

Abstract
Pertussis toxin (PT) moves from the host cell surface to the endoplasmic reticulum (ER) by retrograde vesicular transport. The catalytic PTS1 subunit dissociates from the rest of the toxin in the ER and then shifts to a disordered conformation which may trigger its export to the cytosol through the quality control mechanism of ER-associated degradation (ERAD). Functional roles for toxin instability and ERAD in PTS1 translocation have not been established. We addressed these issues with the use of a surface plasmon resonance system to quantify the cytosolic pool of PTS1 from intoxicated cells. Only 3% of surface-associated PTS1 reached the host cytosol after 3 h of toxin exposure. This represented, on average, 38,000 molecules of cytosolic PTS1 per cell. Cells treated with a proteasome inhibitor contained larger quantities of cytosolic PTS1. Stabilization of the dissociated PTS1 subunit with chemical chaperones inhibited toxin export to the cytosol and blocked PT intoxication. ERAD-defective cell lines likewise exhibited reduced quantities of cytosolic PTS1 and PT resistance. These observations identify the unfolding of dissociated PTS1 as a trigger for its ERAD-mediated translocation to the cytosol.
INTRODUCTION
The AB toxins are composed of a catalytic A moiety and a cell-binding B moiety. Some AB toxins move by retrograde vesicular transport from the cell surface to the endosomes, from the endosomes to the Golgi apparatus, and from the Golgi apparatus to the endoplasmic reticulum (ER), where A/B subunit dissociation occurs (1). Unfolding of the dissociated A chain places the toxin in a translocation-competent conformation and activates the host mechanism of ER-associated degradation (ERAD) (2). This quality control system exports misfolded proteins from the ER to the cytosol through protein-conducting channels in the ER membrane (3). ERAD substrates are usually degraded by the cytosolic ubiquitin-proteasome system, but the A chains of ER-translocating toxins effectively evade this pathway because they lack the lysine residues required for ubiquitin conjugation (4–7).
Pertussis toxin (PT) is an AB5-type, ER-translocating toxin (8, 9). Its A subunit, PTS1, disrupts host signaling events through the ADP-ribosylation of Gα proteins. The B subunit is a hetero-oligomer composed of four proteins (PTS2, PTS3, two copies of PTS4, and PTS5) that bind to specific but largely unidentified glycoconjugates on the host cell (10–14). Holotoxin assembly involves noncovalent positioning of PTS1 above and partially within the central pore of the ring-like PTB oligomer (15). As with other ER-translocating toxins, PT transport from the cell surface to the ER appears to be an inefficient process: fluorescence microscopy, immunoelectron microscopy, and subcellular fractionation can detect internalized toxin in the endosomes and Golgi apparatus but not the ER (16–19). The inhibition of PT intoxication with brefeldin A (BfA), a drug that blocks retrograde vesicular transport to the ER, strongly suggests the functional pool of toxin must move from the Golgi apparatus to the ER before reaching its cytosolic target (17, 20). This interpretation is strengthened by biochemical data demonstrating the acquisition of Golgi apparatus- and ER-specific modifications by a recombinant PTS1 subunit (18, 19).
Displacement of the PTS1 subunit from its noncovalent association with PTB involves a conformational change in PTB that results from its interaction with ATP (8, 19, 21, 22). PTS1 displacement only occurs after the holotoxin reaches the ER, because the ER is the only endomembrane compartment that contains ATP (23, 24). Reduction of the intramolecular PTS1 disulfide bond by ER-localized oxidoreductases could further destabilize the holotoxin and thereby assist PTS1 release from the PTB oligomer (25). Disassembly of the holotoxin leads to the spontaneous unfolding of PTS1, a thermally unstable protein (26) that is otherwise held in a stable conformation by its association with the PTB oligomer (27). This ER-localized unfolding event would likely identify PTS1 as a substrate for ERAD-mediated translocation to the cytosol. Although a role for ERAD in PTS1 translocation has not yet been established, it has been shown that a transfected, ER-localized PTS1 construct can move from the ER to the cytosol (28, 29). It has also been shown that, like other ER-translocating toxins, the lack of lysine residues in PTS1 allows it to avoid ubiquitin-dependent proteasomal degradation in the cytosol (7). However, the unstable nature of free PTS1 may still render it susceptible to ubiquitin-independent degradation by the core 20S proteasome (26). An interaction with NAD, the donor molecule for the ADP-ribosylation reaction, may stabilize the cytosolic pool of PTS1 (26). Other host factors may also stabilize and/or activate PTS1 in the cytosol, similar to the interactions between a variety of host factors and either the ricin A chain (RTA) or cholera toxin A1 chain (CTA1) (30–36).
PTS1 moves from the ER to the cytosol, but the in vivo importance of PTS1 instability and a functional role for ERAD in PTS1 translocation have not been established. We addressed these issues by monitoring PTS1 translocation and PTS1 activity in toxin-treated cells that either (i) had been treated with chemical chaperones which prevented PTS1 unfolding, or (ii) harbored mutations which generate defects in the ERAD system. Using a surface plasmon resonance (SPR) system to quantify the amount of cytosolic PTS1, we estimated that only 3% of cell-associated PTS1 reaches the host cytosol after 3 h of intoxication. Greater quantities of cytosolic PTS1 were detected in cells exposed to a proteasome inhibitor, thus suggesting a role for ubiquitin-independent proteasomal degradation in the PT intoxication process. The inhibition of PTS1 unfolding via chemical chaperones substantially reduced the cytosolic pool of PTS1 and blocked PT intoxication. ERAD-defective cells likewise exhibited reduced quantities of cytosolic PTS1 and resistance to PT. These collective observations identify the thermal unfolding of dissociated PTS1 as a trigger for its ERAD-mediated translocation to the cytosol.
MATERIALS AND METHODS
Materials.
PT and the purified PTS1 subunit were purchased from List Biological Laboratories (Campbell, CA). Antibodies against PTS1 and PTS4 were purchased from Santa Cruz Biotechnology (Dallas, TX) or Abcam (Cambridge, MA). BfA and GD1a were purchased from Sigma (St. Louis, MO), 4-phenylbutyric acid (PBA) was purchased from Calbiochem (Darmstadt, Germany), 6-biotin-17-NAD was purchased from Trevigen (Gaithersburg, MD), the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was purchased from Thermo Scientific (Waltham, MA), the actin antibody was purchased from Santa Cruz Biotechnology, and the protease inhibitor cocktail was purchased from Roche Diagnostics (Indianapolis, IN). Horseradish peroxidase (HRP)-conjugated streptavidin, HRP-conjugated secondary antibodies, and fluorophore-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA).
Structural studies.
A Jasco (Tokyo, Japan) J-810 spectrofluoropolarimeter with a PFD-425S Peltier temperature controller was used to monitor the thermal unfolding of PTS1 over a temperature range of 20°C to 50°C. Experiments were conducted with a 4-mm optical path length quartz cuvette at a protein concentration of 0.18 mg/ml in 20 mM sodium borate buffer (pH 7.0) containing 100 mM NaCl. A final concentration of 10 mM β-mercaptoethanol was added after the first 20°C measurement. Samples were allowed to equilibrate at each temperature for 4 min before measurement. Far- and near-UV circular dichroism (CD) spectra recorded from 200 to 300 nm were averaged from 5 scans. Calculations for mean residue molar ellipticity, thermal unfolding profiles, and melting temperature (Tm) values were performed as previously described (37).
Toxin disassembly assay.
A gold-plated Reichert (Depew, NY) SPR sensor slide was covered in ethanol containing GD1a. The solution was allowed to dry, and the GD1a-coated sensor was placed in a Reichert SR7000 SPR refractometer. Perfusion of 10 μg PT in 1 ml phosphate-buffered saline containing 0.05% Tween 20 (PBST) over the sensor slide for 15 min at a flow rate of 5 μl/min was used to capture PT on the slide. To generate a baseline refractive index unit (RIU) signal corresponding to the mass of the bound PT holotoxin, PBST was perfused over the slide for 10 min at 37°C with a flow rate of 41 μl/min. The time course of the experiment was then initiated by adding various combinations of 100 μM ATP, 0.5% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 10% glycerol, and 100 μM PBA to the perfusion buffer. After ∼200 s, the perfusion buffer was replaced with buffer containing sequential additions of anti-PTS1 and anti-PTS4 antibodies. Glycerol itself affects the RIU, so the background signal produced by 10% glycerol alone was background subtracted from the SPR sensorgram before data presentation.
Toxin detection and quantification by SPR.
Wild-type or mutant CHO cells were seeded into 6-well plates in order to achieve ∼80% confluence after an overnight incubation. The cells were then placed at 4°C for 30 min in 1 ml serum-free medium containing 1 μg of PT. After removing unbound toxin with two washes in Ham's F-12 medium, the cells were returned to 37°C in serum- and toxin-free medium for 3 h of incubation. The chase medium contained 10% glycerol, 100 μM PBA, or 5 μg/ml of BfA, as indicated. For each condition, cells from 3 wells were lifted from the plate with PBS-EDTA and collected in a single microcentrifuge tube. The cell pellet generated from a 5-min 5,000 × g spin was resuspended in 0.1 ml HCN buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 2 mM CaCl2, 10 mM N-ethylmaleimide, and protease inhibitor cocktail) containing 0.04% digitonin for a 10-min incubation at 4°C. This procedure resulted in selective permeabilization of the plasma membrane. A 10-min spin at 16,000 × g separated the semipermeabilized cells into separate fractions: an intact membrane pellet and the cytosol-containing supernatant. The cytosol fraction was diluted in HCN buffer to a final volume of 1 ml, and the organelle-containing membrane pellet was resuspended in 1 ml of HCN buffer containing 1% Triton X-100. Membrane and/or cytosolic fractions were then perfused over SPR sensors coated with anti-PTS1 or anti-PTS4 antibodies as described in detail elsewhere (38). Western blot analysis of membrane and cytosolic fractions with anti-PDI and anti-Hsp90 antibodies was performed as previously described (39).
Several labs have demonstrated that PT binds to specific, although largely unidentified, glycoconjugate receptors on the host plasma membrane (10–14). It should therefore be possible to saturate the surface binding sites for PT with our pulse-load protocol. In support of this assertion, doubling the amount of PT in the pulse medium to 2 μg/ml did not alter the amount of surface-bound PT (see Fig. S1A in the supplemental material). This indicated PT surface binding was saturated by exposure to our standard pulse-load toxin concentration of 1 μg/ml. Excess PT B oligomer in the pulse medium likewise prevented PTS1 in the PT holotoxin from adhering to the cell surface (see Fig. S1B), which was consistent with a previous competition assay (40) and again indicated that PT binding to the plasma membrane is a saturable event. Only 4% of PT in the medium was bound to the cell surface at the end of our pulse-labeling (119 ng from a total pool of 3 μg). Our pulse-load protocol thus placed a defined, saturated amount of PT on the plasma membrane. Excess toxin was removed from the medium before the chase, so there was no contribution of soluble, extracellular toxin to the eventual accumulation of cytosolic PTS1.
Binding data from the SPR sensorgrams were used to determine toxin concentrations in each sample. Initial binding rates were cumulative averages of the rate of binding for each sample over the first 10% of the maximal response. As previously described (41, 42), the initial binding rate is directly proportional to both the association rate constant and the concentration of ligand in the perfusion buffer. The initial binding rates derived from known concentrations of purified PT or PTS1 were thus used to establish a standard curve for calculating the quantities of PT and PTS1 from cytosolic and membrane fractions. Each set of calculations involved data from three independent experiments, with values expressed as averages ± standard deviations. The efficiency of antibody coupling to the SPR sensor determines the level of RIU that can be obtained from subsequent ligand perfusion over the sensor slide. However, it should be noted that the quantity of ligand in the perfusion buffer is not established by the maximal RIU signal from the sensor; the ligand concentration is instead calculated from the association rate constant, which is independent of the absolute RIU value and the efficiency of antibody coupling. RIU signals from multiple sample perfusions over a single sensor can thus provide a relative estimation of ligand concentration, but sensor-to-sensor comparisons require calculations of association rate constants.
Intoxication assay.
Wild-type or mutant CHO cells were seeded into 6-well plates and grown overnight to ∼80% confluence. After a 30-min incubation at 4°C in 1 ml of Ham's F-12 medium containing 250 ng PT, the cells were washed with PBS and returned to 37°C in serum- and toxin-free medium for 3 h. Where indicated, the medium was supplemented with 10% glycerol, 100 μM PBA, or 5 μg/ml of BfA. Cells were harvested with lysis buffer (1% Triton X-100, 10% glycerol, 150 mM NaCl, 20 mM Tris-HCl [pH 7.5], 2 mM EDTA, and protease inhibitor cocktail), and 30 μg total cell lysate was used for an in vitro ADP-ribosylation assay. The reaction was performed at room temperature for 1 h in a final volume of 20 μl assay buffer (10 mM Tris-HCl [pH 7.5], 2 mM dithiothreitol, 10 mM ATP) containing the cell lysate, 5 μM 6-biotin-17-NAD, and 10 ng PTS1. Since the Giα target of PTS1 is only ADP-ribosylated at one location, the in vivo modification of Giα resulting from a productive intoxication prevents its subsequent in vitro modification with biotin-NAD by purified PTS1. At the end of the reaction, samples were mixed with 5 μl of 4× sodium dodecyl sulfate (SDS) sample buffer containing β-mercaptoethanol, boiled for 5 min, and resolved by SDS-polyacrylamide gel electrophoresis with 15% polyacrylamide gels. Proteins were electrotransferred onto polyvinylidene difluoride membranes by using a semidry cell transfer blot (Bio-Rad, Hercules, CA). The membrane was blocked in 2% bovine serum albumin (BSA) in TBST (25 mM Tris-HCl [pH 8], 125 mM NaCl, 0.1% Tween 20) for 2 h at room temperature, followed by a 4-h 4°C incubation with HRP-conjugated streptavidin diluted 1:20,000 in 1% BSA–TBST. The membrane was washed with TBST five times for 1 h per wash, and HRP-streptavidin captured by the biotin-labeled Giα was visualized with West Pico enhanced chemiluminescence (Thermo Scientific, Waltham, MA). Signals were normalized to GAPDH or actin loading controls and then expressed as percentages of the values from the corresponding unintoxicated control cells. Blotting conditions for the loading controls were as follows: mouse anti-GAPDH, 1:3,000 dilution; mouse anti-actin, 1:5,000 dilution; HRP-conjugated goat anti-mouse IgG, 1:15,000 dilution. The residual pool of Giα from PT-treated cells that was still available for in vitro modification with biotin-NAD was consistent with previous studies that used [32P]NAD as the donor molecule for the in vitro ADP-ribosylation reaction (20, 43).
Confocal microscopy.
CHO cells seeded onto glass coverslips in a 24-well plate were incubated with 0.5 μg/ml of PT for 30 min at 4°C. After removing unbound toxin, the cells were chased in toxin-free medium for 3 h at 37°C. The cells were then fixed in 4% paraformaldehyde (30 min at room temperature), permeabilized with 0.25% Triton X-100 (10 min at room temperature), and blocked using PBS containing 1% BSA and 10% normal donkey or goat serum (30 min at room temperature). Primary antibodies were subsequently added in 1% BSA at the following dilutions for an overnight incubation at 4°C: mouse anti-PTS1, 1:200; mouse anti-PTS4, 1:400; rabbit anti-PDI, 1:300; rabbit anti-BiP, 1:300. After washing thoroughly with PBS, secondary antibodies were added in 1% BSA at the following dilutions for 2 h of incubation at room temperature: Cy3-conjugated goat anti-mouse IgG, 1:400; fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG, 1:300; Cy3-conjugated donkey anti-rabbit IgG, 1:400; FITC-conjugated donkey anti-rabbit IgG, 1:300. The coverslips were then washed extensively in PBS and placed on a slide with 4′,6-diamidino-2-phenylindole (DAPI) fluoromount-G (Southern Biotech, Birmingham, AL). Images were captured using a Leica (Buffalo Grove, IL) TCS SP5 confocal microscope with a 60× objective.
RESULTS
Chemical chaperones inhibit the temperature-induced unfolding of PTS1.
Glycerol and PBA are chemical chaperones that block the temperature-induced unfolding of CTA1. This, in turn, disrupts CTA1 translocation to the cytosol and CT intoxication (39, 44). We predicted chemical chaperones would prevent the thermal unfolding of PTS1 as well. Far- and near-UV CD were used to test this prediction (Fig. 1). The far-UV CD spectra of PTS1 displayed a wide minimum around 220 nm at 20°C (Fig. 1A), indicating an α/β secondary structure (45), consistent with its X-ray crystal structure (PDB entry 1PRT) (15). The near-UV CD signal between 270 nm and 280 nm results from the aromatic amino acid residues and reflects the tertiary structural fold of the protein (Fig. 1B) (46). Heating caused a thermal unfolding of the protein, with a secondary structure transition temperature (Tm) of 32°C and a tertiary structure Tm of 29°C (Fig. 1A and B). Glycerol inhibited the thermal unfolding of PTS1, shifting the secondary structure Tm to 40°C and the tertiary structure Tm to 36°C (Fig. 1C and D). PBA also provided a stabilizing effect on the structure of PTS1. In the presence of PBA, PTS1 exhibited a secondary structure Tm of 41°C and a tertiary structure Tm of 38°C (Fig. 1E and F). The thermal unfolding profiles for PTS1, glycerol-treated PTS1, and PBA-treated PTS1 are presented in Fig. 1G and H.
FIG 1.

Chemical chaperones inhibit the thermal unfolding of PTS1. The temperature-induced unfolding of PTS1 was monitored by far-UV CD (A, C, E, and G) and near-UV CD (B, D, F, and H). Both measurements were conducted nearly simultaneously on the same sample. The change in color from blue to red denotes a change in temperature from 20°C to 50°C for untreated PTS1 (A and B), PTS1 treated with 10% glycerol (C and D), or PTS1 treated with 100 μM PBA (E and F). (G and H) Thermal unfolding profiles for untreated PTS1 (red), glycerol-treated PTS1 (blue), and PBA-treated PTS1 (green) derived from the data presented in panels A to F. Tm values for each condition are presented in the corresponding color. For far-UV CD analysis, the mean residue molar ellipticities at 220 nm ([θ]220) were plotted as a function of temperature. For near-UV CD, the mean residue molar ellipticities at 280 nm ([θ]280) were plotted as a function of temperature.
Chemical chaperones do not inhibit disassembly of the PT holotoxin.
The stabilization of PTS1 could potentially inhibit its dissociation from the B oligomer, which is a prerequisite for toxin translocation to the cytosol. To examine this possibility, we appended the PT holotoxin to an SPR sensor coated with GD1a, a ganglioside that binds to the PT B oligomer (11). A baseline measurement corresponding to the mass of the captured PT holotoxin was then recorded. Perfusion of 100 μM ATP alone over this toxin-coated slide did not affect the baseline value, which indicated the PT holotoxin remained stably associated with the sensor slide (Fig. 2A). This conclusion was confirmed with the sequential perfusions of anti-PTS1 and anti-PTS4 antibodies over the slide: both antibodies produced positive signals, thus demonstrating that the A and B subunits of PT were present on the slide. Identical results were obtained when 0.5% CHAPS was perfused over the PT-coated slide (data not shown). In contrast, PT disassembly occurred in the presence of both ATP and CHAPS (Fig. 2B). Under this condition, the RIU dropped to a value below the initial baseline value corresponding to the mass of the PT holotoxin. This indicated ATP and CHAPS had displaced PTS1 from the PT holotoxin. In support of this interpretation, perfusion of an anti-PTS1 antibody over the ATP/CHAPS-treated toxin did not increase the RIU signal. The continued presence of the B oligomer on the sensor slide was confirmed by the positive signal generated in response to perfusion of an anti-PTS4 antibody over the ATP/CHAPS-treated toxin. A combination of ATP and CHAPS has previously been shown to cause PT disassembly (21), so our observations were consistent with published reports. PT disassembly also occurred when ATP and CHAPS were perfused over the toxin-coated slide in the presence of 100 μM PBA (Fig. 2C) or 10% glycerol (Fig. 2D). Chemical chaperones therefore stabilized the PTS1 subunit but did not inhibit ATP/CHAPS-induced disassembly of the PT holotoxin, an event which is triggered by conformational changes in the ATP-bound B oligomer (22).
FIG 2.
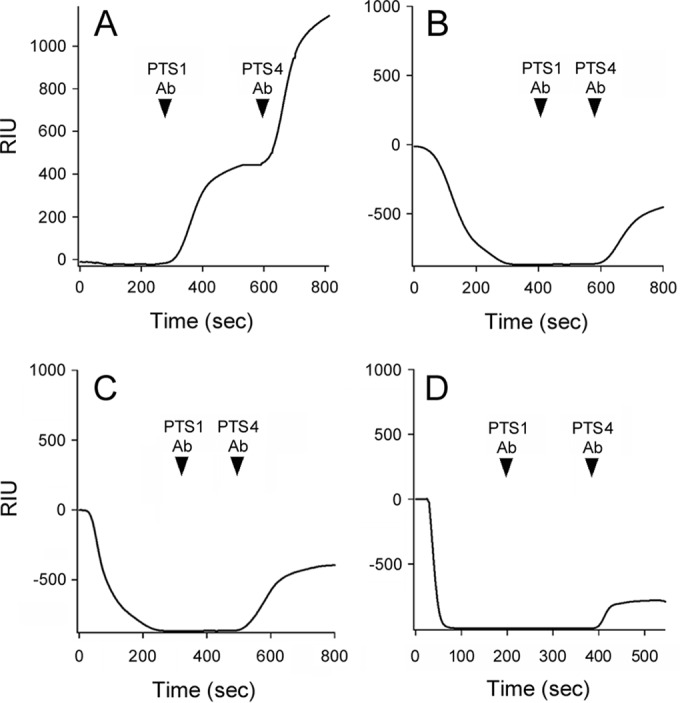
PBA and glycerol do not inhibit disassembly of the PT holotoxin. PT was appended to a GD1a-coated SPR sensor, and a baseline measurement corresponding to the mass of the sensor-bound holotoxin was set as 0 RIU. The experiment was then initiated by adding various reagents to the perfusion buffer at 0 s. The reagents were removed after ∼200 s and replaced with sequential perfusions of PTS1 and PTS4 antibodies (Ab), as indicated by the arrowheads. (A) Addition of ATP; (B) addition of ATP and CHAPS; (C) addition of ATP, CHAPS, and PBA; (D) addition of ATP, CHAPS, and 10% glycerol.
Efficiency of PTS1 delivery to the host cytosol.
An SPR-based assay (38) was used to monitor PTS1 accumulation in the cytosol of intoxicated cultured cells. CHO cells were incubated with PT at 4°C, a temperature that allows toxin binding to the cell surface but prevents toxin endocytosis. Unbound toxin was removed, and the cells were placed in toxin-free medium for 3 h at 37°C. Selective permeabilization of the plasma membrane with digitonin was then used to partition the cells into separate organelle and cytosolic fractions. Western blot analysis demonstrated that protein disulfide isomerase (PDI), a soluble ER resident protein, was only found in the organelle pellet, whereas the vast majority of cytosolic protein Hsp90 was found in the supernatant (Fig. 3A). A strong signal was recorded when the cytosol-containing supernatant fraction was perfused over an SPR sensor coated with an anti-PTS1 antibody (Fig. 3B). This indicated a pool of the initially surface-bound PTS1 had reached the host cytosol after a 3-h chase. No PTS1 was detected in the cytosol of unintoxicated cells or toxin-treated cells incubated with BfA, a drug that blocks PT trafficking to the ER translocation site (17, 20). The unintoxicated control demonstrated that our anti-PTS1 antibody specifically recognized the toxin and not a component of the host cytosol. The intoxicated, BfA-treated control demonstrated that our fractionation protocol did not accidentally release PTS1 into the supernatant fraction through rupture of the intact organelle membranes in the pellet fraction. Furthermore, we did not detect PTS1 in the cytosol collected from cells pulse-loaded at 4°C without a subsequent 37°C chase. The entire pool of pulse-loaded PTS1 was instead, as expected, found in the membrane pellet fraction.
FIG 3.

PTS1 but not PTS4 can be detected in the cytosol and extracellular medium of intoxicated cells. (A) CHO cells treated with digitonin to selectively permeabilize the plasma membrane were separated into intact membrane (pellet [P]) and cytosolic (supernatant [S]) fractions by centrifugation. To demonstrate the fidelity of the fractionation procedure, Western blot analysis was used to track the distributions of PDI, a soluble ER resident protein, and Hsp90, a cytosolic protein, in both fractions. (B and C) CHO cells pulse-loaded at 4°C with 1 μg/ml of PT were chased for 3 h at 37°C in toxin-free medium. Membrane pellet and cytosolic supernatant fractions from digitonin-permeabilized cells were collected at the end of the pulse and the end of the chase. Both fractions were perfused over SPR sensors coated with anti-PTS1 (B) or anti-PTS4 (C) antibodies. The pellet fraction was solubilized in 1% Triton X-100 before perfusion. Unintoxicated cells and cells intoxicated in the presence of BfA were used as negative controls. Ligand was removed from the perfusion buffer after 200 s. (D and E) CHO cells pulse-loaded at 4°C with 1 μg/ml of PT were chased for 3 h at 37°C in toxin-free medium. The extracellular medium collected at the end of the pulse and the end of the chase was perfused over SPR sensors coated with anti-PTS1 (D) or anti-PTS4 (E) antibody. Unintoxicated cells and cells intoxicated in the presence of BfA were used as negative controls. Ligand was removed from the perfusion buffer after 200 s. Each individual condition tested in panels B to E was derived from the same cell population. Data are presented in multiple panels due to the use of different antibodies and different signal strengths for intracellular versus extracellular PTS1.
Additional control conditions utilized an SPR sensor coated with an anti-PTS4 antibody (Fig. 3C). With this sensor, we detected the PTB pentamer in the pellet fraction of pulse-loaded cells. As expected, the PTB pentamer was absent from the cytosol of pulse-loaded cells, unintoxicated cells, and toxin-treated cells chased for 3 h at 37°C in either the absence or presence of BfA. These observations indicated that only the PTS1 subunit enters the host cytosol and confirmed, along with the BfA control of Fig. 3B, that membrane-trapped toxin was not released from intracellular organelles during permeabilization of the plasma membrane.
To ensure the translocated pool of PTS1 does not remain associated with the cytosolic face of the ER or other organelles, we used 0.8 M NaCl to strip peripheral membrane proteins from the external face of the organelles collected in our membrane pellet. As shown in Fig. S2 in the supplemental material, this procedure was sufficient to remove Hsp90 from the membrane fraction but did not, as detected by SPR, a release a pool of PTS1 into the supernatant. Translocated PTS1, like CTA1 (47), thus appears to be soluble in the host cytoplasm.
After toxin disassembly in the ER, a substantial pool of free CTA1 is secreted from CT-challenged cells (39). To determine if PTS1 was secreted from PT-treated cells, we collected media samples from cells used in the experiments of Fig. 3B and C. These media samples were perfused over SPR sensors coated with anti-PTS1 (Fig. 3D) or anti-PTS4 (Fig. 3E) antibodies. A small pool of secreted PTS1 was detected in the medium of intoxicated cells at the end of the 3-h chase (Fig. 3D). No PTS1 was detected in the medium of unintoxicated cells, pulse-loaded cells, or cells intoxicated in the presence of BfA. PTS4 was not detected in any medium sample (Fig. 3E). These results indicated that the extracellular pool of PTS1 was free, rather than holotoxin-associated, PTS1. The BfA control of Fig. 3D further indicated that toxin transport to the ER was a prerequisite for secretion of free PTS1 into the medium, which was consistent with the ER-localized displacement of PTS1 from its holotoxin.
The initial binding rate calculated from an SPR sensorgram is directly proportional to the concentration of the ligand in the perfusion buffer (41, 42). We therefore calculated the cytosolic and extracellular concentrations of PTS1 from a standard curve of the initial binding rates obtained from known quantities of PTS1 (see Fig. S3A and B in the supplemental material). These PTS1 standards were perfused over the same anti-PTS1 sensor slides used for the assays represented in Fig. 3B and D. To calculate the amount of holotoxin-associated PTS1 on the surface of pulse-loaded cells, we also generated a standard curve from known quantities of PT holotoxin that were perfused over an anti-PTS4 sensor slide (see Fig. S3C and D). Because cell counts were recorded for each experiment (n = 3), we could also estimate the average number of cytosolic PTS1 molecules per cell.
As shown in Table 1, 119 ng of PTS1 was present on the plasma membrane of ∼2.25 × 106 pulse-loaded cells. After a 3-h chase, 3.7 ng of the initially surface-associated PTS1 had reached the cytosol. Another 0.3 ng of the initially surface-associated PTS1 had been secreted into the medium. The majority of PTS1 released from the PT holotoxin in the ER was therefore delivered to the cytosol, with a minor pool of dissociated PTS1 entering the secretory pathway for export back into the extracellular medium. Overall, only 3.1% of surface-associated PTS1 was present in the cytosol after 3 h of chase. We further estimated that, on average, an intoxicated cell contains 38,000 molecules of cytosolic PTS1 after 3 h of toxin exposure. To the best of our knowledge, this represents the first direct measurement for the quantity of toxin A chain in the cytosol.
TABLE 1.
Quantity of cell-associated PTS1 and efficiency of toxin delivery to the cytosol
| Condition | PTS1 (ng) (mean ± SD) | No. of PTS1 molecules/cell | % surface-associated PTS1 |
|---|---|---|---|
| Pulse | |||
| Membrane | 119 ± 29a | 1,200,000 | 100 |
| Chase | |||
| Cytosol | 3.7 ± 0.4 | 38,000 | 3.1 |
| Medium | 0.3 ± 0.1 | 3,000 | 0.25 |
PT standards (see Fig. S3C and D in the supplemental material) were used to calculate the amount of PT on the plasma membrane of pulse-loaded cells (Fig. 3C). This value was then corrected for the molar ratio of PTS1 in the PT holotoxin.
Chemical chaperones inhibit PTS1 passage into the cytosol and PT intoxication of cultured cells.
According to our model, thermal unfolding of the dissociated PTS1 subunit will activate the ERAD translocation mechanism. The inhibition of PTS1 unfolding by glycerol or PBA would therefore block toxin export to the cytosol. We monitored the appearance of PTS1 in the cytosol of untreated, glycerol-treated, or PBA-treated cells to test this prediction (Fig. 4). Our previous studies have shown glycerol and PBA do not block CT transport from the cell surface to the ER (39, 44), which was further confirmed for PT by confocal microscopy; PT could not be seen in the ER under control conditions (18, 19) (see Fig. S4A in the supplemental material), but it colocalized with PDI or BiP in the ER of cells treated with PBA (see Fig. S4B) or glycerol (see Fig. S4C). An inhibition of toxin export from the ER to the cytosol likely promoted toxin accumulation in the ER, thereby facilitating the visualization of ER-localized PT in glycerol- or PBA-treated cells. We recently reported a similar phenomenon with a mutant PT holotoxin that could not undergo disassembly in the ER (19).
FIG 4.

Chemical chaperones inhibit PTS1 delivery to the cytosol and PT intoxication. (A and B) CHO cells pulse-loaded at 4°C with PT were chased for 3 h at 37°C in toxin-free medium containing 10% glycerol (A) or 100 μM PBA (B). Intoxicated cells chased in the absence of drug treatment were used as positive controls, while unintoxicated cells and cells intoxicated in the presence of BfA were used as negative controls. To detect the translocated toxin, cytosolic fractions from digitonin-permeabilized cells were perfused over an SPR slide coated with an anti-PTS1 antibody. PTS1 standards were perfused over the sensor as additional controls; only the 1-ng and 0.1-ng controls are shown (for scaling purposes). Ligand was removed from the perfusion buffer after 200 s. (C) CHO cells pulse-loaded at 4°C with PT were chased for 3 h at 37°C in toxin-free medium containing 10% glycerol or 100 μM PBA. Unintoxicated cells, intoxicated cells chased in the absence of drug treatment, and cells intoxicated in the presence of BfA were used as controls. Cell extracts generated by detergent lysis were incubated at 25°C for 1 h with purified PTS1 and biotin-NAD, the donor molecule for the ADP-ribosylation reaction. Since Giα can only be ADP-ribosylated by PTS1 at one site, its in vivo modification due to productive intoxication will prevent subsequent in vitro modification with biotin-labeled ADP-ribose. Western blot analysis was used to detect biotin-labeled Giα or the GAPDH loading control. Lane 1, intoxicated cells; lane 2, unintoxicated cells; lane 3, cells intoxicated in the presence of BfA; lane 4, cells intoxicated in the presence of glycerol; lane 5, cells intoxicated in the presence of PBA. (D) Signals for biotin-labeled Giα were normalized to those for the GAPDH loading control and then expressed as percentages of the values from the unintoxicated control cells. The averages ± standard deviations of results from 3 to 5 independent experiments per condition are shown.
Using the aforementioned SPR-based translocation assay, we found glycerol-treated (Fig. 4A) and PBA-treated (Fig. 4B) cells had substantially smaller pools of cytosolic PTS1 than did the untreated control cells. Unintoxicated cells and cells intoxicated in the presence of BfA were used as negative controls. The levels of cytosolic toxin in untreated and drug-treated cells were again calculated from a plot of the initial binding rates for the PTS1 standards. Glycerol-treated cells contained 16-fold less cytosolic PTS1, and PBA-treated cells contained 5.6-fold less cytosolic PTS1 than the untreated control cells (Table 2). Thus, chemical chaperones inhibited both the thermal unfolding and ER-to-cytosol export of PTS1. These observations suggested a functional role for PTS1 unfolding in triggering the translocation event.
TABLE 2.
Thermal stabilization of PTS1 limits toxin delivery to the cytosol
| Condition | Cytosolic PTS1 (ng) (mean ± SD) | Change from control |
|---|---|---|
| Untreated cells | 2.8 ± 0.1 | |
| Glycerol-treated cells | 0.18 ± 0.01 | 16-fold less |
| PBA-treated cells | 0.50 ± 0.01 | 5.6-fold less |
An inhibition of PTS1 translocation to the cytosol should generate a toxin-resistant phenotype. To test this prediction, we performed intoxication assays with untreated, glycerol-treated, and PBA-treated cells. Extracts generated from cells pulse-chased with PT for 3 h were supplemented with 10 ng purified PTS1 and 5 μM biotin-NAD (the donor molecule for the ADP-ribosylation reaction). The Giα target of PT can only be ADP-ribosylated at one site, so the in vivo modification of Giα in PT-challenged cells prevents its subsequent in vitro modification when PTS1 is added to the cell extract. In contrast, the unmodified Giα resulting from a block of intoxication will be accessible for the in vitro PTS1-catalyzed addition of biotin-conjugated ADP-ribose (20, 43).
Western blot analysis with HRP-conjugated streptavidin was used to detect biotin-labeled Giα from our intoxication assay (Fig. 4C). Productive intoxication was demonstrated by the greatly reduced level of biotinylated Giα from PT-treated cells (lane 1) in comparison to the amount of biotinylated Giα from unintoxicated cells (lane 2). A strong signal for biotinylated Giα was also obtained from cells intoxicated in the presence of BfA (lane 3). This control was indicative of a toxin-resistant phenotype. Likewise, a strong signal for biotinylated Giα was obtained from cells intoxicated in the presence of glycerol (lane 4) or PBA (lane 5). Quantification of the normalized Giα signals from BfA-, glycerol-, or PBA-treated cells indicated that the full cellular pool of Giα was available for in vitro modification by PTS1 and, thus, had not been modified during the incubation with exogenous PT (Fig. 4D). Both glycerol-treated and PBA-treated cells were therefore resistant to PT. Control conditions established that (i) addition of both PTS1 and biotin-NAD to the cell extracts was required to generate the biotin-labeled pool of Giα and (ii) glycerol and PBA did not directly interfere with the ADP-ribosylation reaction (data not shown).
ERAD mediates PTS1 export to the cytosol.
Unfolding of the dissociated PTS1 subunit should identify the toxin as a substrate for ERAD-mediated export to the cytosol, but a functional role for ERAD in PTS1 translocation has not yet been established. To address this issue, we performed our SPR-based translocation assay on a subset of CHO mutant cell lines with established defects in ERAD (Fig. 5). Mutants 16 and 46 exhibited a defect in the ER-to-cytosol export of ERAD substrates, which limits toxin access to the cytosol (48). Less PTS1 entered the cytosol of mutants 16 and 46 than the wild-type parental CHO cells, which was consistent with the predicted role of ERAD in PTS1 translocation (Fig. 5A). Prolonged toxin residence in the ER of mutants 16 and 46 allowed a larger pool of free PTS1 to enter the secretory pathway for release into the extracellular medium (see Fig. S5 in the supplemental material). Addition of proteasome inhibitor ALLN to the parental CHO cells resulted in elevated levels of cytosolic PTS1, suggesting proteasomal degradation is a normal part of the intoxication process. Elevated levels of PTS1 were likewise found in the cytosol of mutants 16 and 46 after ALLN treatment (see Fig. S6 in the supplemental material). The quantity of cytosolic PTS1 thus appears to represent a balance between toxin delivery to the cytosol and toxin degradation in the cytosol.
FIG 5.
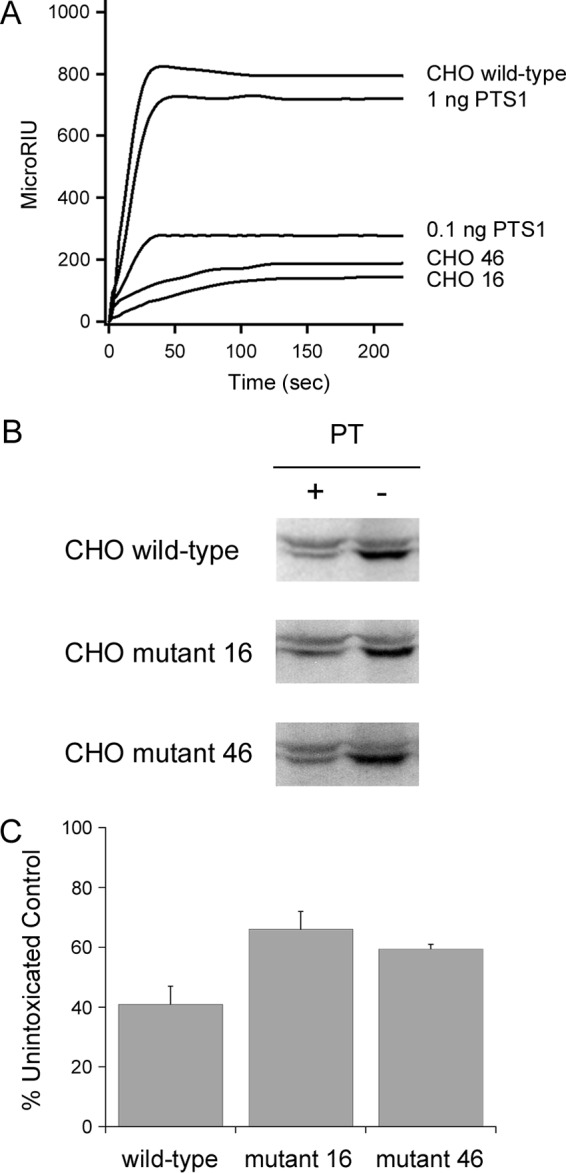
Attenuated ERAD activity inhibits PTS1 translocation and PT intoxication. (A) Wild-type CHO cells and two mutant CHO cell lines with attenuated ERAD-mediated translocation to the cytosol (CHO 16 and CHO 46) were incubated with PT at 4°C for 30 min. Unbound toxin was removed, and the cells were chased for 3 h at 37°C in toxin-free medium. Cytosolic fractions from digitonin-permeabilized cells and known quantities of PTS1 standards were then perfused over an SPR slide coated with an anti-PTS1 antibody. (B and C) Wild-type and mutant CHO cells incubated at 4°C for 30 min in the absence or presence of PT were chased for 3 h at 37°C in toxin-free medium. Cell extracts generated by detergent lysis were incubated at 25°C for 1 h with purified PTS1 and biotin-NAD, the donor molecule for the ADP-ribosylation reaction. (B) Western blot analysis was used to detect biotin-labeled Giα (lower band) or the actin loading control (upper band). (C) Signals for biotin-labeled Giα were normalized to the actin loading control and then expressed as percentages of the values from the corresponding unintoxicated control cells. The averages ± ranges for results from 2 to 4 independent experiments per condition are shown.
The limited pools of cytosolic toxin in our CHO mutants should protect those cells from the cytopathic activity of PTS1. We performed a PT intoxication assay to test this prediction. As shown in Fig. 5B and C, both mutant cell lines were partially resistant to PT: a 50% stronger signal for biotinylated Giα was obtained from PT-challenged CHO mutants 16 and 46 than from the intoxicated wild-type parental cells. Thus, attenuated ERAD activity affected the PTS1 translocation event and inhibited PT intoxication. These collective observations provided experimental evidence for the functional role of ERAD in PTS1 translocation.
DISCUSSION
Instability in the toxin A chain appears to be a common property of ERAD-exploiting toxins (2). The A chain maintains a stable conformation during transit from the cell surface to the ER because of its covalent and/or noncovalent interactions with the holotoxin (27, 49, 50). However, the structural constraints on A chain unfolding are removed after holotoxin disassembly occurs in the ER. Spontaneous or phospholipid-induced unfolding of the dissociated A chain would then identify the toxin as a substrate for ERAD-mediated translocation to the cytosol (39, 51–53). The paucity of lysine residues for ubiquitin conjugation allows the translocated A chain to evade the ubiquitin-dependent proteasomal degradation which usually follows export to the cytosol, although the labile nature of the isolated A chain could still render it susceptible to ubiquitin-independent degradation by the core 20S proteasome (4–7). This model has been derived from studies focused mainly on CTA1 and RTA (54, 55). Given that the isolated PTS1 subunit is a thermally unstable protein with a disordered conformation at the physiological temperature of 37°C (26), we predicted PTS1 would follow the general CTA1/RTA translocation pathway as well. Methods in biophysics and cell biology were used to test this hypothesis and provided molecular insights into many unresolved issues related to the PT intoxication process.
Toxin transport from the cell surface to the ER is an inefficient process: the majority of internalized toxin is diverted to the lysosomes for degradation, so only a minor pool of cell-associated toxin reaches the Golgi apparatus and ER (56–60). The efficiency of PT trafficking to the ER has not been calculated, although it is thought that only a minor pool of cell-associated PT reaches the ER as well (18). Moreover, no study has yet determined the efficiency of PTS1 delivery to the cytosol. This is not equivalent to toxin delivery to the ER, as a portion of the dissociated, ER-localized A chain enters the secretory pathway and is released into the medium (Fig. 3D) (39). With our SPR-based translocation assay, we were able to calculate the efficiency of PTS1 delivery to the cytosol. Only 3% of surface-bound toxin was present in the cytosol after a 3-h chase (Table 1). Thus, as with other ER-translocating toxins, the bulk of internalized PT appears to be routed to the lysosomes rather than to the ER translocation site (16, 17). Despite this inefficiency, the approximately 38,000 molecules of PTS1 which accumulate (on average) in the cytosol of an intoxicated cell are sufficient to elicit a cytopathic response (Fig. 4 and 5; Table 1). Given that some cytosolic toxin is degraded by the proteasome (see Fig. S6 in the supplemental material), our calculations likely represent a slight underestimate of the total amount of translocated toxin. Still, this is the first report to establish the quantity of cytosolic PTS1 and the efficiency of toxin passage into the cytosol.
Far- and near-UV CD measurements demonstrated that chemical chaperones can block the thermal unfolding of free PTS1 at the physiological temperature of 37°C. Both glycerol and PBA raised the secondary structure Tm of PTS1 by 8 to 9°C and the tertiary structure Tm of PTS1 by 7 to 9°C (Fig. 1). Although glycerol- or PBA-treated PTS1 would still be in a partially disordered conformation at physiological temperature, the extent of toxin stabilization was sufficient to impede toxin translocation to the cytosol; glycerol- or PBA-treated cells contained 16-fold and 5-fold less cytosolic PTS1 than the untreated control cells, respectively (Fig. 4A and B; Table 2). This, in turn, produced a toxin-resistant phenotype (Fig. 4C and D). Our collective data suggest that, after its ER-localized displacement from the PT holotoxin, PTS1 must shift to a highly disordered state in order to trigger its export to the cytosol.
Our studies have now shown that PBA inhibits the thermal disordering of PTS1 (Fig. 1), CTA1, and RTA (44, 61). PBA also blocks the cytopathic effects of PT (Fig. 4C and D) and CT, but it does not confer resistance to ricin or Shiga toxin (ST), another ER-translocating AB toxin (44, 61). This difference correlates to the relative stabilities of the toxin A chains: PTS1 and CTA1 are less stable than RTA (26, 31, 53) and are predicted to be less stable than the catalytic STA1 subunit (61). Thus, to attain a translocation-competent state, RTA and possibly STA1 use an interaction with the negatively charged phospholipids of the ER membrane to induce further A chain unfolding (51, 52, 62, 63). The destabilizing effect of anionic phospholipids is dominant over the stabilizing effect of PBA but does not negate the stabilizing effect of glycerol, so in vivo ricin intoxication is blocked by glycerol but not PBA (61, 64). The ability of PBA to inhibit PT intoxication suggests that, unlike RTA, additional host interactions are not required to place the dissociated PTS1 subunit in a translocation-competent conformation. This is consistent with the relative stabilities of the toxins, as RTA is substantially more stable than either CTA1 or PTS1.
Our data indicate that the cytosolic pool of PTS1 is degraded by the proteasome. Elevated levels of cytosolic PTS1 were recovered from wild-type and mutant cells treated with a proteasome inhibitor (see Fig. S6 in the supplemental material), which indicated that the steady-state quantity of cytosolic PTS1 represents a balance between toxin translocation to the cytosol and toxin degradation in the cytosol. Under normal conditions, the rate of PTS1 turnover is apparently insufficient to protect against intoxication. This may be due, in part, to the absence of PTS1 lysine residues, which allows the toxin to avoid ubiquitination and subsequent efficient degradation by the 26S proteasome (4, 7). However, as demonstrated with lysine-containing PTS1 subunits (7), cells can withstand PT exposure when PTS1 is rapidly removed from the cytosol. Under these conditions, PTS1 still enters the cytosol but does not accumulate to the extent required for productive intoxication. The mutations in CHO clones 16 and 46 likewise generated a PT-resistant phenotype by restricting the accumulation of cytosolic toxin without completely blocking toxin translocation to the cytosol (Fig. 5). Clones 16 and 46 limit the rate of ERAD-mediated toxin translocation to the cytosol (48), which apparently facilitates proteasomal clearance of the cytosolic toxin before it can accumulate to a substantial degree.
Our ERAD-defective cell lines were originally isolated from a screen that challenged mutagenized CHO cells with a combination of ricin and exotoxin A, two ER-translocating toxins that bind to separate surface receptors, follow distinct trafficking routes to the ER, and modify different cytosolic targets (48). Defective ERAD activity, which was an unselected phenotype, most likely accounted for the simultaneous acquisition of resistance to ricin and exotoxin A in clones 16 and 46. Additional experiments demonstrated that the clones had also gained unselected resistance to three other ER-translocating toxins: CT, Escherichia coli heat-labile toxin, and plasmid-encoded toxin (48, 65, 66). Toxin resistance was limited to ER-translocating toxins, as the mutant cell lines exhibited wild-type sensitivity to two toxins that move from the endosomes to the cytosol: saporin and diphtheria toxin (48, 65). This specificity was consistent with the role of ERAD in facilitating only ER-to-cytosol toxin transport. As such, PT resistance and reduced quantities of cytosolic PTS1 in our ERAD-defective cell lines strongly suggest PTS1 translocation utilizes an ERAD mechanism. Our collective observations thus indicate that the spontaneous unfolding of dissociated PTS1 serves as a trigger for its ERAD-mediated translocation to the cytosol.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI099493 to K.T.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00732-16.
REFERENCES
- 1.Spooner RA, Smith DC, Easton AJ, Roberts LM, Lord JM. 2006. Retrograde transport pathways utilised by viruses and protein toxins. Virol J 3:26. doi: 10.1186/1743-422X-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teter K. 2013. Toxin instability and its role in toxin translocation from the endoplasmic reticulum to the cytosol. Biomolecules 3:997–1029. doi: 10.3390/biom3040997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brodsky JL, Skach WR. 2011. Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol 23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hazes B, Read RJ. 1997. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 36:11051–11054. doi: 10.1021/bi971383p. [DOI] [PubMed] [Google Scholar]
- 5.Deeks ED, Cook JP, Day PJ, Smith DC, Roberts LM, Lord JM. 2002. The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry 41:3405–3413. doi: 10.1021/bi011580v. [DOI] [PubMed] [Google Scholar]
- 6.Rodighiero C, Tsai B, Rapoport TA, Lencer WI. 2002. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep 3:1222–1227. doi: 10.1093/embo-reports/kvf239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worthington ZE, Carbonetti NH. 2007. Evading the proteasome: absence of lysine residues contributes to pertussis toxin activity by evasion of proteasome degradation. Infect Immun 75:2946–2953. doi: 10.1128/IAI.02011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carbonetti NH. 2010. Pertussis toxin and adenylate cyclase toxin: key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol 5:455–469. doi: 10.2217/fmb.09.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Locht C, Coutte L, Mielcarek N. 2011. The ins and outs of pertussis toxin. FEBS J 278:4668–4682. doi: 10.1111/j.1742-4658.2011.08237.x. [DOI] [PubMed] [Google Scholar]
- 10.Brennan MJ, David JL, Kenimer JG, Manclark CR. 1988. Lectin-like binding of pertussis toxin to a 165-kilodalton Chinese hamster ovary cell glycoprotein. J Biol Chem 263:4895–4899. [PubMed] [Google Scholar]
- 11.Hausman SZ, Burns DL. 1993. Binding of pertussis toxin to lipid vesicles containing glycolipids. Infect Immun 61:335–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Witvliet MH, Burns DL, Brennan MJ, Poolman JT, Manclark CR. 1989. Binding of pertussis toxin to eucaryotic cells and glycoproteins. Infect Immun 57:3324–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.el Baya A, Bruckener K, Schmidt MA. 1999. Nonrestricted differential intoxication of cells by pertussis toxin. Infect Immun 67:433–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armstrong GD, Howard LA, Peppler MS. 1988. Use of glycosyltransferases to restore pertussis toxin receptor activity to asialoagalactofetuin. J Biol Chem 263:8677–8684. [PubMed] [Google Scholar]
- 15.Stein PE, Boodhoo A, Armstrong GD, Cockle SA, Klein MH, Read RJ. 1994. The crystal structure of pertussis toxin. Structure 2:45–57. doi: 10.1016/S0969-2126(00)00007-1. [DOI] [PubMed] [Google Scholar]
- 16.Kugler S, Bocker K, Heusipp G, Greune L, Kim KS, Schmidt MA. 2007. Pertussis toxin transiently affects barrier integrity, organelle organization and transmigration of monocytes in a human brain microvascular endothelial cell barrier model. Cell Microbiol 9:619–632. doi: 10.1111/j.1462-5822.2006.00813.x. [DOI] [PubMed] [Google Scholar]
- 17.el Baya A, Linnemann R, von Olleschik-Elbheim L, Robenek H, Schmidt MA. 1997. Endocytosis and retrograde transport of pertussis toxin to the Golgi complex as a prerequisite for cellular intoxication. Eur J Cell Biol 73:40–48. [PubMed] [Google Scholar]
- 18.Plaut RD, Carbonetti NH. 2008. Retrograde transport of pertussis toxin in the mammalian cell. Cell Microbiol 10:1130–1139. doi: 10.1111/j.1462-5822.2007.01115.x. [DOI] [PubMed] [Google Scholar]
- 19.Plaut RD, Scanlon KM, Taylor M, Teter K, Carbonetti NH. 2016. Intracellular disassembly and activity of pertussis toxin require interaction with ATP. Pathog Dis 74:ftw065. doi: 10.1093/femspd/ftw065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Barbieri JT. 1995. Pertussis toxin-mediated ADP-ribosylation of target proteins in Chinese hamster ovary cells involves a vesicle trafficking mechanism. Infect Immun 63:825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burns DL, Manclark CR. 1986. Adenine nucleotides promote dissociation of pertussis toxin subunits. J Biol Chem 261:4324–4327. [PubMed] [Google Scholar]
- 22.Hazes B, Boodhoo A, Cockle SA, Read RJ. 1996. Crystal structure of the pertussis toxin-ATP complex: a molecular sensor. J Mol Biol 258:661–671. doi: 10.1006/jmbi.1996.0277. [DOI] [PubMed] [Google Scholar]
- 23.Braakman I, Helenius J, Helenius A. 1992. Role of ATP and disulphide bonds during protein folding in the endoplasmic reticulum. Nature 356:260–262. doi: 10.1038/356260a0. [DOI] [PubMed] [Google Scholar]
- 24.Clairmont CA, De Maio A, Hirschberg CB. 1992. Translocation of ATP into the lumen of rough endoplasmic reticulum-derived vesicles and its binding to luminal proteins including BiP (GRP 78) and GRP 94. J Biol Chem 267:3983–3990. [PubMed] [Google Scholar]
- 25.Burns DL, Manclark CR. 1989. Role of cysteine 41 of the A subunit of pertussis toxin. J Biol Chem 264:564–568. [PubMed] [Google Scholar]
- 26.Pande AH, Moe D, Jamnadas M, Tatulian SA, Teter K. 2006. The pertussis toxin S1 subunit is a thermally unstable protein susceptible to degradation by the 20S proteasome. Biochemistry 45:13734–13740. doi: 10.1021/bi061175+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krell T, Greco F, Nicolai MC, Dubayle J, Renauld-Mongenie G, Poisson N, Bernard I. 2003. The use of microcalorimetry to characterize tetanus neurotoxin, pertussis toxin and filamentous haemagglutinin. Biotechnol Appl Biochem 38:241–251. doi: 10.1042/BA20030089. [DOI] [PubMed] [Google Scholar]
- 28.Veithen A, Raze D, Locht C. 2000. Intracellular trafficking and membrane translocation of pertussis toxin into host cells. Int J Med Microbiol 290:409–413. doi: 10.1016/S1438-4221(00)80053-3. [DOI] [PubMed] [Google Scholar]
- 29.Castro MG, McNamara U, Carbonetti NH. 2001. Expression, activity and cytotoxicity of pertussis toxin S1 subunit in transfected mammalian cells. Cell Microbiol 3:45–54. doi: 10.1046/j.1462-5822.2001.00092.x. [DOI] [PubMed] [Google Scholar]
- 30.Ampapathi RS, Creath AL, Lou DI, Craft JW Jr, Blanke SR, Legge GB. 2008. Order-disorder-order transitions mediate the activation of cholera toxin. J Mol Biol 377:748–760. doi: 10.1016/j.jmb.2007.12.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Argent RH, Parrott AM, Day PJ, Roberts LM, Stockley PG, Lord JM, Radford SE. 2000. Ribosome-mediated folding of partially unfolded ricin A-chain. J Biol Chem 275:9263–9269. doi: 10.1074/jbc.275.13.9263. [DOI] [PubMed] [Google Scholar]
- 32.Ray S, Taylor M, Banerjee T, Tatulian SA, Teter K. 2012. Lipid rafts alter the stability and activity of the cholera toxin A1 subunit. J Biol Chem 287:30395–30405. doi: 10.1074/jbc.M112.385575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spooner RA, Hart PJ, Cook JP, Pietroni P, Rogon C, Hohfeld J, Roberts LM, Lord JM. 2008. Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc Natl Acad Sci U S A 105:17408–17413. doi: 10.1073/pnas.0809013105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banerjee T, Taylor M, Jobling MG, Burress H, Yang Z, Serrano A, Holmes RK, Tatulian SA, Teter K. 2014. ADP-ribosylation factor 6 acts as an allosteric activator for the folded but not disordered cholera toxin A1 polypeptide. Mol Microbiol 94:898–912. doi: 10.1111/mmi.12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burress H, Taylor M, Banerjee T, Tatulian SA, Teter K. 2014. Co- and post-translocation roles for Hsp90 in cholera intoxication. J Biol Chem 289:33644–33654. doi: 10.1074/jbc.M114.609800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pietroni P, Vasisht N, Cook JP, Roberts DM, Lord JM, Hartmann-Petersen R, Roberts LM, Spooner RA. 2013. The proteasome cap RPT5/Rpt5p subunit prevents aggregation of unfolded ricin A chain. Biochem J 453:435–445. doi: 10.1042/BJ20130133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerjee T, Pande A, Jobling MG, Taylor M, Massey S, Holmes RK, Tatulian SA, Teter K. 2010. Contribution of subdomain structure to the thermal stability of the cholera toxin A1 subunit. Biochemistry 49:8839–8846. doi: 10.1021/bi101201c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor M, Banerjee T, VanBennekom N, Teter K. 3 January 2012. Detection of toxin translocation into the host cytosol by surface plasmon resonance. J Vis Exp doi: 10.3791/3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Massey S, Banerjee T, Pande AH, Taylor M, Tatulian SA, Teter K. 2009. Stabilization of the tertiary structure of the cholera toxin A1 subunit inhibits toxin dislocation and cellular intoxication. J Mol Biol 393:1083–1096. doi: 10.1016/j.jmb.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamura M, Nogimori K, Yajima M, Ase K, Ui M. 1983. A role of the B-oligomer moiety of islet-activating protein, pertussis toxin, in development of the biological effects on intact cells. J Biol Chem 258:6756–6761. [PubMed] [Google Scholar]
- 41.Edwards PR, Leatherbarrow RJ. 1997. Determination of association rate constants by an optical biosensor using initial rate analysis. Anal Biochem 246:1–6. doi: 10.1006/abio.1996.9922. [DOI] [PubMed] [Google Scholar]
- 42.Homola J. 2003. Present and future of surface plasmon resonance biosensors. Anal Bioanal Chem 377:528–539. doi: 10.1007/s00216-003-2101-0. [DOI] [PubMed] [Google Scholar]
- 43.Xu Y, Barbieri JT. 1996. Pertussis toxin-catalyzed ADP-ribosylation of Gi-2 and Gi-3 in CHO cells is modulated by inhibitors of intracellular trafficking. Infect Immun 64:593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor M, Banerjee T, Navarro-Garcia F, Huerta J, Massey S, Burlingame M, Pande AH, Tatulian SA, Teter K. 2011. A therapeutic chemical chaperone inhibits cholera intoxication and unfolding/translocation of the cholera toxin A1 subunit. PLoS One 6:e18825. doi: 10.1371/journal.pone.0018825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sreerama N, Woody RW. 2000. Circular dichroism of peptides and proteins, p 601–620. In Berova N, Nakanishi K, Woody RW (ed), Circular dichroism: principles and applications, 2nd ed John Wiley & Sons, Inc., Hoboken, NJ. [Google Scholar]
- 46.Woody RW, Dunker AK. 1996. Aromatic and cysteine side-chain circular dichroism in proteins, p 109–157. In Fasman GD. (ed), Circular dichroism and the conformational analysis of biomolecules. Plenum Press, New York, NY. [Google Scholar]
- 47.Teter K, Jobling MG, Holmes RK. 2004. Vesicular transport is not required for the cytoplasmic pool of cholera toxin to interact with the stimulatory alpha subunit of the heterotrimeric G protein. Infect Immun 72:6826–6835. doi: 10.1128/IAI.72.12.6826-6835.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teter K, Holmes RK. 2002. Inhibition of endoplasmic reticulum-associated degradation in CHO cells resistant to cholera toxin, Pseudomonas aeruginosa exotoxin A, and ricin. Infect Immun 70:6172–6179. doi: 10.1128/IAI.70.11.6172-6179.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goins B, Freire E. 1988. Thermal stability and intersubunit interactions of cholera toxin in solution and in association with its cell-surface receptor ganglioside GM1. Biochemistry 27:2046–2052. doi: 10.1021/bi00406a035. [DOI] [PubMed] [Google Scholar]
- 50.Jackson LS, Tolleson WH, Chirtel SJ. 2006. Thermal inactivation of ricin using infant formula as a food matrix. J Agric Food Chem 54:7300–7304. doi: 10.1021/jf061199n. [DOI] [PubMed] [Google Scholar]
- 51.Day PJ, Pinheiro TJ, Roberts LM, Lord JM. 2002. Binding of ricin A-chain to negatively charged phospholipid vesicles leads to protein structural changes and destabilizes the lipid bilayer. Biochemistry 41:2836–2843. doi: 10.1021/bi012012i. [DOI] [PubMed] [Google Scholar]
- 52.Mayerhofer PU, Cook JP, Wahlman J, Pinheiro TT, Moore KA, Lord JM, Johnson AE, Roberts LM. 2009. Ricin A chain insertion into endoplasmic reticulum membranes is triggered by a temperature increase to 37°C. J Biol Chem 284:10232–10242. doi: 10.1074/jbc.M808387200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pande AH, Scaglione P, Taylor M, Nemec KN, Tuthill S, Moe D, Holmes RK, Tatulian SA, Teter K. 2007. Conformational instability of the cholera toxin A1 polypeptide. J Mol Biol 374:1114–1128. doi: 10.1016/j.jmb.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spooner RA, Lord JM. 2012. How ricin and Shiga toxin reach the cytosol of target cells: retrotranslocation from the endoplasmic reticulum. Curr Top Microbiol Immunol 357:19–40. doi: 10.1007/82_2011_154. [DOI] [PubMed] [Google Scholar]
- 55.Teter K. 2013. Cholera toxin interactions with host cell stress proteins, p 323–338. In Henderson B. (ed), Moonlighting cell stress proteins in microbial infections. Springer, New York, NY. [Google Scholar]
- 56.Lencer WI, de Almeida JB, Moe S, Stow JL, Ausiello DA, Madara JL. 1993. Entry of cholera toxin into polarized human intestinal epithelial cells. Identification of an early brefeldin A sensitive event required for A1-peptide generation. J Clin Invest 92:2941–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orlandi PA, Curran PK, Fishman PH. 1993. Brefeldin A blocks the response of cultured cells to cholera toxin. Implications for intracellular trafficking in toxin action. J Biol Chem 268:12010–12016. [PubMed] [Google Scholar]
- 58.Guimaraes CP, Carette JE, Varadarajan M, Antos J, Popp MW, Spooner E, Brummelkamp TR, Ploegh HL. 2011. Identification of host cell factors required for intoxication through use of modified cholera toxin. J Cell Biol 195:751–764. doi: 10.1083/jcb.201108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Deurs B, Sandvig K, Petersen OW, Olsnes S, Simons K, Griffiths G. 1988. Estimation of the amount of internalized ricin that reaches the trans-Golgi network. J Cell Biol 106:253–267. doi: 10.1083/jcb.106.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sandvig K, Prydz K, Hansen SH, van Deurs B. 1991. Ricin transport in brefeldin A-treated cells: correlation between Golgi structure and toxic effect. J Cell Biol 115:971–981. doi: 10.1083/jcb.115.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ray S, Taylor M, Burlingame M, Tatulian SA, Teter K. 2011. Modulation of toxin stability by 4-phenylbutyric acid and negatively charged phospholipids. PLoS One 6:e23692. doi: 10.1371/journal.pone.0023692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saleh MT, Ferguson J, Boggs JM, Gariepy J. 1996. Insertion and orientation of a synthetic peptide representing the C-terminus of the A1 domain of Shiga toxin into phospholipid membranes. Biochemistry 35:9325–9334. doi: 10.1021/bi960177z. [DOI] [PubMed] [Google Scholar]
- 63.Menikh A, Saleh MT, Gariepy J, Boggs JM. 1997. Orientation in lipid bilayers of a synthetic peptide representing the C-terminus of the A1 domain of Shiga toxin. A polarized ATR-FTIR study. Biochemistry 36:15865–15872. [DOI] [PubMed] [Google Scholar]
- 64.Sandvig K, Madshus IH, Olsnes S. 1984. Dimethyl sulphoxide protects cells against polypeptide toxins and poliovirus. Biochem J 219:935–940. doi: 10.1042/bj2190935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Geden SE, Gardner RA, Fabbrini MS, Ohashi M, Phanstiel O IV, Teter K. 2007. Lipopolyamine treatment increases the efficacy of intoxication with saporin and an anticancer saporin conjugate. FEBS J 274:4825–4836. doi: 10.1111/j.1742-4658.2007.06008.x. [DOI] [PubMed] [Google Scholar]
- 66.Navarro-Garcia F, Canizalez-Roman A, Burlingame KE, Teter K, Vidal JE. 2007. Pet, a non-AB toxin, is transported and translocated into epithelial cells by a retrograde trafficking pathway. Infect Immun 75:2101–2109. doi: 10.1128/IAI.01515-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.