

Abstract
Filifactor alocis is a recently recognized periodontal pathogen; however, little is known regarding its interactions with the immune system. As the first-responder phagocytic cells, neutrophils are recruited in large numbers to the periodontal pocket, where they play a crucial role in the innate defense of the periodontium. Thus, in order to colonize, successful periodontal pathogens must devise means to interfere with neutrophil chemotaxis and activation. In this study, we assessed major neutrophil functions, including degranulation and cell migration, associated with the p38 mitogen-activated protein kinase (MAPK) signaling pathway upon challenge with F. alocis. Under conditions lacking a chemotactic gradient, F. alocis-challenged neutrophils had increased migration compared to uninfected cells, indicating that F. alocis increases chemokinesis in human neutrophils. In addition, neutrophil chemotaxis induced by interleukin-8 was significantly enhanced when cells were challenged with F. alocis, compared to noninfected cells. Similar to live bacteria, heat-killed F. alocis induced both random and directed migration of human neutrophils. The interaction of F. alocis with Toll-like receptor 2 induced granule exocytosis along with a transient ERK1/2 and sustained p38 MAPK activation. Moreover, F. alocis-induced secretory vesicle and specific granule exocytosis were p38 MAPK dependent. Blocking neutrophil degranulation with TAT-SNAP23 fusion protein significantly reduced the chemotactic and random migration induced by F. alocis. Therefore, we propose that induction of random migration by F. alocis will prolong neutrophil traffic time in the gingival tissue, and subsequent degranulation will contribute to tissue damage.
INTRODUCTION
Periodontitis is a multifactorial chronic inflammatory disease induced by a dysbiotic polymicrobial community of bacteria (1, 2). It is the sixth most common infectious disease worldwide, and over half the U.S. population will experience some form of periodontal disease (3). Additionally, accumulating epidemiological and mechanistic studies established an association between periodontal disease, periodontal pathogens, and serious systemic conditions, including pneumonia, cardiovascular disease, preterm-low-birthweight delivery, and some forms of cancer (2, 4, 5).
Recent human oral microbiome studies reveal an abundance and diversity of fastidious and yet-to-be cultivated taxons, many of which show a strong correlation with disease severity (6, 7). Nonetheless, the contribution to disease by these newly appreciated organisms has been overshadowed by the more readily cultivable species, and appreciation of their pathogenicity is just beginning to emerge (8, 9). Filifactor alocis is a slow-growing, Gram-positive anaerobe that is consistently found at increased frequency and in elevated numbers at periodontal disease sites compared to healthy sites in culture-independent studies (7, 10–20). In vivo, F. alocis is found in subgingival biofilms (21), and the organism positively correlates with other periodontal pathogens, such as Porphyromonas gingivalis, forming a co-occurrence group that is enriched across different oral habitats (22). In vitro, the organism also participates in synergistic community formation with other common periodontal bacteria (12). Notably, F. alocis is relatively resistant to oxidative stress (16), can produce trypsin-like proteases (16), and can invade and induce the secretion of proinflammatory cytokines from gingival epithelial cells (11), properties which could contribute to its pathogenicity in the periodontal pocket.
Neutrophils are the core phagocytic defenders of the periodontal pocket and are recruited in large numbers after adhesion to, and transmigration through, blood vessel walls (23). As a major component of the innate host response, neutrophils contribute to the maintenance of periodontal health by protecting the tissue against bacterial infection (24). Indeed, defects in neutrophil recruitment or function, such as neutropenia, leukocyte adhesion deficiency (LAD), and Chediak-Higashi syndrome, strongly predispose the affected individuals to periodontitis (25). Directional movement, or chemotaxis, of neutrophils toward sites of injury and inflammation occurs by sensing and deciphering a chemoattractant gradient (26–30). Neutrophils are able to respond to signals from both intermediary chemoattractants (such as interleukin-8 [IL-8]), which are encountered upon travel to sites of infection and inflammation, and from end-target cellular chemoattractants (such as formylated bacterial peptides, like fMLF), operating at the site of infection (28, 30, 31). A high concentration of chemoattractants is an indication to neutrophils that the cells had arrived at their final destination, and the process of phagocytosis and oxygen-independent and -dependent killing begins (28).
In the inflamed periodontal tissue, chemotactic factors like IL-8, as well as bacterially derived products, such as fMLF, will be abundant and guide neutrophils from the blood vessels and through the gingival tissue toward the periodontal pocket. During this cell migration process, neutrophil granule exocytosis will take place and contribute to gingival tissue damage. Given the importance of neutrophils in innate defense of the periodontium, successful periodontal pathogens, both individually and in the context of dysbiotic communities, have devised means to interfere with neutrophil chemotaxis and/or bacterial killing abilities (9, 23).
In this study, we show for the first time that F. alocis interaction with human neutrophils results in enhanced random migration and chemotaxis toward IL-8. In addition, F. alocis, through Toll-like receptor 2 (TLR2) activation, induced extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinase (MAPK) phosphorylation, which preceded the enhanced random migration and stimulated neutrophil granule exocytosis. Moreover, F. alocis-mediated neutrophil migration was dependent on the bacterium-induced degranulation.
MATERIALS AND METHODS
Neutrophil isolation.
Neutrophils were isolated from blood of healthy donors using plasma-Percoll gradients as previously described (32) and in accordance with the guidelines approved by the Institutional Review Board of the University of Louisville. Microscopic evaluation of the isolated cells showed that >95% of the cells were neutrophils. Trypan blue exclusion indicated that >97% of cells were viable.
Bacterial strains and growth conditions.
F. alocis ATCC 38596 was cultured in brain heart infusion (BHI) broth supplemented with l-cysteine (0.1%) and arginine (0.05%) for 7 days anaerobically at 37°C as previously described (27, 33). Heat-killed F. alocis was generated by incubation at 90°C for 60 min.
TAT-SNAP23 fusion protein.
Fusion proteins were created as previously described (32). Escherichia coli BL21-AI cells (Invitrogen) were transformed to overexpress the recombinant TAT fusion proteins. Purification of TAT-SNAP23 was performed by sonication and lysis of the bacterial pellet with a denaturing buffer (7 M urea, 0.5 M NaCl, 50 mM NaPO4 [pH 8], 20 mM imidazole), followed by protein separation from the supernatant by nickel-nitrilotriacetic acid (Ni-NTA) beads (Invitrogen). Protein eluted from the beads was dialyzed against 10% glycerol, 0.01% Triton X-100 in phosphate-buffered saline (PBS), pH 7.4, and stored at −80°C until use.
Neutrophil chemotaxis.
Freshly isolated neutrophils (4 × 106 cells/ml) were left unstimulated, challenged with F. alocis (multiplicity of infection [MOI] of 10), challenged with zymosan (2 to 3 particles per cell; Life Technologies), pretreated with TAT-SNAP23 (0.9 μg/ml, 10 min) followed by F. alocis challenge, or pretreated with TAT-SNAP23 followed by zymosan challenge at 37°C for 30 min. After appropriate treatment, 100 μl of cell suspension was added to the upper chamber of the transwell inserts contained in 24-well plates (VWR, Corning). Chemotaxis was initiated by adding 600 μl of chemoattractants into the lower chamber. The chemoattractants used were fMLF (100 nM; Sigma) and IL-8 (100 ng/ml; Sigma), along with supernatants collected from unstimulated or F. alocis-challenged neutrophils (MOI of 10; 1, 4, and 20 h). After 30 min, the transwell membranes were stained with a HEMA 3 stain set kit by following the manufacturer's instructions (Thermo Fisher Scientific). Chemotaxis was assessed by light microscopic examination (magnification, ×100; VWR compound trinocular microscope) of the underside of the membrane. The average number of cells from a total of 10 fields was determined, and data were normalized by the area of the membrane circle and field of view.
ERK1/2 and p38 MAPK phosphorylation.
Phosphorylation of ERK1/2 and p38 MAPK was determined as previously described (32, 34). In brief, neutrophils (1 × 107 cells/ml) were left unstimulated, stimulated with fMLF (300 nM, 1 min) or F. alocis (MOI of 10; 5, 15, 30, and 60 min), or, for some experiments, pretreated with anti-TLR2 antibody (50 μg/ml; clone TL2.1; BioLegend) or isotype control IgG2a kappa (50 μg/ml; clone MOPC-173; BioLegend) followed by F. alocis (MOI of 10, 15 min). After the different experimental procedures, cells were centrifuged at 6,000 × g for 30 s and lysed in ice-cold lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% [vol/vol] Triton X-100, 0.5% [vol/vol] Nonidet P-40, 20 mM NaF, 20 mM NaVO3, 1 mM EDTA, 1 mM EGTA, 5 mM phenylmethylsulfonyl fluoride [PMSF], 21 μg/ml aprotinin, and 5 μg/ml leupeptin). Cell lysates were separated by 4 to 12% gradient or 10% SDS-PAGE and immunoblotted with antibodies to phospho-ERK1/2, total ERK1/2, phospho-p38 MAPK, or total p38 MAPK (Cell Signaling), all at 1:500. The appropriate secondary antibodies were used at 1:5,000 (Santa Cruz). The ECL system (Amersham Pharmacia Biotech) was used to visualize antigen-antibody reactions. Densitometric values of each band were calculated using ImageJ software.
Neutrophil granule exocytosis.
Neutrophils (4 × 106 cell/ml) were incubated with buffer (basal) or with F. alocis at an MOI of 10 for 30 min, with fMLF (300 nM, 5 min; Sigma) or tumor necrosis factor alpha (TNF-α; 2 ng/ml, 10 min; R&D Systems) followed by fMLF (300 nM, 5 min), or with latrunculin-A (1 μM, 30 min; Sigma) or latrunculin-A followed by fMLF. Inhibitors, with which cells were pretreated for 30 min, were SB-203580 (3 μM), anti-TLR2 antibody (50 μg/ml; clone TL2.1; BioLegend), anti-TLR4 antibody (50 μg/ml; clone HTA125; BioLegend), and isotype control IgG2a kappa (50 μg/ml; clone MOPC-173; BioLegend). For evaluating the potential role of TLR4 in F. alocis-induced exocytosis, neutrophils were left unstimulated, stimulated with ultrapure lipopolysaccharide (LPS; 100 ng/ml, 60 min; ENZO Life Sciences), or pretreated with TLR4 signaling inhibitor CLI-095 (1 μg/ml, 15 min; Invivogen) followed by challenge with either LPS (100 ng/ml, 60 min) or F. alocis (MOI of 10, 30 min). For blocking granule exocytosis, neutrophils were pretreated with TAT-SNAP23 for 10 min, followed by stimulation with fMLF, F. alocis, or zymosan. Exocytosis of secretory vesicles, specific granules, and azurophil granules was determined by measuring the increase in plasma membrane expression of phycoerythrin (PE)-conjugated anti-human CD35 (clone E11; BioLegend), fluorescein isothiocyanate (FITC)-conjugated anti-human CD66b (clone G10F5; BioLegend), and FITC-conjugated anti-human CD63 (clone AHN16.1/46-4-5; Ancell Corporation). Antibody reactivity was measured with a BD FACSCalibur flow cytometer (Becton Dickinson). Extracellular release of myeloperoxidase (MPO) was determined as the NaN3-inhibitable isoluminol chemiluminescence in the presence of an exogenous source of hydrogen peroxide. Chemiluminescence was continuously measured under the different experimental conditions at 37°C for 10 min as previously described (32). Supernatants from the different experimental conditions were collected, and the extracellular release of albumin and lactoferrin was determined by enzyme-linked immunosorbent assay (ELISA) (Abcam).
Statistical analysis.
For all of the experimental conditions tested in this study, the statistical analysis used was a one-way analysis of variance (ANOVA) with the Tukey-Kramer multiple-comparison test (GraphPad Prism Software, San Diego, CA, USA). Differences were considered statistically significant at a P value of <0.05.
RESULTS
F. alocis challenge of human neutrophils resulted in stimulated cell migration with and without directionality.
Neutrophil migration toward sites of infection is a key early event in the process of protection against pathogenic microorganisms, and in periodontal disease, neutrophils are recruited in high numbers into the gingival tissues and crevicular fluid to control and combat the bacterial infection (35). However, neutrophils from chronic periodontitis patients show dysfunctional chemotactic function which increases the cell transit time in the gingival connective tissue, promoting collateral tissue damage (36). Using a mouse chamber model, we recently showed that F. alocis infection resulted in rapid neutrophil infiltration to the site of infection (10). To determine whether F. alocis interaction with human neutrophils can impact neutrophil migration, chemotaxis assays were performed using the transwell system. Assessed by light microscopic analysis and cell migration quantification, unstimulated cells showed minimal cell migration in the absence of a chemotactic source, whereas there was significant neutrophil migration toward the potent chemotactic formylated peptide fMLF, as expected (Fig. 1A). Interestingly, F. alocis-challenged neutrophils showed significant migration toward buffer alone compared to unstimulated cells (Fig. 1A), which indicates that the bacterial challenge increased random cell migration in the absence of a chemoattractant source. However, migration toward fMLF was similar under all conditions regardless of bacterial challenge. Thus, we sought to determine if the random migration induced by F. alocis would have no impact on directed migration independent of chemotactic source. Since IL-8 is an important chemotactic factor involved in neutrophil recruitment to the site of periodontal infection (23), we examined the impact of F. alocis challenge on IL-8-dependent neutrophil chemotaxis. Unlike what was observed with fMLF, the F. alocis-challenged cells showed a significant increase in chemotaxis toward IL-8 compared to unstimulated cells (Fig. 1B).
FIG 1.

Effect of F. alocis stimulation on neutrophil chemotaxis. Neutrophils were left unchallenged (control), challenged with F. alocis (30 min), or challenged with heat-killed F. alocis (HK-F. alocis; 30 min). (A to F) Following the bacterial challenge, cells were placed in the upper chamber of the transwell system, and after 30 min of incubation, the membrane was stained with a HEMA 3 stain set kit. Chemotaxis was assessed by light microscopic examination (magnification, ×100). (A) Buffer or fMLF (100 nM) was placed in the lower well. Data are expressed as mean numbers ± standard errors of the mean (SEM) of migrated cells/insert from 9 independent experiments. (B) Buffer or IL-8 (100 ng/ml) was placed in the lower well. Data are expressed as mean numbers ± SEM of migrated cells/insert from 5 independent experiments. (C) Unstimulated cells were placed in the upper chamber of the transwell plate, and buffer, conditioned supernatant collected from unstimulated cells (UT-cond-sup), or conditioned supernatant collected after 60 min of stimulation with F. alocis (F. alocis-cond-sup), IL-8 (100 ng/ml), or fMLF (100 nM) was placed in the lower well. Data are means ± SEM from 6 independent experiments. (D to F) Buffer (D), fMLF (E), or IL-8 (F) was placed in the lower well. Data are expressed as mean numbers ± SEM of migrated cells/insert from 5 independent experiments.
Neutrophils can also be a source of IL-8 as they have preformed IL-8, which is stored in rapid mobilized vesicles (37), and can also synthesize the chemokine upon stimulation (38). To determine if the enhanced migration observed after F. alocis stimulation could be related to the release of IL-8 from the stored pools, neutrophil supernatants were collected after 60 min of bacterial challenge and added to the lower chamber of the transwell system. As shown in Fig. 1C, supernatant collected after 60 min of F. alocis challenge did not induce neutrophil migration, whereas both fMLF and IL-8 induced significant neutrophil chemotaxis. In addition, we were unable to detect IL-8 levels in the supernatant collected after 60 min of F. alocis challenge (data not shown), arguing against the possibility of an autocrine IL-8 effect induced by F. alocis responsible for the increased chemotaxis.
We next examined if bacterial viability is critical for inducing neutrophil motility by challenging cells with viable or heat-killed F. alocis. Both viable and heat-killed F. alocis-challenged neutrophils displayed significantly enhanced random migration in the absence of a chemotactic source (Fig. 1D), no difference in directed migration toward fMLF compared to unchallenged control cells (Fig. 1E), and enhanced chemotaxis toward IL-8 (Fig. 1F). Collectively these results indicate that the heat-stable cell wall components from F. alocis stimulate random migration in the absence of a chemotactic source and enhance the migration of neutrophils with directionality toward intermediary chemoattractants, such as IL-8.
F. alocis interaction with human neutrophils induced granule exocytosis through TLR2 with ERK and p38 MAPK activation.
Components of Gram-positive bacteria are usually recognized by Toll-like receptor 2 (TLR2), which is expressed by neutrophils (39). Moreover, TLR2 agonists, such as N-palmitoyl-S-[2,3-bis (palmitoyloxy)-(2RS)-propyl]-(R)-cysteinyl-seryl-(lysyl) (3)-lysine (P3CSK4), can induce neutrophil random migration by triggering extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinase (MAPK) signal transduction pathways (40). To examine the ability of F. alocis to activate the MAPK signaling pathways, phosphorylation of ERK1/2 and p38 MAPK was measured by immunoblot analysis. Figure 2A and B shows that F. alocis infection increased the levels of ERK1/2 and p38 MAPK phosphorylation in neutrophils, with maximum activation reached at 15 min for ERK1/2 and 30 min for p38. By 60 min after F. alocis challenge, a decrease in both ERK1/2 and p38 MAPK phosphorylation was observed. To further characterize the dependence of F. alocis-induced MAPK signaling on TLR2 recognition, the bacterium-receptor interaction was blocked by a TLR2 monoclonal antibody (MAb). Figure 3, lane 3, shows that blocking TLR2 significantly inhibited F. alocis-induced phosphorylation of ERK1/2. A similar inhibitory trend, although not reaching statistical significance, was observed for p38 MAPK phosphorylation. These results indicate that the F. alocis-induced ERK1/2 signaling pathway is TLR2 dependent.
FIG 2.
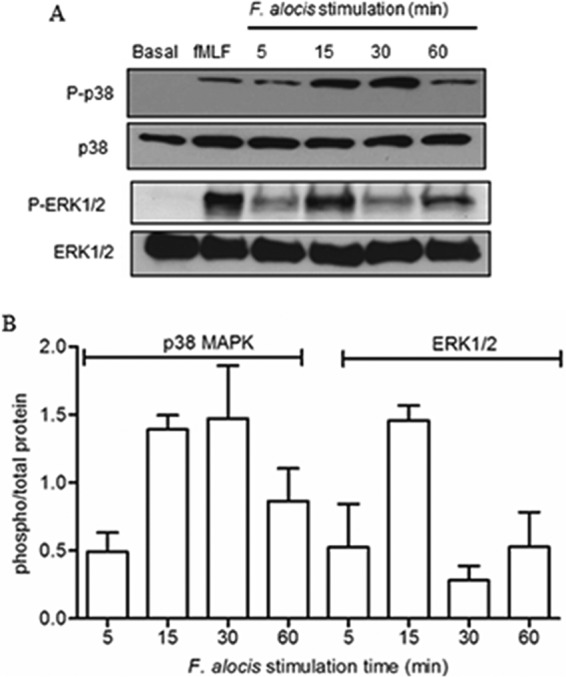
F. alocis-induced ERK1/2 and p38 MAPK activation in human neutrophils. Neutrophils were unchallenged (basal), stimulated with fMLF (300 nM, 1 min), or challenged with F. alocis for the indicated times. Cells were lysed, and proteins were separated by SDS-PAGE and immunoblotted for phospho-p38 (P-p38) or phospho-ERK1/2 (P-ERK1/2). Blots were stripped and reblotted for total p38 (p38) or total ERK1/2 (ERK1/2), respectively. (A) Representative immunoblot of 5 independent experiments. (B) Densitometric analysis of the 5 immunoblots for P-p38 or P-ERK1/2 normalized to the total amount of p38 or ERK1/2, respectively. Data are expressed as the mean ratio ± SEM of phosphorylated to total kinase.
FIG 3.

TLR2 activation is involved in F. alocis-induced phosphorylation of both ERK1/2 and p38 MAPK. Neutrophils were unchallenged (basal), challenged with F. alocis (MOI of 10, 15 min), or pretreated with either anti-TLR2 MAb or isotype control (isotype-Ctrol), followed by F. alocis challenge. Cells were lysed and proteins separated by SDS-PAGE and immunoblotted for phospho-p38 (P-p38) or phospho-ERK1/2 (P-ERK1/2). Blots were stripped and reblotted for total p38 (p38) or total ERK1/2 (ERK1/2), respectively. (A) Representative immunoblot of 4 independent experiments. (B) Densitometric analysis of the 4 immunoblots for P-ERK1/2/total ERK1/2. (C) Densitometric analysis of the 4 immunoblots for P-p38 MAPK/total p38 MAPK. Data are expressed as mean fold changes ± SEM over the basal level of the phosphorylated/total kinase ratio.
In addition to its role in MAPK signaling pathways, TLR activation can also induce other neutrophil functions, such as exocytosis of neutrophil granules, which has been linked to chemotaxis (33, 39, 41, 42). The exocytosis of secretory vesicles and specific granules increases not only the number but also the diversity of the receptor repertoire on neutrophil plasma membrane and facilitates cell firm adhesion and extravasation from the blood to the tissue (43). Hence, to examine whether F. alocis-enhanced random migration in the absence of a chemotactic source could be linked to granule mobilization, the increases on the cell plasma membrane of secretory vesicles and specific granule markers were determined by flow cytometry. Stimulation of neutrophils for 30 min with F. alocis resulted in a significant secretory vesicle release similar to the exocytosis induced by fMLF (Fig. 4A). In addition, specific granule exocytosis, as measured by expression of CD66b on the plasma membrane, was significantly increased by F. alocis stimulation (Fig. 4B). Activation of p38 MAPK has been associated with cell migration and with TNF- and LPS-induced neutrophil granule exocytosis (33). In order to test the involvement of p38 MAPK signaling in F. alocis-stimulated granule exocytosis, neutrophils were pretreated with the p38 inhibitor SB-203580 before bacterial challenge. Figure 4C and D shows that blocking p38 MAPK resulted in a significant decrease of secretory vesicles and specific granule exocytosis. To confirm that F. alocis-induced upregulation of CD35 and CD66b at the plasma membrane was accompanied by the release of granule content, the extracellular release of albumin and lactoferrin, respectively, was determined by ELISA. Figure 4E and F show that F. alocis induced significant release of both albumin and lactoferrin in a p38 MAPK-dependent manner. These results demonstrate that F. alocis challenge triggered secretory vesicles and specific granule exocytosis, and that the process was p38 MAPK dependent.
FIG 4.

F. alocis stimulation of secretory vesicle and specific granule exocytosis is p38 MAPK dependent. Neutrophils were left unchallenged (basal), challenged with fMLF (300 nM, 5 min), challenged with F. alocis (MOI of 10, 30 min), or pretreated for 30 min with SB203580 followed by F. alocis challenge (SB + F. alocis). (A to D) Secretory vesicle and specific granule exocytosis were determined by the increase in plasma membrane expression of the CD35 or CD66b marker, respectively, by flow cytometry. Data are expressed as the mean channel of fluorescence (mcf) ± SEM from 5 independent experiments. (E and F) Supernatants from all of the different experimental conditions were collected, and the release of albumin or lactoferrin to determine secretory vesicle or specific granule exocytosis, respectively, was measured by ELISA. Data from albumin or lactoferrin release are expressed as means ± SEM in ng/4 × 106 cells from 5 independent experiments for albumin and 6 independent experiments for lactoferrin.
Chemotactic factors can induce the release of azurophil granule components such as β-glucuronidase (44), and the contribution of lysosome exocytosis and fusion with the plasma membrane, through the regulation of Rab27a, to the promotion of cell migration has been established (42). Hence, exocytosis of azurophil granules upon F. alocis challenge was measured by both the increase of plasma membrane expression of the granule marker CD63 by flow cytometry and the release of the granule component myeloperoxidase (MPO). Figure 5A and B show that F. alocis challenge did not induce azurophil granule exocytosis. Moreover, increasing the amount of bacteria per neutrophil from an MOI of 10 to 50 did not result in azurophil granule mobilization, measured both by upregulation of the granule marker (Fig. 5A) and by the release of MPO (Fig. 5B).
FIG 5.
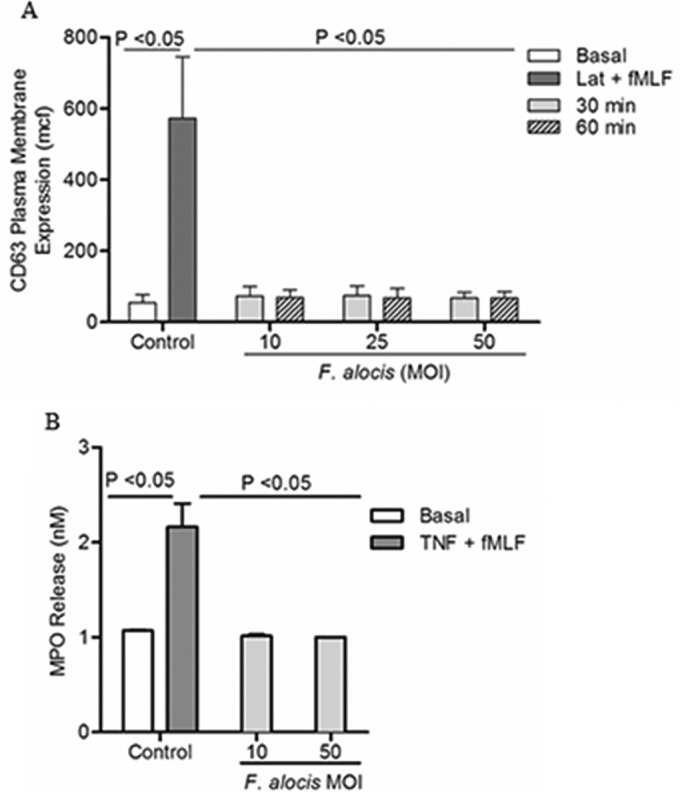
F. alocis challenge does not induce azurophil granule exocytosis. Neutrophils were left unchallenged (basal), were pretreated with latrunculin-A (1 μM, 30 min) followed by fMLF stimulation (Lat + fMLF, 300 nM, 5 min), challenged with TNF (2 ng/ml, 10 min) followed by fMLF stimulation (TNF + fMLF, 300 nM, 5 min), or challenged with F. alocis (MOI of 10, 25, and 50 for 30 or 60 min). (A) Azurophil granule exocytosis was determined by the increase in plasma membrane expression of CD63 by flow cytometry. Data are expressed as mean mcf ± SEM from 3 independent experiments. (B) Extracellular release of myeloperoxidase (MPO), to determine azurophil granule exocytosis, was measured as described in Materials and Methods. Data from MPO release are expressed as means ± SEM in nM from 3 independent experiments.
Our data thus far show that F. alocis interaction with human neutrophils results in TLR2-mediated phosphorylation of ERK1/2 and p38 MAPK, although the latter may not be solely dependent on TLR2. Moreover, F. alocis induced the exocytosis of secretory vesicles and specific granules through activation of the p38 MAPK signaling pathway. Hence, we wanted to determine if upstream of the p38 MAPK signaling pathway, F. alocis interaction with TLR receptors could be the initial trigger that induces granule release. Figure 6A shows that blocking TLR2 with MAb resulted in a significant inhibition of F. alocis-induced secretory vesicle exocytosis, as measured by expression of CD35 on the plasma membrane, whereas TLR4 MAb had a minimal inhibitory effect. Similarly, F. alocis-induced specific granule exocytosis was inhibited to the same extent in anti-TLR2 treated cells (data not shown). To provide additional evidence for a lack of TLR4 activation by F. alocis, a pharmacologic inhibitor, CLI-095, which blocks the intracellular domain of TLR4 (45), was used with lipoprotein-free LPS as a control. Figure 6B shows that, as expected, blocking TLR4 signaling resulted in a significant inhibition of LPS-induced secretory vesicle exocytosis. In contrast, and concordant with the anti-TLR4 data, chemical blocking of TLR4 resulted in a minimal inhibition of F. alocis-induced secretory vesicle exocytosis. These results show that F. alocis stimulation of both secretory vesicles and specific granule exocytosis is TLR2 and p38 MAPK dependent.
FIG 6.
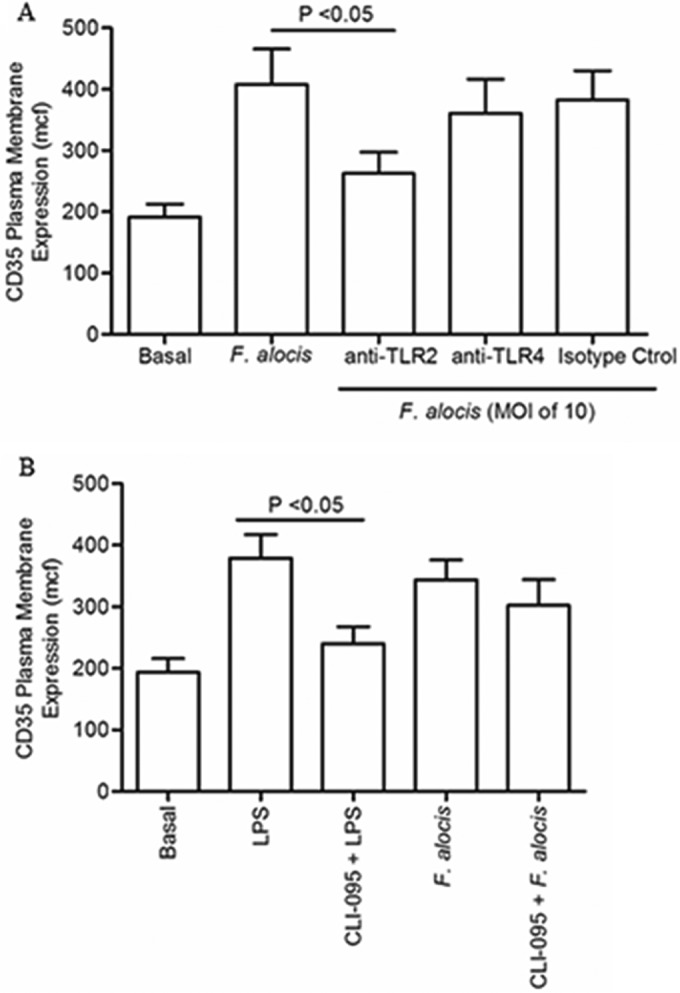
F. alocis interaction with TLR2 triggered secretory vesicle exocytosis. (A) Neutrophils were left unchallenged (basal), challenged with F. alocis (M0I of 10, 30 min), or pretreated for 30 min with either anti-TLR2 MAb, anti-TLR4 MAb, or isotype control (isotype ctrol) followed by F. alocis challenge. (B) Neutrophils were left unchallenged (basal), challenged with LPS (100 ng/ml, 60 min), pretreated for 15 min with CLI-095 followed by LPS challenge, challenged with F. alocis, or pretreated with CLI-095 followed by F. alocis challenge. In both panels, secretory vesicle exocytosis was determined by the increase in plasma membrane expression of the CD35 marker by flow cytometry. Data are expressed as mean mcf ± SEM from 5 independent experiments.
Blocking neutrophil degranulation inhibits F. alocis-induced neutrophil cell migration.
Both p38 MAPK activation and neutrophil granule exocytosis play a key role in early-stage neutrophil responses, such as diapedesis and chemotaxis (33, 42). Therefore, we sought to assess whether neutrophil granule exocytosis played a role in F. alocis-induced neutrophil migration in the presence or in the absence of a chemotactic source. Neutrophil degranulation can be blocked, without affecting neutrophil phagocytic ability and activation of p38 MAPK, by using the TAT-SNAP23 fusion protein (32). First, we wanted to confirm that the TAT-SNAP23 pretreatment would block F. alocis-induced granule exocytosis. Figure 7A and B shows that the TAT-SNAP23 fusion protein, as previously characterized (32), significantly blocked fMLF-stimulated secretory vesicles and specific granule exocytosis. Moreover, our data showed that the TAT-SNAP23 fusion protein also significantly blocked F. alocis-induced secretory vesicles and specific granule exocytosis (Fig. 7A and B) and had no nonspecific effect when using a stimulation procedure, in this case with zymosan, that did not induce granule exocytosis. Upon verification that TAT-SNAP23 was significantly blocking F. alocis-induce granule exocytosis, we tested the effect on cell migration. Figure 7C shows that treatment of unstimulated cells with the TAT-SNAP23 fusion protein reduced the number of neutrophils crossing the membrane toward fMLF, which further emphasizes the role of granule exocytosis in neutrophil chemotaxis. On the contrary, when zymosan was used to stimulate neutrophils, the particulate stimuli induced chemotaxis toward IL-8, but pretreatment with the TAT-SNAP23 fusion protein had no effect on zymosan-induced chemotaxis (Fig. 7D). However, as shown in Fig. 7E, blocking neutrophil degranulation with TAT-SNAP23 resulted in a significant inhibition of F. alocis-induced random migration in the absence of a chemotactic source. In addition, blocking granule release significantly reduced the ability of F. alocis-challenged neutrophils to migrate toward both chemotactic sources, fMLF and IL-8 (Fig. 7E and F). These results demonstrate that when granule exocytosis is involved in cell migration, pretreatment with the TAT-SNAP23 fusion protein prevents exocytosis-mediated cell migration. Collectively, these data suggest that the enhanced migration of F. alocis-challenged neutrophils observed in the presence or in the absence of a chemotactic source is due in part to the p38 MAPK-dependent granule exocytosis induced by the bacterial challenge.
FIG 7.

Blocking neutrophil granule exocytosis inhibits F. alocis-induced random and directed migration. Neutrophils were left unchallenged (control), stimulated with fMLF (300 nM, 5 min), treated with TAT-SNAP23 (10 min), pretreated with TAT-SNAP23 followed by fMLF stimulation, challenged with F. alocis (30 min), challenged with zymosan (Zy; 30 min), pretreated with TAT-SNAP23 (10 min) followed by F. alocis challenge (TAT-SNAP23 + F. alocis), or pretreated with TAT-SNAP23 followed by zymosan challenge (TAT-SNAP23 + Zy). (A and B) Secretory vesicle and specific granule exocytosis were determined by the increase in plasma membrane expression of the CD35 or CD66b marker, respectively, by flow cytometry. Data are expressed as mean mcf ± SEM from 5 independent experiments. (C to F) Following cell stimulation or bacterial challenge, cells were placed in the upper chamber of the transwell system. After 30 min of incubation, the membrane was stained with a HEMA 3 stain set kit. Chemotaxis was assessed by light microscopic examination (magnification, ×100). (C and E) Buffer or fMLF (100 nM) was placed in the lower well. Data are means ± SEM from 5 independent experiments. (D and F) Buffer or IL-8 (100 ng/ml) was placed in the lower well. Data are expressed as mean (±SEM) number of migrated cells/insert from 5 independent experiments (D) and from 7 independent experiments (F).
DISCUSSION
Given the importance of neutrophils in innate defense of the periodontium, successful periodontal pathogens, both individually and in the context of dysbiotic communities, have devised means to interfere with neutrophil chemotaxis and killing (9, 23). Furthermore, congenital diseases, such as leukocyte adhesion deficiency, that impair neutrophil chemotaxis result in severe periodontitis at an early stage in life (46, 47). In the mouse subcutaneous chamber model of infection, the newly appreciated periodontal pathogen F. alocis elicits a local inflammatory response with extensive neutrophil recruitment as well as spread to remote tissues, inducing lung edema with neutrophil recruitment and causing acute kidney injury (10). However, very little is known about the pathogenic nature of F. alocis and its interaction with the innate immune system. In the present study, we showed that F. alocis interaction with human neutrophils, through TLR2 recognition, resulted in enhanced random and directed migration and degranulation via activation of the p38 MAPK signaling pathway. Degranulation and p38 MAPK activation induced by F. alocis were major contributors to the enhanced cell migration. Based on our data, a schematic model for neutrophil degranulation and enhanced migration in response to F. alocis challenge is proposed in Fig. 8.
FIG 8.
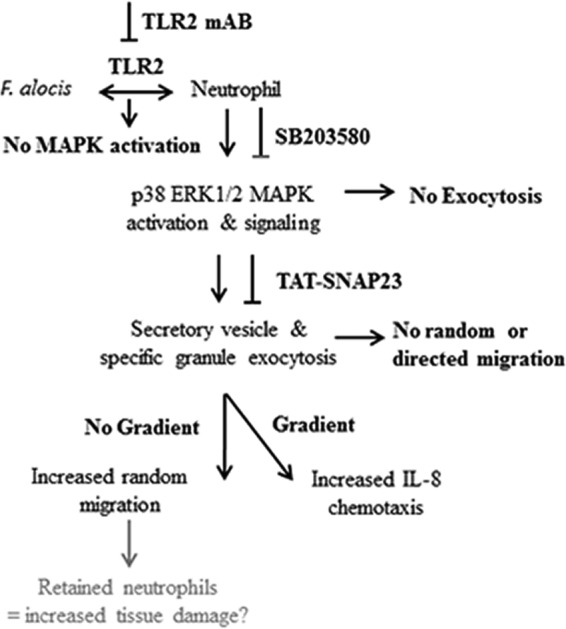
Schematic representation of F. alocis-induced neutrophil granule exocytosis, random and directed migration. F. alocis binding to TLR2 on the neutrophil plasma membrane induces phosphorylation of ERK1/2 and p38 MAPK. Activation of p38 MAPK promotes the exocytosis of secretory vesicles and specific granules, which contribute to F. alocis-induced random and directed migration. F. alocis-induced neutrophil granule exocytosis, enhanced random migration and chemotaxis toward IL-8, could retain these activated professional phagocytes in the gingival tissue and increase tissue damage.
In the periodontal pocket there is a continual influx of neutrophils that decipher and migrate through a chemotactic gradient. Our results showed that F. alocis interaction with human neutrophils resulted in a significant increase in random migration. Preexposure to F. alocis did not affect the ability of the cells to migrate with directionality toward fMLF, which is consistent with previous observations of human neutrophils challenged with either TLR2/1 or TLR4 agonists such as P3CSK4 or LPS, respectively (40). In an inflamed tissue, different neutrophil chemotactic products, such as IL-8 and formylated peptides, and complement products, such as C5a, are produced by different sources; however, neutrophils efficiently transit toward the end target by migrating in sequence from one chemotactic source to another (48). Fan and Malik showed that neutrophils activated by the TLR4 agonist LPS display enhanced migration toward IL-8 by modulating CXCR2 expression and preventing receptor desensitization (49). We found here that F. alocis significantly enhanced neutrophil chemotaxis toward IL-8 through TLR2 activation. Hence, both TLR2 and TLR4 activation of neutrophils can result in enhanced migration toward IL-8. Another important signaling mechanism linked to neutrophil chemotaxis toward IL-8 or fMLF is differential activation of the two integrin molecules MAC-1 and LFA-1 (50). Activation of LFA-1 or MAC-1 enhances neutrophil chemotaxis toward IL-8 or fMLF, respectively (50). Therefore, it is possible that F. alocis-enhanced chemotaxis toward IL-8 is due to LFA-1 activation. In addition, F. alocis-induced secretory vesicle exocytosis would increase the number and variety of receptors on the cell plasma membrane, making neutrophils more prone to mounting an enhanced response upon subsequent stimulation. Moreover, the F. alocis-induced release of specific granule content, which, among other proteins, includes members of the matrix metalloprotease family, such as collagenase and gelatinase, will contribute to tissue damage.
The MAP kinase family signaling components ERK and p38 MAPK play important roles in the regulation of fMLF-induced neutrophil migration (29, 51–53), whereas IL-8 stimulation results in activation of the phosphatidyl inositol 3-kinase (PI3K) signaling pathway (54). Stimulation of neutrophils with TLR2 or TLR4 agonists signals through ERK and p38 MAPK to control random migration and chemotactic activity (40). Hence, the role of the different kinases in human neutrophil migration is dependent on the agonist. Our study showed that F. alocis triggered activation of both ERK and p38 MAPK but with temporal differences, with ERK activation, which occurred through TLR2, peaking at 15 min, whereas p38 MAPK showed a different phosphorylation pattern, increasing with time and peaking at 30 min. Neutrophils migrate with directionality to sites of infection following increasing concentrations of a chemoattractant, but when the chemoattractant concentrations are elevated, it is an indication that the cells have reached their final destination, so a stop signal is triggered to prevent more migratory movement (55). The balance between ERK and p38 MAPK activation fine-tunes neutrophil chemotaxis, as ERK regulates the stop signal and p38 MAPK promotes constant migration by suppression of the stop mechanism (55). Thus, the phosphorylation pattern of p38 MAPK, along with the transient phosphorylation of ERK induced by F. alocis, would lead to enhanced random migration and chemotaxis toward IL-8 as the result of the constant suppression of the stop signal by p38 MAPK, allowing sustained migration. In addition, fMLF-induced chemotaxis is regulated by ERK and p38 MAPK, and similarly, F. alocis challenge results in activation of both MAP kinases, which suggests that the oral pathogen induces signaling pathways similar to those of fMLF to stimulate neutrophil chemotaxis.
Upon stimulation, neutrophils will mobilize their granules, which will fuse with either the cytoplasmic membrane or the phagosomal membrane, ultimately resulting in functional responses, including exocytosis, extravasation, phagocytosis, and elimination of various microorganisms (56–58). Several neutrophil responses, including exocytosis, chemotaxis, respiratory burst activity, and chemokine synthesis, are mediated through the p38 MAPK pathway (33). Activation of p38 MAPK signaling has also been associated with permitting neutrophils to sense and interpret the chemotactic sources by controlling the surface expression of adhesion molecules, like CD11b and CD66b, and chemoattractant receptors for fMLF and IL-8 (9). In this study, F. alocis induced secretory vesicle and specific granule exocytosis, which was mediated through TLR2 activation and was dependent on p38 MAPK. Secretory vesicles are organelles that are easy to mobilize and are involved in augmenting the number of receptors and adhesion molecules, like CD11b/CD18, which participate in the adhesion and transmigration process. F. alocis also induced a significant increase in the plasma membrane expression of CD66b, a specific granule marker used to evaluate granule exocytosis, and is involved in adhesion to fibronectin and E-selectin (51). When granule exocytosis was blocked with TAT-SNAP23 fusion protein (32), both random and directed migration induced by F. alocis challenge were impeded. It is plausible that F. alocis-induced secretory vesicle and specific granule exocytosis contribute to the enhanced chemotaxis toward IL-8 by increasing the availability of CXCR2 receptors as well as neutrophil adhesion capabilities by increasing CD66b plasma membrane expression. Moreover, we can speculate that in the context of periodontitis, besides the role of granule exocytosis on cell migration, the release of granule content to the extracellular space induced by F. alocis will contribute to tissue damage and disease progression.
Successful periodontal bacteria employ a variety of strategies to compromise neutrophil function. The major outer sheath protein (Msp) of Treponema denticola alters the balance of intracellular phosphoinositide, causing impairment of neutrophil directional migration toward fMLF and inhibition of downstream events, leading to chemotactic responses (59). Msp does not form a pore in neutrophils but remains associated with the plasma membrane, and it triggers “outside-in” signaling that results in inhibition of PI3-kinase activity and an increase in the activity of the phosphatase PTEN. The Msp virulence factor favors neutrophil PTEN activity over PI3K, resulting in a decrease in the amount of the phosphoinositide PIP3, which compromises actin dynamics, preventing the cell from having proper directed chemotaxis (59). The keystone periodontal pathogen, P. gingivalis, generates a local and transient chemokine paralysis by antagonizing the synthesis and release of IL-8 from gingival epithelial cells (60). The transient suppression of neutrophil recruitment to the gingival tissue facilitates the colonization of the tissue by P. gingivalis and other oral bacteria (61). Interaction between F. alocis and gingival epithelial cells results in release of IL-8 (11), and the current study shows that when F. alocis interacts with human neutrophils, there is no significant difference in cell migration toward fMLF compared to unstimulated cells, but there is significantly enhanced migration toward IL-8. Hence, F. alocis may function in obstructing the neutrophil from distinguishing between intermediary (IL-8) and end-target (fMLF) chemoattractants. As it is necessary for neutrophils to eventually migrate toward end-target chemoattractants in order to reach sites of infection, F. alocis manipulation of neutrophils could lead to defective deciphering abilities between chemoattractant sources, leading to constant migration and cell activation, which could contribute to dysregulated and sustained inflammation as well as tissue damage.
In conclusion, we showed that F. alocis significantly induces random and directed migration of human neutrophils toward IL-8. Activation of TLR2 by F. alocis incites a transient ERK1/2 activation and a sustained p38 MAPK activation, which results in exocytosis of secretory vesicles and specific granules. Ultimately, the p38 MAPK-dependent degranulation was responsible for F. alocis-enhanced neutrophil migration, which may contribute to dysbiotic host responses in the periodontium and promote tissue damage by activated neutrophils (Fig. 8).
ACKNOWLEDGMENTS
We thank Terri Manning for neutrophil isolation and for her expert technical help.
We have no conflicts of interest to declare.
REFERENCES
- 1.Hajishengallis G, Lamont RJ. 2014. Breaking bad: manipulation of the host response by Porphyromonas gingivalis. Eur J Immunol 44:328–338. doi: 10.1002/eji.201344202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajishengallis G. 2015. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 15:30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eke PI, Dye BA, Wei L, Slade GD, Thornton-Evans GO, Borgnakke WS, Taylor GW, Page RC, Beck JD, Genco RJ. 2015. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol 86:611–622. doi: 10.1902/jop.2015.140520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maddi A, Scannapieco FA. 2013. Oral biofilms, oral and periodontal infections, and systemic disease. Am J Dent 26:249–254. [PubMed] [Google Scholar]
- 5.Kumar PS. 2013. Oral microbiota and systemic disease. Anaerobe 24:90–93. doi: 10.1016/j.anaerobe.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML, Leys EJ. 2003. New bacterial species associated with chronic periodontitis. J Dent Res 82:338–344. doi: 10.1177/154405910308200503. [DOI] [PubMed] [Google Scholar]
- 7.Kumar PS, Griffen AL, Moeschberger ML, Leys EJ. 2005. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J Clin Microbiol 43:3944–3955. doi: 10.1128/JCM.43.8.3944-3955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hajishengallis G, Lamont RJ. 2016. Dancing with the stars: how choreographed bacterial interactions dictate nososymbiocity and give rise to keystone pathogens, accessory pathogens, and pathobionts. Trends Microbiol 24:477–489. doi: 10.1016/j.tim.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hajishengallis G. 2014. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol 35:3–11. doi: 10.1016/j.it.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Jotwani R, Le J, Krauss JL, Potempa J, Coventry SC, Uriarte SM, Lamont RJ. 2014. Filifactor alocis infection and inflammatory responses in the mouse subcutaneous chamber model. Infect Immun 82:1205–1212. doi: 10.1128/IAI.01434-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moffatt CE, Whitmore SE, Griffen AL, Leys EJ, Lamont RJ. 2011. Filifactor alocis interactions with gingival epithelial cells. Mol Oral Microbiol 26:365–373. doi: 10.1111/j.2041-1014.2011.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q, Wright CJ, Dingming H, Uriarte SM, Lamont RJ. 2013. Oral community interactions of Filifactor alocis in vitro. PLoS One 8:e76271. doi: 10.1371/journal.pone.0076271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aruni W, Chioma O, Fletcher HM. 2014. Filifactor alocis: the newly discovered kid on the block with special talents. J Dent Res 93:725–732. doi: 10.1177/0022034514538283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colombo AP, Boches SK, Cotton SL, Goodson JM, Kent R, Haffajee AD, Socransky SS, Hasturk H, Van Dyke TE, Dewhirst F, Paster BJ. 2009. Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol 80:1421–1432. doi: 10.1902/jop.2009.090185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahlen G, Leonhardt A. 2006. A new checkerboard panel for testing bacterial markers in periodontal disease. Oral Microbiol Immunol 21:6–11. doi: 10.1111/j.1399-302X.2005.00243.x. [DOI] [PubMed] [Google Scholar]
- 16.Aruni AW, Roy F, Fletcher HM. 2011. Filifactor alocis has virulence attributes that can enhance its persistence under oxidative stress conditions and mediate invasion of epithelial cells by porphyromonas gingivalis. Infect Immun 79:3872–3886. doi: 10.1128/IAI.05631-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aruni AW, Zhang K, Dou Y, Fletcher H. 2014. Proteome analysis of coinfection of epithelial cells with Filifactor alocis and Porphyromonas gingivalis shows modulation of pathogen and host regulatory pathways. Infect Immun 82:3261–3274. doi: 10.1128/IAI.01727-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aruni AW, Roy F, Sandberg L, Fletcher HM. 2012. Proteome variation among Filifactor alocis strains. Proteomics 12:3343–3364. doi: 10.1002/pmic.201200211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI. 2013. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J 7:1016–1025. doi: 10.1038/ismej.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oliveira RRDS, Fermiano D, Feres M, Figueiredo LC, Teles FRF, Soares GMS, Faveri M. 2016. Levels of candidate periodontal pathogens in subgingival biofilm. J Dent Res 95:711–718. doi: 10.1177/0022034516634619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlafer S, Riep B, Griffen AL, Petrich A, Hubner J, Berning M, Friedmann A, Gobel UB, Moter A. 2010. Filifactor alocis–involvement in periodontal biofilms. BMC Microbiol 10:66. doi: 10.1186/1471-2180-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Liu Y, Zhang M, Wang G, Qi Z, Bridgewater L, Zhao L, Tang Z, Pang X. 2015. A Filifactor alocis-centered co-occurrence group associates with periodontitis across different oral habitats. Sci Rep 5:9053. doi: 10.1038/srep09053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott DA, Krauss J. 2012. Neutrophils in periodontal inflammation. Front Oral Biol 15:56–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hart TC, Atkinson JC. 2007. Mendelian forms of periodontitis. Periodontol 2000 45:95–112. doi: 10.1111/j.1600-0757.2007.00233.x. [DOI] [PubMed] [Google Scholar]
- 25.Sima C, Gastfreund S, Sun C, Glogauer M. 2014. Rac-Null leukocytes are associated with increased inflammation-mediated alveolar bone loss. Am J Pathol 184:472–482. doi: 10.1016/j.ajpath.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 26.Yin X, Knecht DA, Lynes MA. 2005. Metallothionein mediates leukocyte chemotaxis. BMC Immunol 6:21. doi: 10.1186/1471-2172-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srinivasan S, Wang F, Glavas S, Ott A, Hofmann F, Aktories K, Kalman D, Bourne HR. 2003. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol 160:375–385. doi: 10.1083/jcb.200208179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selvatici R, Falzarano S, Mollica A, Spisani S. 2006. Signal transduction pathways triggered by selective formylpeptide analogues in human neutrophils. Eur J Pharmacol 534:1–11. doi: 10.1016/j.ejphar.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 29.Wong CH, Heit B, Kubes P. 2010. Molecular regulators of leucocyte chemotaxis during inflammation. Cardiovasc Res 86:183–191. doi: 10.1093/cvr/cvq040. [DOI] [PubMed] [Google Scholar]
- 30.Gambardella L, Vermeren S. 2013. Molecular players in neutrophil chemotaxis–focus on PI3K and small GTPases. J Leukoc Biol 94:603–612. doi: 10.1189/jlb.1112564. [DOI] [PubMed] [Google Scholar]
- 31.Heit B, Tavener S, Raharjo E, Kubes P. 2002. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol 159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uriarte SM, Rane MJ, Luerman GC, Barati MT, Ward RA, Nauseef WM, McLeish KR. 2011. Granule exocytosis contributes to priming and activation of the human neutrophil respiratory burst. J Immunol 187:391–400. doi: 10.4049/jimmunol.1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coxon PY, Rane MJ, Uriarte S, Powell DW, Singh S, Butt W, Chen Q, McLeish KR. 2003. MAPK-activated protein kinase-2 participates in p38 MAPK-dependent and ERK-dependent functions in human neutrophils. Cell Signal 15:993–1001. doi: 10.1016/S0898-6568(03)00074-3. [DOI] [PubMed] [Google Scholar]
- 34.Uriarte SM, Jog NR, Luerman GC, Bhimani S, Ward RA, McLeish KR. 2009. Counterregulation of clathrin-mediated endocytosis by the actin and microtubular cytoskeleton in human neutrophils. Am J Physiol Cell Physiol 296:C857–C867. doi: 10.1152/ajpcell.00454.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nussbaum G, Shapira L. 2011. How has neutrophil research improved our understanding of periodontal pathogenesis? J Clin Periodontol 38:49–59. doi: 10.1111/j.1600-051X.2010.01678.x. [DOI] [PubMed] [Google Scholar]
- 36.Roberts HM, Ling MR, Insall R, Kalna G, Spengler J, Grant MM, Chapple ILC. 2015. Impaired neutrophil directional chemotactic accuracy in chronic periodontitis patients. J Clin Periodontol 42:1–11. doi: 10.1111/jcpe.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellmé S, Mörgelin M, Tapper H, Mellqvist U-H, Dahlgren C, Karlsson A. 2006. Localization of human neutrophil interleukin-8 (CXCL-8) to organelle(s) distinct from the classical granules and secretory vesicles. J Leukoc Biol 79:564–573. [DOI] [PubMed] [Google Scholar]
- 38.Bazzoni F, Cassatella MA, Rossi F, Ceska M, Dewald B, Baggiolini M. 1991. Phagocytosing neutrophils produce and release high amounts of the neutrophil-activating peptide 1/interleukin 8. J Exp Med 173:771–774. doi: 10.1084/jem.173.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayashi F, Means TK, Luster AD. 2003. Toll-like receptors stimulate human neutrophil function. Blood 102:2660–2669. doi: 10.1182/blood-2003-04-1078. [DOI] [PubMed] [Google Scholar]
- 40.Aomatsu K, Kato T, Fujita H, Hato F, Oshitani N, Kamata N, Tamura T, Arakawa T, Kitagawa S. 2008. Toll-like receptor agonists stimulate human neutrophil migration via activation of mitogen-activated protein kinases. Immunology 123:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward RA, Nakamura M, McLeish KR. 2000. Priming of the neutrophil respiratory burst involves p38 mitogen-activated protein kinase-dependent exocytosis of flavocytochrome b 558-containing granules. J Biol Chem 275:36713–36719. doi: 10.1074/jbc.M003017200. [DOI] [PubMed] [Google Scholar]
- 42.Colvin RA, Means TK, Diefenbach TJ, Moita LF, Friday RP, Sever S, Campanella GSV, Abrazinski T, Manice LA, Moita C, Andrews NW, Wu D, Hacohen N, Luster AD. 2010. Synaptotagmin-mediated vesicle fusion regulates cell migration. Nat Immunol 11:495–502. doi: 10.1038/ni.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soehnlein O, Weber C, Lindbom L. 2009. Neutrophil granule proteins tune monocytic cell function. Trends Immunol 30:538–546. doi: 10.1016/j.it.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 44.Becker EL, Showell HJ, Henson PM, Hsu LS. 1974. The ability of chemotactic factors to induce lysosomal enzyme release. I. The characteristics of the release, the importance of surfaces and the relation of enzyme release to chemotactic responsiveness. J Immunol 112:2047–2054. [PubMed] [Google Scholar]
- 45.Glushkova OV, Parfenyuk SB, Khrenov MO, Novoselova TV, Lunin SM, Fesenko EE, Novoselova EG. 2013. Inhibitors of TLR-4, NF-κB, and SAPK/JNK signaling reduce the toxic effect of lipopolysaccharide on RAW 2647 cells J Immunotoxicol 10:133–140. [DOI] [PubMed] [Google Scholar]
- 46.Hajishengallis E, Hajishengallis G. 2014. Neutrophil homeostasis and periodontal health in children and adults. J Dent Res 93:231–237. doi: 10.1177/0022034513507956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hajishengallis G, Moutsopoulos NM, Hajishengallis E, Chavakis T. 2016. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin Immunol 28:146–158. doi: 10.1016/j.smim.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foxman EF, Campbell JJ, Butcher EC. 1997. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J Cell Biol 139:1349–1360. doi: 10.1083/jcb.139.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan J, Malik AB. 2003. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat Med 9:315–321. doi: 10.1038/nm832. [DOI] [PubMed] [Google Scholar]
- 50.Heit B, Colarusso P, Kubes P. 2005. Fundamentally different roles for LFA-1, Mac-1 and alpha4-integrin in neutrophil chemotaxis. J Cell Sci 118:5205–5220. doi: 10.1242/jcs.02632. [DOI] [PubMed] [Google Scholar]
- 51.Kim D, Haynes CL. 2013. The role of p38 MAPK in neutrophil functions: single cell chemotaxis and surface marker expression. Analyst 138:6826–6833. doi: 10.1039/c3an01076g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heit B, Liu L, Colarusso P, Puri KD, Kubes P. 2008. PI3K accelerates, but is not required for, neutrophil chemotaxis to fMLP. J Cell Sci 121:205–214. doi: 10.1242/jcs.020412. [DOI] [PubMed] [Google Scholar]
- 53.Zu YL, Qi J, Gilchrist A, Fernandez GA, Vazquez-Abad D, Kreutzer DL, Huang CK, Sha'afi RI. 1998. p38 mitogen-activated protein kinase activation is required for human neutrophil function triggered by TNF-alpha or FMLP stimulation. J Immunol 160:1982–1989. [PubMed] [Google Scholar]
- 54.Knall C, Worthen GS, Johnson GL. 1997. Interleukin 8-stimulated phosphatidylinositol-3-kinase activity regulates the migration of human neutrophils independent of extracellular signal-regulated kinase and p38 mitogen-activated protein kinases. Pro Natl Acad Sci U S A 94:3052–3057. doi: 10.1073/pnas.94.7.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Ma B, Malik AB, Tang H, Yang T, Sun B, Wang G, Minshall RD, Li Y, Zhao Y, Ye RD, Xu J. 2012. Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nat Immunol 13:457–464. doi: 10.1038/ni.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rorvig S, Ostergaard O, Heegaard NH, Borregaard N. 2013. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. J Leukoc Biol 94:711–721. doi: 10.1189/jlb.1212619. [DOI] [PubMed] [Google Scholar]
- 57.Catz SD. 2014. The role of Rab27a in the regulation of neutrophil function. Cell Microbiol 16:1301–1310. doi: 10.1111/cmi.12328. [DOI] [PubMed] [Google Scholar]
- 58.Dib K, Perecko T, Jenei V, McFarlane C, Comer D, Brown V, Katebe M, Scheithauer T, Thurmond RL, Chazot PL, Ennis M. 2014. The histamine H4 receptor is a potent inhibitor of adhesion-dependent degranulation in human neutrophils. J Leukoc Biol 96:411–418. doi: 10.1189/jlb.2AB0813-432RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Visser MB, Sun C-X, Koh A, Ellen RP, Glogauer M. 2013. Treponema denticola major outer sheath protein impairs the cellular phosphoinositide balance that regulates neutrophil chemotaxis. PLoS One 8:e66209. doi: 10.1371/journal.pone.0066209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takeuchi H, Hirano T, Whitmore SE, Morisaki I, Amano A, Lamont RJ. 2013. The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-kappaB RelA/p65. PLoS Pathog 9:e1003326. doi: 10.1371/journal.ppat.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zenobia C, Hajishengallis G. 2015. Porphyromonas gingivalis virulence factors involved in subversion of leukocytes and microbial dysbiosis. Virulence 6:236–243. doi: 10.1080/21505594.2014.999567. [DOI] [PMC free article] [PubMed] [Google Scholar]