ for average fiber diameters (Fig. 1C, D). Average fiber diameter and standard deviation reflects measurements taken from 100 different fibers from three separate electrospun samples.
FIG. 1.
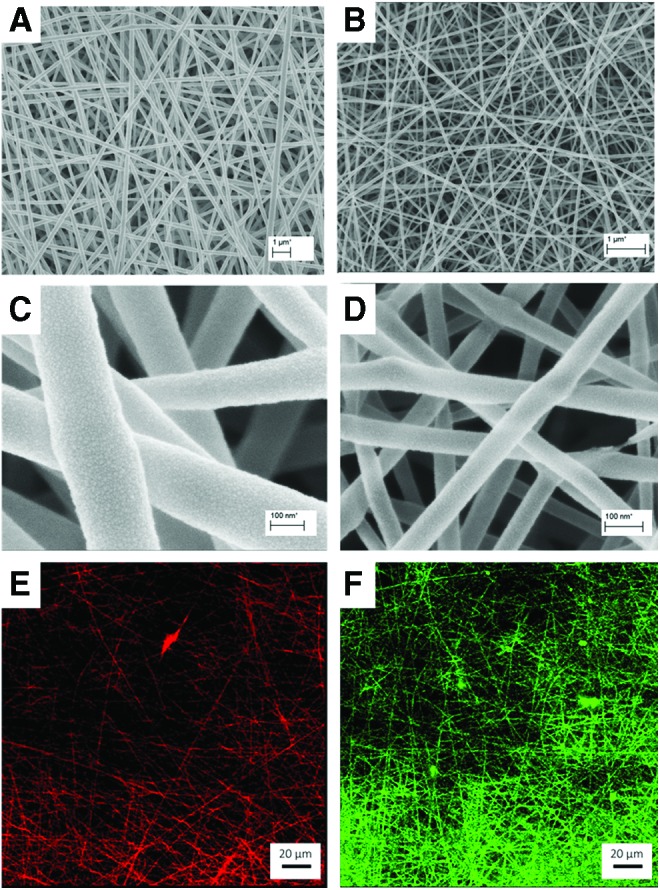
Diameter characterization and conjugation confirmation of scaffolds used in this study. (A) and (B) low magnification SEM images of 250 nm (247 ± 39 nm) and sub-100 nm (58 ± 39 nm) diameter PLGA nanofibers, respectively. Scale, 1 μm. (C) and (D) high-magnification SEM images of 250 nm and sub-100 nm diameter PLGA nanofibers, respectively. Scale, 100 nm. (E) Fluorescence microscopy of unmodified 250 nm diameter PLGA nanofibers. (F) Confocal X-Y images of laminin-111-modified (green) 250 nm diameter PLGA nanofibers. Scale, 20 μm. SEM, scanning electron microscopy; PLGA, poly lactic-co-glycolic acid. Color images available online at www.liebertpub.com/tec
Nanofiber modification
Laminin-111 conjugation was performed using EDC-NHS chemistry.16,23 Scaffolds were placed in an activating solution for 1 h at room temperature to activate the carboxyl groups on PLGA nanofibers.24 The activating solution consisted of 4 mM EDC, 100 mM NHS, and 10 mM MES buffer in deionized water, as reported previously.16 Next, scaffolds were incubated in 10 mg/mL laminin-111 solution in cold 1 × PBS (pH 7.4) at 4°C overnight. Samples were subsequently washed twice with PBS. To confirm attachment of laminin-111, fiber scaffolds were immunostained with anti-laminin antibody (secondary antibody, AlexaFluor-488 anti-rabbit) and observed with confocal microscopy (Leica TCS SP5) (Fig. 1E, F).
Immunocytochemistry
All samples for confocal microscopy were subject to immunocytochemistry to detect cell nuclei, nanofibers, the tight junction protein, ZO-1, and actin. Samples, kept on ice for 20 min, were fixed in 4% paraformaldehyde and 5% sucrose in PBS. Samples were washed twice in PBS-Tween (PBS-T), permeabilized for 15 min in 0.1% Triton X-100, and then blocked for 2 h in PBS-T with 20% donkey serum. The primary antibody solution was incubated overnight at 4°C on a rocker and prepared as follows: a rabbit anti-ZO-1 (Cat No. 402200) was diluted 1:400 in a PBS-T-3% bovine serum albumin (BSA) solution. Samples were washed four times for 10 min with PBS-T. The secondary antibody solution was rocked at room temperature for 2 h and prepared as follows: 1:200 DAPI (to detect nuclei), 1:250 AlexFluor-647donkey anti-rabbit (to detect ZO-1), and a 1:400 dilution of AlexFluor-488 phalloidin (to detect actin) added to a PBS-T-3% BSA solution. The samples were again washed four times for 10 min each, and mounted on glass slides using Fluoro-gel mounting media with 1:100 PPD antifade solution. Samples were sealed with clear nail polish and dried before imaging.
Confocal microscopy
LSCM was performed using a Leica SP5 microscope (Leica Microscope Systems), and images were acquired with a 63 × oil-immersion objective and 512 × 512 image resolution. Z slices of 0.5 μm in thickness were acquired for all samples. Three-dimensional reconstructions (shown in Fig. 3 below) were constructed using IMARIS software (Bitplane).
FIG. 3.

Overview of Bio-LIME capabilities for quantification of protein localization in three-dimensional image datasets. (A) An example of an XY maximum projection image of cells seeded on a nanofiber scaffold. Nanofibers (red), ZO-1 (cyan), cell nucleus (blue), actin cytoskeleton (green). (B) X-Z cross-section of a stack of X confocal images, immunostained as in (A). (C) Schematic of how the program quantifies protein localization from confocal Z-stack images. (D) Bio-LIME final analysis, left to right: nucleus height and width, cell count, protein localization, and average cell monolayer heights. Three-dimensional reconstruction preformed using IMARIS software. Bio-LIME, Bio-LabVIEW Image Matrix Evaluation. Color images available online at www.liebertpub.com/tec
Image processing using Bio-LIME
Each confocal stack was converted into a tagged image file format (.tiff) in the Leica software. The .tiff images of all channels, including, DAPI, ZO-1, actin, and scaffold fluorescence (SRB), were loaded into Bio-LIME. All Z-stack images were saved in numerical order between channels: 0, 1, 2, and 3, respectively.
Statistical analysis
One-way analysis of variance followed by unpaired t-tests were carried out to identify significant differences between data sets within each experiment in GraphPad Prism 6 software. A value of p ≤ 0.05 was considered to be statistically significant.
Results and Discussion
To illustrate the morphological differences between cells grown on various scaffolds and the importance of characterization by additional image processing, PLGA (85:15) nanofibers of sub-100 nm and 250 nm in diameter and laminin-111 surface-functionalized 250 nm PLGA nanofibers were analyzed.
To identify statistical relevance from qualitative observations, a platform capable of interpreting a 3D environment is necessary. Taking a 2D snapshot of a 3D X-Z slice is not a precise way to understand an entire surrounding area. For example, sampling individual X-Z slices with each containing only 6–10 cells in a frame leads to a large source of error when there are ∼300 cells in each 63 × confocal Z-stack. These 300 cells are also interacting with neighboring cells, which makes it important to observe the complete volume of space. Quantifying an entire 3D image allows for a more robust and substantiated conclusion.
Bio-LIME was developed to quantify confocal images of tissue constructs. For proof of concept, typical images expected to be seen per channel were produced in Microsoft Paint (Fig. 2). Case 1 shows a scenario where the cell nucleus and protein fluorescence are detected above the absolute bottom of the engineered scaffold. One can note that the protein fluorescence is detected in the most favorable spot, or localized apically. Case 2 depicts an event, where the cell nucleus and protein fluorescence are again detected above the scaffold base; however, the protein fluorescence is located laterally through the apical and basal ends with respect to the cell nucleus. Case 3, is a nonideal case, where the protein fluorescence, due to noise within the samples, is located below the established bottom of the engineered scaffold. These cases were used as a starting point to see how well Bio-LIME detects binary images for cell count, scaffold thickness, cell widths/heights, and protein localization. Case 1 would be a scenario, where the percent localization of fluorescence expressed at the top 25% of the cell nucleus and above would be 100%, whereas in Case 2, the percent fluorescence detected at the top 25% and above would be 25%.
FIG. 2.

Calibration images produced in Microsoft paint to portray the possible scenarios of protein localization in cells grown on different scaffolds. X and Y axes refer to the number of pixels in frame and Z-stack number, respectively. (Case 1) protein is localized to apical end with respect to cell fluorescence, (Case 2) protein is laterally expressed throughout the cell with respect to DAPI fluorescence, and (Case 3) protein that is expressed throughout the samples, even passed the lower limit of the fiber mat. The absolute bottom of the fiber mat is represented in the highlighted portion of the scaffold image (white), nanofiber mat (red), cell nucleus (blue), and tight junction protein, ZO-1 (cyan). DAPI, 4′,6-diamidino-2-phenylindole; ZO-1, zonula occludens-1. Color images available online at www.liebertpub.com/tec
Confocal Z-stack images are collected and processed using all X-Z/Y-Z (Fig. 3A, B) projection data to measure actin height, nucleus height/width, and protein localization (Fig. 3D). Bio-LIME begins by identifying the base of the scaffold. It measures the thickness of the scaffold by pixels and scores the lower-most pixel portion of the scaffold as the absolute bottom of the overall volume of space. This feature allows the user to be able to vertically count pixels in every other channel based on the scaffold and the continuous path of fluorescence while moving through Z height stacks (Fig. 3C). For these experiments, all cells were oriented above the scaffold base in a monolayer formation. Figure 4A shows a maximum projection X-Y image of the whole acquired confocal image. Figure 4B and C shows X-Z/Y-Z cross-sections for each channel (scaffold, nucleus, and protein) with the addition of the highlighted scaffold base so that users can see where fluorescence for each channel is detected with respect to the bottom.
FIG. 4.

Schematic showing the (A) Maximum projection, X and Y axes are number of pixels per line and lines per frame, respectively. (B) Y-Z cross-sections and (C) X-Z cross-sections that users can see for each channel while processing. The base of the scaffold is detected and highlighted (dotted lines at the bottom with red arrows). X and Z axes refer to the number of pixels in frame and Z-stack number, respectively. This is the same for the Y-Z axes as well. Color images available online at www.liebertpub.com/tec
After the scaffold thickness and lower limits are established, users can analyze cell counts in the image frame by converting fluorescence into binary images based on the user-defined threshold value. This is done to remove the low-intensity noise pixels (Fig. 5A). In thresholded images, the program looks for connected regions and counts them as individual cells. To block out any cell debris or cell clumps from being calculated incorrectly, a minimum and maximum cell area needs to be established by the user. Using a histogram, the user is able to collect information on the average size of debris, single cells, and cell clumps (Fig. 5B). Then, the regions with an area less than the established minimum can be discarded as debris and regions with the area above maximum are counted as multiple cells (Fig. 5C, D).
FIG. 5.

Image cleanup process flow diagram, demonstrating how single nuclei are identified. Images were produced in Microsoft paint and processed through Bio-LIME. (A) Raw X-Y image before cleanup. (B) Histogram developed based on the distribution of pixel areas found in the entire raw image. X and Y axes are pixel area and amplitude, respectively. (C) Debris cleaning step: based on the histogram the user can identify the objects that are smaller than a single nucleus. The user can input the minimum area and monitor the output. The output should resemble all objects that were greater than or equal to the minimum pixel area of a single nucleus. (D) Clump sorter step: using the histogram the user can see the distribution of all objects that are above a single nucleus pixel area. Once the maximum area is input into the program the user can observe the output. The output should be everything less than or equal to a size of a single nucleus. Red dotted circles indicate clumps that were sorted out into single cells. Debris cleaner and clump sorter are algorithms independent from one another, and can be done in any order. (E) After maximum and minimum pixel areas are analyzed, a final data image with the cell count and cell width is given. X and Y axes are number of pixels per line and lines per frame, respectively. Color images available online at www.liebertpub.com/tec
To find the maximum and minimum cell area, one can display a histogram of the areas of connected regions and observe the peaks at the typical cell size, as well as double, triple, quadruple etc. sizes (the latter is due to two, three, four, etc. cells located close together, so they are represented by one connecting region). After all of the processing is done, Bio-LIME prompts the user with the cell count and average nucleus width. An example of a final data image is shown in Figure 5E, where the resulting cell count was 13 nuclei in the frame. Conversion of pixel area into physical dimensions (microns) was performed using scalar information recorded during image capture. Once the cell count and average nucleus widths are complete, nucleus height and actin height analysis can be performed. This step is completed based on the already established scaffold base.
To determine average monolayer heights, Bio-LIME finds the z-distance between the lower boundary of the nucleus and the upper boundary of the actin (Fig. 6A). This function was incorporated because actin filaments tend to interact and redistribute apically with tight junction proteins in epithelial cell monolayers.25 Once the cell polarizes, the actin fluorescence is no longer distributed vertically from the bottom to the top of the cell, but rather is enriched at the apical end of the cell. The protein distribution is determined by specifying a pixel offset based on the established nuclei heights (Fig. 6B). Due to the spatial resolution of fluorescence in the Z direction, an apical localization of protein was assumed to be represented by the protein expression within the top 25% of nuclei and above the average height of the nucleus channel. The program then computes the total amount of pixels detected above and below the pixel offset in percentages. For the remainder of this article, pixels detected at or above the offset made at the top 25% of the average cell nuclei height will be designated as “apical,” and everything below that offset will be designated as “basolateral.”
FIG. 6.

Diagram of X-Z slices shown in the program. (A) For analysis of average monolayer heights, sections are used by collecting information from the actin and nuclear stains. The z-distance between them is how Bio-LIME computes the average cell monolayer heights. The arrows indicate the distance in between the top most actin fluorescence and the bottom most nuclear fluorescence. (B) For detection of protein localization, the scaffold and nuclear stains are used. The base of the scaffold is highlighted (dotted line, bottom) and the continuous vertical path of nuclear stain is collected as the nucleus height. The offset is found by taking the average nucleus height and creating a boundary (red) at the top 25% and above. All fluorescence in the protein channel above this boundary is defined as apical and fluorescence below is defined as basolateral. X and Y axes refer to the number of pixels in frame and Z-stack number, respectively. Color images available online at www.liebertpub.com/tec
Two applications required to determine scaffold performance were executed using Bio-LIME. The first application was comparing 250 nm PLGA fibers with and without the addition of laminin-111 covalently conjugated to the fiber surface. Laminin-111 is a protein found in the basement membrane of epithelial cells in vivo and is known for its role in polarizing tight junction proteins, including ZO-1 and occludin, as previously reported.16,26 The second application is the investigation of cell morphology on 250 nm diameter fibers and a more physiologically relevant fiber diameter of sub-100 nm. Since previous work suggested that 250 nm PLGA nanofiber scaffolds promote salivary epithelial cells apicobasal polarity,16 investigation of whether sub-100 nm fibers are more effective in promoting apicobasal polarity was performed. SIMS cells were cultured on the scaffolds for 6 days and stained for nuclei, actin, and ZO-1. Using confocal microscopy, Z-stack images were obtained and subsequently run through Bio-LIME. Samples with comparable cellular confluence levels were investigated further for cell morphology quantification.
When investigating the effect of cellular morphology with the addition of laminin-111 conjugation to 250 nm diameter PLGA nanofibers, SIMS cells were seeded on nanofiber scaffolds and stained to detect nuclei, actin, and ZO-1. The nuclear counts were determined after going through the cleanup steps for debris and clumps (Fig. 5A). Taken from a n = 3 set, PLGA scaffolds on average had 453 ± 41 cells and PLGA + laminin scaffolds had, on average, 439 ± 57 cells. Figure 7A and B shows representative maximum projections and X-Z slices assembled from confocal images for both scaffold types. Cells on the laminin-conjugated scaffolds exhibited increased actin heights and slightly increased nuclear widths; 4.77 and 6.21 μm, respectively (Fig. 7C). Cells grown on PLGA nanofiber scaffolds had average nuclear heights, widths, and actin heights of 2.54, 4.42, and 3.76 μm, respectively (Fig. 7C). Cells grown on the PLGA + laminin scaffolds also showed increased apical localization of tight junction protein ZO-1; 75.7 ± 4.54% (Fig. 7D). The PLGA nanofiber control displayed only 43.9 ± 1.72% of the total fluorescence at the apical end (Fig. 7D). These quantifications are consistent with previously published confocal image data.
FIG. 7.

Quantification of cell morphology and apicobasal polarization of cells grown on PLGA nanofiber scaffolds with or without conjugated laminin using Bio-LIME. Confocal maximum projection and X-Z slice of SIMS cells for 6 days on scaffolds. (A) PLGA scaffold, (B) PLGA + laminin scaffold. Scale bar, 10 μm. Nanofibers (red), DAPI (blue), ZO-1 tight junction protein (cyan), (C) SIMS cell morphology, (D) Localization of tight junction protein, ZO-1, of SIMS cells cultured on PLGA scaffolds, and PLGA + laminin scaffolds. ns, not significant; *p < 0.05, ***p < 0.001; unpaired t test. SIMS, submandibular immortalized mouse salivary gland epithelial. Color images available online at www.liebertpub.com/tec
When comparing SIMS cells grown on 250 nm PLGA scaffolds and sub-100 nm PLGA scaffolds, the average cell counts were 380 ± 37 and 363 ± 22, respectively. Figure 8A and B shows a representative maximum projection with a X-Z slice acquired from confocal images captured of cells grown on both scaffold types and labeled to detect nuclei, actin, and ZO-1 on labeled nanofibers. This analysis was acquired from a n = 4 set for each. For the 250 nm PLGA scaffolds, SIMS cell morphology for nucleus height, width, and actin height were 2.28, 4.81, and 3.78 μm, respectively (Fig. 8C). For the sub-100 nm PLGA scaffolds, the SIMS cell morphology was 2.12, 5.25, and 3.65 μm for nucleus height, width, and actin height (Fig. 8C). When determining the percent apical localization of the tight junction protein ZO-1, the 250 nm PLGA scaffold and sub-100 nm PLGA scaffolds showed 40.5 ± 3.31% and 38.7 ± 4.42%, respectively (Fig. 8D).
FIG. 8.

Comparison of cell morphology and apicobasal polarization of cells grown on 250 nm versus sub-100 nm PLGA nanofiber scaffolds using Bio-LIME. Confocal maximum projection and X-Z slice of SIMS cells cultured for 6 days on scaffolds. (A) 250 nm PLGA scaffold, (B) sub-100 nm PLGA scaffold. Scale bar, 10 μm. Nanofibers (red), DAPI (blue), ZO-1 tight junction protein (cyan). (C) SIMS cells morphology, (D) Protein localization analysis of cells cultured on 250 nm diameter PLGA scaffolds and sub-100 nm diameter PLGA scaffolds. ns, not significant, *p < 0.05; unpaired t test. Color images available online at www.liebertpub.com/tec
The average cell monolayer height was determined using actin staining. This method was based on prior work,9 where ImageJ and multiple X-Z projections were used to determine SIMS cell monolayer heights. The results obtained using the method described here is similar to the 4 μm monolayer heights of cells on PLGA scaffolds determined by Soscia et al.9 We report here that laminin-111-conjugated scaffolds showed increased localization of tight junction protein, ZO-1, compared with the control PLGA scaffolds, 75% and 43%, respectively. This quantification along with the visual identification of the ZO-1 stain is consistent with prior qualitative findings that laminin promotes tight junction protein apical localization16 and demonstrates the functionality of Bio-LIME to provide quantitative comparisons between cells grown on different scaffolds (Fig. 7). In the test case of the physical difference in fiber diameter, 250 nm versus sub-100 nm, the fiber size seemed to have little effect on cell morphology and tight junction localization. This conclusion was inferred by the visual inspection of fluorescent microscopy and confirmed from the quantification of protein distribution in the scaffolds (Fig. 8).
Conclusion
The ability to quantify protein localization in 3D from confocal images of tissue constructs was demonstrated through the use of an image processing platform developed from custom LabVIEW scripts. In the program's initial phase, Bio-LIME has provided a platform for analyzing the development of single cell monolayers for scaffold performance investigation. The results obtained from Bio-LIME and reported in this study support previous work, namely that laminin-111-conjugated PLGA nanofiber scaffolds promote cell polarity while providing a quantitative comparison of the entire sample in 3D. Additionally, Bio-LIME analysis suggested that sub-100 nm nanofibers provide no significant benefit on cell height or apicobasal polarization in comparison with 250 nm nanofibers in salivary epithelial cells. The quantification of confocal images reported here allows the investigation of the performance of a scaffold used for TE by quantifying differences in cellular morphology and protein localization that are critical for evaluating scaffold efficacy in promoting tissue assembly. Bio-LIME software not only reduces user bias, but also allows quantification of confocal images in a high-throughput manner.
Acknowledgments
This work was supported by the University at Albany, SUNY, NIH R01DE022467 (to M.L. and J.C.), NIH C06 RR015464 (to University at Albany, SUNY), and NSF DV10922830 (to J.C.). The authors thank Daniel Malamud, NYU, for the gift of SIMS cells.
Disclosure Statement
No competing financial interests exist.
References
- 1.Chen Q., Liang S., and Thouas G.A. Elastomeric biomaterials for tissue engineering. Prog Polym Sci 38, 584, 2013 [Google Scholar]
- 2.Aframian D.J., Cukierman E., Nikolovski J., Mooney D.J., Yamada K.M., and Baum B.J. The growth and morphological behavior of salivary epithelial cells on matrix protein-coated biodegradable substrata. Tissue Eng 6, 209, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Stachewicz U., Qiao T., Rawlinson S.C.F., et al. 3D imaging of cell interactions with electrospun PLGA nanofiber membranes for bone regeneration. Acta Biomater 27, 88, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Chen F., and Liu X. Advancing biomaterials of human origin for tissue engineering. Prog Polym Sci 53, 86, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang P.-C., and Mahmood T. Western blot: technique, theory, and trouble shooting. N Am J Med Sci 4, 429, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gingras A.C., Aebersold R., and Raught B. Advances in protein complex analysis using mass spectrometry. J Physiol 563(Pt 1), 11, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bankhead P. Analyzing fluorescence microscopy images with ImageJ. ImageJ 1, 195, 2014 [Google Scholar]
- 8.Gonzales R.C., and Woods R.E. Digital Image Processing. Reading, Massachusetts: Addison-Wesley, 1993. [Google Scholar]
- 9.Soscia D.A., Sequeira S.J., Schramm R.A., et al. Salivary gland cell differentiation and organization on micropatterned PLGA nanofiber craters. Biomaterials 34, 6773, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rytlewski J.A., Geuss L.R., Anyaeji C.I., Lewis E.W., and Suggs L.J. Three-dimensional image quantification as a new morphometry method for tissue engineering. Tissue Eng Part C Methods 18, 507, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xavier J.B., White D.C., and Almeida J.S. Automated biofilm morphology quantification from confocal laser scanning microscopy imaging. Water Sci Technol 47, 31, 1991 [PubMed] [Google Scholar]
- 12.Kozlowski C., and Weimer R.M. An automated method to quantify microglia morphology and application to monitor activation state longitudinally in vivo. PLoS One 7, e31814, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoo C., Vines J.B., Alexander G., Murdock K., Hwang P., and Jun H.-W. Adult stem cells and tissue engineering strategies for salivary gland regeneration: a review. Biomater Res 18, 9, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sequeira S.J., Soscia D.A., Oztan B., et al. The regulation of focal adhesion complex formation and salivary gland epithelial cell organization by nanofibrous PLGA scaffolds. Biomaterials 33, 3175, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters S.B., Nelson D.A., Kwon H.R., Koslow M., DeSantis K.A., and Larsen M. TGFβ signaling promotes matrix assembly during mechanosensitive embryonic salivary gland restoration. Matrix Biol 43, 109, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cantara S.I., Soscia D.A., Sequeira S.J., Jean-Gilles R.P., Castracane J., and Larsen M. Selective functionalization of nanofiber scaffolds to regulate salivary gland epithelial cell proliferation and polarity. Biomaterials 33, 8372, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X., Ding B., and Li B. Biomimetic electrospun nanofibrous structures for tissue engineering. Mater Today 16, 229, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teixeira A.I., Abrams G.A., Bertics P.J., Murphy C.J., and Nealey P.F. Epithelial contact guidance on well-defined micro- and nanostructured substrates. J Cell Sci 116(Pt 10), 1881, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jean-Gilles R., Soscia D., Sequeira S., et al. Novel modeling approach to generate a polymeric nanofiber scaffold for salivary gland cells. J Nanotechnol Eng Med 1, 31008, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manninen A. Epithelial polarity—generating and integrating signals from the ECM with integrins. Exp Cell Res 334, 337, 2015 [DOI] [PubMed] [Google Scholar]
- 21.Bhattarai N., Edmondson D., Veiseh O., Matsen F.A., and Zhang M. Electrospun chitosan-based nanofibers and their cellular compatibility. Biomaterials 26, 6176, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Laoide B.M., Courty Y., Gastinne I., Thibaut C., and Kellermann O. Immortalised mouse submandibular epithelial cell lines retain polarised structural and functional properties. J Cell Sci 109, 2789, 1996 [DOI] [PubMed] [Google Scholar]
- 23.Koh H.S., Yong T., Chan C.K., and Ramakrishna S. Enhancement of neurite outgrowth using nano-structured scaffolds coupled with laminin. Biomaterials 29, 3574, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Bai M.-Y., and Liu S.-Z. A simple and general method for preparing antibody-PEG-PLGA sub-micron particles using electrospray technique: an in vitro study of targeted delivery of cisplatin to ovarian cancer cells. Colloids Surf B Biointerfaces 117, 346, 2014 [DOI] [PubMed] [Google Scholar]
- 25.Fanning A.S., Jameson B.J., Jesaitis L.A., and Melvin J. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem 273, 29745, 1998 [DOI] [PubMed] [Google Scholar]
- 26.Ogawa M., Oshima M., Imamura A., et al. Functional salivary gland regeneration by transplantation of a bioengineered organ germ. Nat Commun 4, 1, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]