

Abstract
Streptococcus gordonii and related species of oral viridans group streptococci (VGS) are common etiological agents of infective endocarditis (IE). We explored vaccination as a strategy to prevent VGS-IE, using a novel antigen-presenting system based on non-genetically modified Lactococcus lactis displaying vaccinogens on its surface. Hsa and PadA are surface-located S. gordonii proteins implicated in platelet adhesion and aggregation, which are key steps in the pathogenesis of IE. This function makes them ideal targets for vaccination against VGS-IE. In the present study, we report the use of nonliving L. lactis displaying at its surface the N-terminal region of Hsa or PadA by means of the cell wall binding domain of Lactobacillus casei A2 phage lysine LysA2 (Hsa-LysA2 and PadA-LysA2, respectively) and investigation of their ability to elicit antibodies in rats and to protect them from S. gordonii experimental IE. Immunized and control animals with catheter-induced sterile aortic valve vegetations were inoculated with 106 CFU of S. gordonii. The presence of IE was evaluated 24 h later. Immunization of rats with L. lactis Hsa-LysA2, L. lactis PadA-LysA2, or both protected 6/11 (55%), 6/11 (55%), and 11/12 (91%) animals, respectively, from S. gordonii IE (P < 0.05 versus controls). Protection correlated with the induction of high levels of functional antibodies against both Hsa and PadA that delayed or totally inhibited platelet aggregation by S. gordonii. These results support the value of L. lactis as a system for antigen delivery and of Hsa and PadA as promising candidates for a vaccine against VGS-IE.
INTRODUCTION
The viridans group streptococci (VGS) are commensal bacteria of the human oral cavity but can cause infective endocarditis (IE) when they enter the bloodstream (1). VGS-IE accounts for ca. 20% of IE cases (1) and generally results from cumulative exposure to recurrent bouts of transient low-grade bacteremia, occurring during normal day-to-day activities, including tooth brushing, flossing, and chewing (2–4). Under these circumstances, antibiotic prophylaxis regimens cannot be recommended to prevent VGS-IE. Based upon this assumption, the American Heart Association (AHA) and the European Society of Cardiology (ESC) drastically restricted the use of antibiotic prophylaxis for IE in at-risk patients undergoing dental procedures (5, 6). The British National Institute for Health and Clinical Excellence (NICE) went even further and suggested the total abolition of antibiotic-based prophylaxis (7). However, since the AHA guidelines' revision in 2007, a significant increase in the incidence of VGS-IE has been reported in the United States (8). This suggests that the development of an effective prophylactic strategy against VGS-IE is an unmet medical need.
A number of immunization strategies for the prevention of VGS-IE have been explored in the past and have been shown to protect animal models from IE (9–13). However, no further step has been made toward the development of vaccines against oral streptococci, and no vaccine currently exists against VGS-IE in the market.
The oral VGS bacterium Streptococcus gordonii is a major etiological agent of IE (14). S. gordonii is well known for its ability to interact with human platelets, a step that is considered crucial for the initiation and progression of IE (15, 16). S. gordonii adheres to platelets via the surface-anchored proteins Hsa (hemagglutinin salivary antigen) and PadA (platelet adherence protein A). Hsa mediates the initial interactions with platelets by binding the membrane glycoprotein GPIbα (17–20). The high on-off rate of GPIbα allows rapid loss and formation of new interactions between platelets and the immobilized bacteria, leading to platelets rolling over the microorganisms. This process, which slows down platelets from the high shear stress experienced in the bloodstream, is then followed by the interaction of PadA with the platelet receptor GPIIβIIIα, which promotes firm bacterium-platelet adhesion and ultimately leads to platelet aggregation (21, 22).
Due to their role in platelet aggregation, Hsa and PadA (18, 22) represent intuitively logical candidates for vaccine development against IE induced by VGS. In the present study, we employed a recently developed antigen display system (23) to immunize rats with both adhesins. This system is based on nonliving, non-genetically modified Lactococcus lactis cells displaying on the cell wall the functional N-terminal region (directly involved in platelet activation) of Hsa or PadA fused to the C-terminal domain of Lactobacillus casei A2 phage lysine (LysA2), which was previously shown to bind to the cell wall of a wide spectrum of lactic acid bacteria (24). The immunizations with L. lactis displaying Hsa-LysA2 (L. lactis Hsa-LysA2) and L. lactis displaying PadA-LysA2 (L. lactis PadA-LysA2), individually or after coimmunization, were evaluated for their ability to induce specific antibodies in rats and to protect against S. gordonii experimental IE.
Our results indicate that immunization of rats with L. lactis Hsa-LysA2 and/or L. lactis PadA-LysA2, individually or together, was effective in inducing functional Hsa- and PadA-specific antibodies that inhibited platelet aggregation and protected against S. gordonii experimental IE. Taken together, these results support the suitability of Hsa and PadA as potential candidates for the development of an anti-VGS-IE vaccine.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
L. lactis (strain MG1363) (25) was grown at 30°C in M17 broth medium (Difco-Becton Dickinson, Sparks, MD) containing 1% glucose (GM17). S. gordonii Challis (strain DL1) (19) was grown at 37°C in brain infusion broth (Difco-Becton Dickinson) in the presence of 5% CO2. Escherichia coli DH5α (Invitrogen, Carlsbad, CA) and E. coli BL21(DE3)pLysS (24) were grown in Luria-Bertani (LB) broth (Difco-Becton Dickinson).
Construction of the plasmids carrying Hsa-LysA2 and PadA-LysA2 fusion cassettes.
Genomic DNA was extracted from S. gordonii using a genomic DNA purification kit (Thermo Fisher Scientific, Waltham, MA), according to the manufacturer's instructions. The N-terminal regions coding for amino acids (aa) 39 to 449 of hsa and 35 to 1327 of padA were PCR amplified from S. gordonii genomic DNA using forward primers hsa-fw (ATATGGATCCTTTAAGATTAATGAAGGGTGCTG) and padA-fw (ATATGGATCCTGATCCTCTGAATATTGAGACG) containing BamHI recognition sites (underlined) and reverse primers hsa-rv (TATACGGCCGTACTAATCTGAAGATCTTTAAC) and padA-rv (TATACGGCCGCTTCCAAGGTATCAGAAAGTACA) containing EagI recognition sites (underlined). The resulting products of 1,258 bp and 3,901 bp were digested with BamHI and EagI (Promega, Madison, WI) and ligated into the pLysA2 vector, a plasmid containing the gene coding for bacteriophage A2 of L. casei (24), previously digested with the same enzymes. The resulting constructs, namely, pHsa-LysA2 and pPadA-LysA2, encoded the N-terminal regions of Hsa and PadA with an N-terminal 6-His tag and Xpress epitope and fused to the cell-binding domain of L. casei LysA2 at the C terminus (Fig. 1). The correct insertion of the genes and the absence of mutations were verified by commercial DNA sequencing. Plasmids containing solely the N-terminal regions coding for amino acids 39 to 449 of hsa (pHsa) and 35 to 1327 of padA (pPadA) were generated using the Q5 site-directed mutagenesis kit (New England BioLabs, Ipswich, MA) according to the manufacturer's instructions. Forward primers hsa-fw2 (TAAGCATATTACAAAAATGCC) and padA-fw2 (TAAGCATATTACAAAAATGCC) and reverse primers hsa-rv2 (CGTACTAATCTGAAGATCTTTAAC) and padA-rv2 (TTCCAAGGTATCAGAAAG) were used to delete the LysA2 coding region from pHsa-LysA2 and pPadA-LysA2, respectively, thereby generating pHsa and pPadA. All the plasmids were propagated in E. coli DH5α and purified with the Wizard Plus SV Minipreps DNA purification system (Promega), according to the manufacturer's instructions.
FIG 1.

Schematic representations of PadA, Hsa, LysA2, and the chimeric proteins Hsa-LysA2 and PadA-LysA2. PadA is composed of a leader peptide (in black), a unique N-terminal region (in gray) which comprises a von Willebrand factor-A1-like domain, 14 repeat blocks with unknown function (in white), and a C-terminal cell wall anchoring domain (with diagonal lines). The primary sequence of Hsa comprises a leader peptide (in black), two serine-rich regions (in white), one located in the N-terminal region and the other in the C-terminal region (in gray), and a C-terminal cell wall-anchoring domain (with diagonal lines). LysA2 contains an N-terminal catalytic domain (with vertical lines) and a C-terminal cell wall binding domain (in light gray). Molecular weights at right are in thousands.
Purification of Hsa, PadA, Hsa-LysA2, and PadA-LysA2.
Hsa (aa 39 to 449), PadA (aa 35 to 1327), Hsa-LysA2, and PadA-LysA2 were purified as previously described (24). Briefly, E. coli BL21(DE3)pLysS containing pHsa, pPadA, pHsa-LysA2, and pPadA-LysA2 was grown aerobically at 37°C in LB containing ampicillin (100 μg/ml) until the mid-exponential phase (optical density at 600 nm [OD600], ∼0.5). Then, cells were induced with 1 mM isopropyl-β-d-thiogalactoside (Roche Diagnostics, Basel, Switzerland), and the growth was continued overnight at 18°C. The cells were then harvested by centrifugation, washed with buffer P (100 mM phosphate buffer, pH 5.5) with 5 mM imidazole, and resuspended in 20 ml of the same buffer. Cells were then broken by three passages through a French press at 40 MPa, and cell debris was removed by centrifugation at 4°C. The resulting supernatant was incubated with 0.5 ml nickel-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen, Germantown, MD) for 2 h at 4°C in an end-over-end tube rotator. The suspension was then loaded on a column, and the resin was washed with 10 volumes of buffer P, followed by 10 volumes of the same buffer with 30 mM imidazole. The proteins were eluted with 5 ml buffer P containing 300 mM imidazole. The eluates were analyzed on a 12% acrylamide gel and dialyzed against 100 volumes of phosphate-buffered saline (PBS) twice for 2 h at 4°C. The protein concentrations were determined with the Bio-Rad protein assay, using bovine serum albumin (BSA) as standard.
Binding of Hsa-LysA2 and PadA-LysA2 fusion proteins on the surface of L. lactis.
L. lactis was grown in MG17 until the exponential phase (OD600, ∼0.5). Then, cells were harvested by centrifugation, washed two times with PBS, and resuspended in the same buffer to obtain a concentration of 109 CFU/ml. Bacteria were then killed by exposure to UV light for 1 h at 1 J/cm2 (26). UV-induced killing was confirmed by the absence of viable organisms. The cells (100 μl) were then mixed with increasing amounts of Hsa-LysA2 or PadA-LysA2 in order to determine the amounts of proteins necessary for the complete saturation of the L. lactis surface and incubated for 1 h at 30°C to promote binding of the proteins. L. lactis cells were also incubated with BSA (control). Cell protein suspensions were then centrifuged, washed with PBS, and resuspended into 100 μl of the same buffer.
Assessment of Hsa-LysA2 and PadA-LysA2 levels on L. lactis surface by enzyme-linked immunosorbent assay (ELISA).
One hundred microliters of UV-treated L. lactis cells (final concentration, 108 CFU/ml) preincubated with different amounts of Hsa-LysA2 and PadA-LysA2 was adsorbed on an F96 MaxiSorp microplate (Thermo Fisher Scientific) for 18 h at 4°C. The presence of Hsa-LysA2 and PadA-LysA2 on the surface of immobilized L. lactis cells was detected using a mouse anti-Xpress antibody (Invitrogen) and a peroxidase-conjugated goat anti-mouse secondary antibody (Dako, Baar, Switzerland). Fifteen minutes after the addition of the tetramethylbenzidine (TMB) solution (Thermo Fisher Scientific), the reactions were stopped with 2 M H2SO4. The absorbance at 450 nm was then measured with the Infinite 200 Pro microplate reader (Tecan, Maennedorf, Switzerland). The assays were carried out in triplicate.
Western blot analysis for the detection of surface-associated Hsa-LysA2 and PadA-LysA2.
Twenty microliters of UV-inactivated L. lactis used for the immunization studies (see below) was mixed with 10 μl NuPAGE lithium dodecyl sulfate (LDS) sample buffer (4×; Invitrogen) and boiled for 10 min at 99°C. The resulting protein samples together with 1 ng of each purified protein were resolved by 10% SDS-polyacrylamide gel electrophoresis. Western blotting (WB) was performed using a standard procedure (27). Blots were incubated with a 1:1,000 dilution of a mouse anti-Xpress antibody (Invitrogen) and successively with a 1:3,000 dilution of a peroxidase-conjugated goat anti-mouse secondary antibody (Dako). The bands were detected by chemiluminescence with the enhanced chemiluminescence (ECL) Western blotting detection reagents (Amersham Biosciences. Piscataway, NJ).
Animal studies.
All animal protocols were reviewed and approved by the Cantonal Committee on Animal Experiments of the State of Vaud, Switzerland (permit number 879.9). A mixture of ketamine (75 mg/kg of body weight) and midazolam (5 mg/kg) anesthetics was administered to the animals before any surgical procedure.
Immunization studies.
UV-killed L. lactis (108 CFU) mixed with 2 μg of Hsa-LysA2 (L. lactis Hsa-LysA2) or 5 μg of PadA-LysA2 (L. lactis PadA-LysA2), the smallest amounts of proteins sufficient to saturate the L. lactis surface (see Results), was used for immunization.
Rats (6-week-old female Wistar rats) were injected subcutaneously with an initial dose of L. lactis Hsa-LysA2, L. lactis PadA-LysA2, or the combination of the two emulsified with complete Freund's adjuvant (Sigma-Aldrich, Buchs, Switzerland), followed by two booster doses in incomplete Freund's adjuvant (Sigma-Aldrich) at 14-day intervals. Control groups of rats were sham immunized with L. lactis plus the adjuvant. Blood samples were collected at days 0 (before immunization) and 45 (at the time of catheter insertion, 14 days after the third immunization dose; see below). Following centrifugation (3,000 rpm for 15 min at 4°C), sera were stored in aliquots at −80°C until use.
Animal model of endocarditis.
The production of catheter-induced aortic vegetations and the installation of an intravenous (i.v.) line to deliver the bacterial inocula were performed in rats 2 weeks after the last immunization dose, i.e., on day 45, as described previously (28).
Twenty-four hours later, immunized and control rats were inoculated i.v. by continuous infusion (0.0017 ml/min over 10 h) of 106 CFU of S. gordonii, to simulate cumulative exposure to low-grade bacteremia (28). Rats were euthanized 24 h after the end of the inoculation. The cardiac vegetations and the spleen were removed, weighed, homogenized, serially diluted, and plated on blood agar plates. Plates were incubated for 48 h at 37°C to determine the number of viable organisms in the tissues.
ELISA for detection of antibodies.
The presence of polyclonal antibodies specific for Hsa and PadA in sequentially diluted (from 1:1,000 to 1:100,000) rat sera was determined by ELISA as described above. Binding of antibodies to the immobilized N-terminal regions of Hsa and PadA absorbed for 18 h at 4°C on the microplate was detected using peroxidase-conjugated goat anti-rat secondary antibodies. The reactions were stopped with 2 M H2SO4, and the absorbance at 450 nm was measured with Infinite 200 Pro microplate reader (Tecan). The assays were performed in triplicate on two independent occasions.
Characterization of functional antibodies in sera.
The presence of functional antibodies was determined by testing the ability of sera from immunized rats to inhibit platelet aggregation by S. gordonii.
Platelet-rich plasma (PRP) and platelet-poor plasma (PPP) for platelet aggregation tests were obtained from anticoagulated human blood as described previously (29).
The ability of sera from immunized rats to inhibit S. gordonii-induced platelet aggregation was evaluated in vitro by conventional light aggregometry as previously described (29). Briefly, 20 μl of S. gordonii (final concentration of 1 × 109 CFU/ml in Tyrode's buffer) was preincubated for 1 h at room temperature with 2 μl of control or anti-Hsa-, anti-PadA-, and anti-Hsa- plus anti-PadA-containing sera and then added to 180 μl of PRP in siliconized flat-bottom cuvettes. Sera from sham-immunized rats were used as a control. ADP, a known inducer of platelet aggregation, was used as a positive control. Platelet aggregation was assessed by monitoring light transmission at 37°C with stirring at 120 rpm using a LaBiTec Apact 4004 aggregometer (LaBiTec GmbH, Ahrensburg, Germany). The light transmission of PRP without added bacteria and the light transmission of PPP were defined as 0% and 100% light transmission, respectively. Aggregation was recorded over 20 min. Platelet aggregation was expressed as the interval between the addition of organism-serum samples to the PRP suspension and the onset of aggregation response (lag time) and the number of platelets (percent) that were aggregated in the presence of the bacterium-serum suspension (maximal aggregation). The final values represent results of three independent experiments performed in quadruplicate.
Statistical analysis.
Data for antibody titers, platelet aggregation assays, and mean bacterial densities in vegetations and spleens were evaluated by the Student t test. The incidences of valve infection were compared by the chi-square test. A value of P < 0.05 was considered significant by using two-tailed significance levels. All statistical analyses were performed with the GraphPad Prism 6.0 program (GraphPad, Inc., La Jolla, CA).
RESULTS
Generation of chimeric Hsa-LysA2 and PadA-LysA2.
LysA2, the L. casei bacteriophage A2 lysine, consists of an N-terminal catalytic domain and a C-terminal cell wall binding domain (CBD), which was previously shown to bind with high affinity to L. lactis (23). By virtue of this property, LysA2 CBD was here used to efficiently immobilize Hsa and PadA on the surface of L. lactis, which served as an antigen-presenting system. Since Hsa and PadA are large surface-associated proteins composed of 2,178 and 3,646 amino acid residues, respectively, only the functional surface-exposed N-terminal regions were used to generate the fusion proteins. Hsa contains two serine-rich regions (SRs), which are associated with the binding to the sialylated N-terminal region of GPIbα on platelets. SR1 is relatively short (81 amino acid residues) and is located in the N-terminal region between two nonrepetitive regions (NR1 and NR2). In contrast, SR2 is a long serine-rich region containing 113 dodecapeptide repeats, which are comprised between the N-terminal region and the C-terminal cell wall anchoring domain (Fig. 1). Since SR1, NR1, and NR2, but not SR2, were previously shown to be indispensable for the binding to sialoglycoconjugates (18), the N-terminal region spanning SR1, NR1, and NR2 was used to generate Hsa-LysA2. PadA consists of a leader peptide, an N-terminal region comprising a von Willebrand factor-like domain, a long C-terminal domain with 14 repeat blocks of 148 to 152 amino acid residues, and a cell wall anchor domain (Fig. 1). Analogously to Hsa, the N-terminal region was used for the generation of PadA-LysA2, as it was shown to be directly involved in the binding to platelets (21).
Binding of surface-saturating levels of Hsa-LysA2 and PadA-LysA2 on L. lactis.
The ability of the Hsa-LysA2 and PadA-LysA2 fusion proteins to bind to the L. lactis cell wall and the optimal protein/cell ratio for antigen presentation were assessed by ELISA and Western blotting (WB). As shown in Fig. 2A, the absorbance increased with the amounts of proteins added to L. lactis cells, with maximal signal intensities obtained upon addition of 2 μg of Hsa-LysA2 and 5 μg of PadA-LysA2. Interestingly, further addition of the fusion proteins did not result in increased chemiluminescent signals, suggesting that the binding capacity of the L. lactis cells was saturated at those concentrations. Consistently, bands corresponding to Hsa-LysA2 and PadA-LysA2 appeared in L. lactis preincubated with 2 μg of Hsa-LysA2 and 5 μg of PadA-LysA2 (Fig. 2B), further corroborating the idea that the chimeric proteins were firmly attached on the cell wall.
FIG 2.
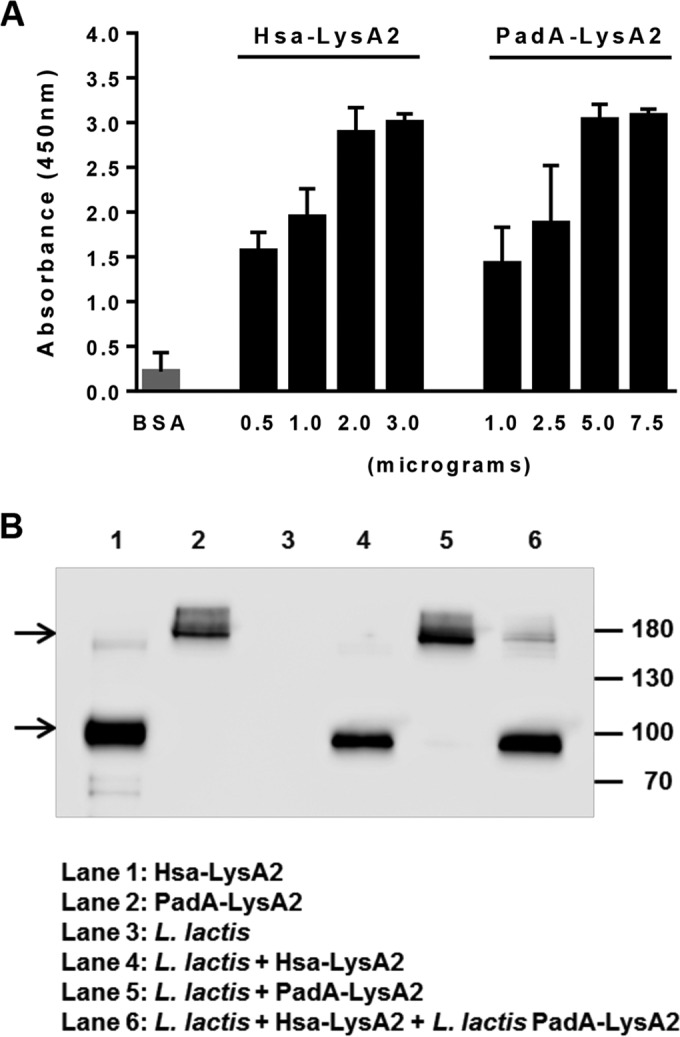
(A) Relative levels of cell wall-associated Hsa-LysA2 and PadA-LysA2. The indicated amounts of Hsa-LysA2 and PadA-LysA2 were incubated with 108 CFU of L. lactis, and levels of surface-associated proteins were quantified by ELISA. Data were produced from three independent experiments. (B) Detection of Hsa-LysA2 (66 kDa) and PadA-LysA2 (165 kDa) bound to L. lactis. Crude extracts of equivalent amounts of L. lactis used for the immunization were separated by polyacrylamide gel electrophoresis, followed by Western blotting and development with a monoclonal antibody that recognizes the N-terminal Xpress epitope present on Hsa-LysA2 and PadA-LysA2. Molecular masses in kilodaltons are indicated at the right of the gel. The arrows indicate the bands corresponding to PadA-LysA2 (top) and Hsa-LysA2 (bottom).
Vaccination with L. lactis Hsa-LysA2, L. lactis PadA-LysA2, and L. lactis Hsa-LysA2 plus L. lactis PadA-LysA2 efficiently elicited anti-Hsa and anti-PadA antibodies in rats.
Animals were immunized with three subcutaneous doses of L. lactis Hsa-LysA2, L. lactis PadA-LysA2, or the combination of the two bacteria given at intervals of 15 days. Antibody titers of Hsa and PadA in sera were assessed after the third dose. As shown in Fig. 3, sera obtained from rats immunized with L. lactis Hsa-LysA2 or L. lactis PadA-LysA2 showed an approximately 10- to 15-fold increase in specific antibodies compared to those of control rats and animals before immunization. Coimmunization with L. lactis Hsa-LysA2 and L. lactis PadA-LysA2 induced antibody levels comparable to those of single-antigen-immunized animals.
FIG 3.
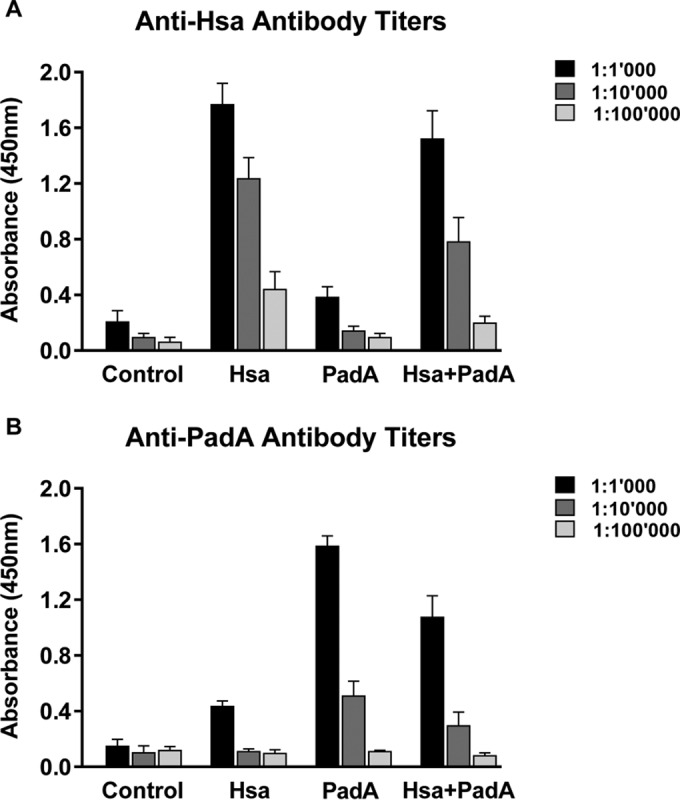
Relative anti-Hsa (A) and anti-PadA (B) antibody levels in the sera of rats immunized with 108 CFU of either L. lactis (control), L. lactis Hsa-LysA2 (Hsa), L. lactis PadA-LysA2 (PadA), or the combination of L. lactis Hsa-LysA2 and L. lactis PadA-LysA2 (Hsa+PadA) as described in Materials and Methods. Antibodies were quantified by ELISA. Data represent the mean ± standard deviation from six samples from six individual rats per group.
Anti-Hsa and anti-PadA antibodies inhibited S. gordonii-induced platelet aggregation.
As shown in Fig. 4, sera from L. lactis Hsa-LysA2- or L. lactis PadA-LysA2-immunized animals significantly delayed S. gordonii-induced platelet aggregation (lag time of 14.0 ± 1.6 min and 15.0 ± 1.2 min, respectively), compared to sera from sham-immunized rats (lag time of 8.0 ± 0.8 min; P < 0.0005). Moreover, upon preincubation with anti-Hsa or anti-PadA, S. gordonii exhibited comparable maximum platelet aggregation percentages (64% and 56%, respectively), which were significantly lower than that induced after the addition of control sera (73%; P < 0.005). Remarkably, sera taken from animals coimmunized with L. lactis Hsa-LysA2 and L. lactis PadA-LysA2 completely prevented S. gordonii-induced platelet aggregation (lag time, >20 min; P < 0.0001) and reduced maximum aggregation to 13% (P < 0.0001).
FIG 4.
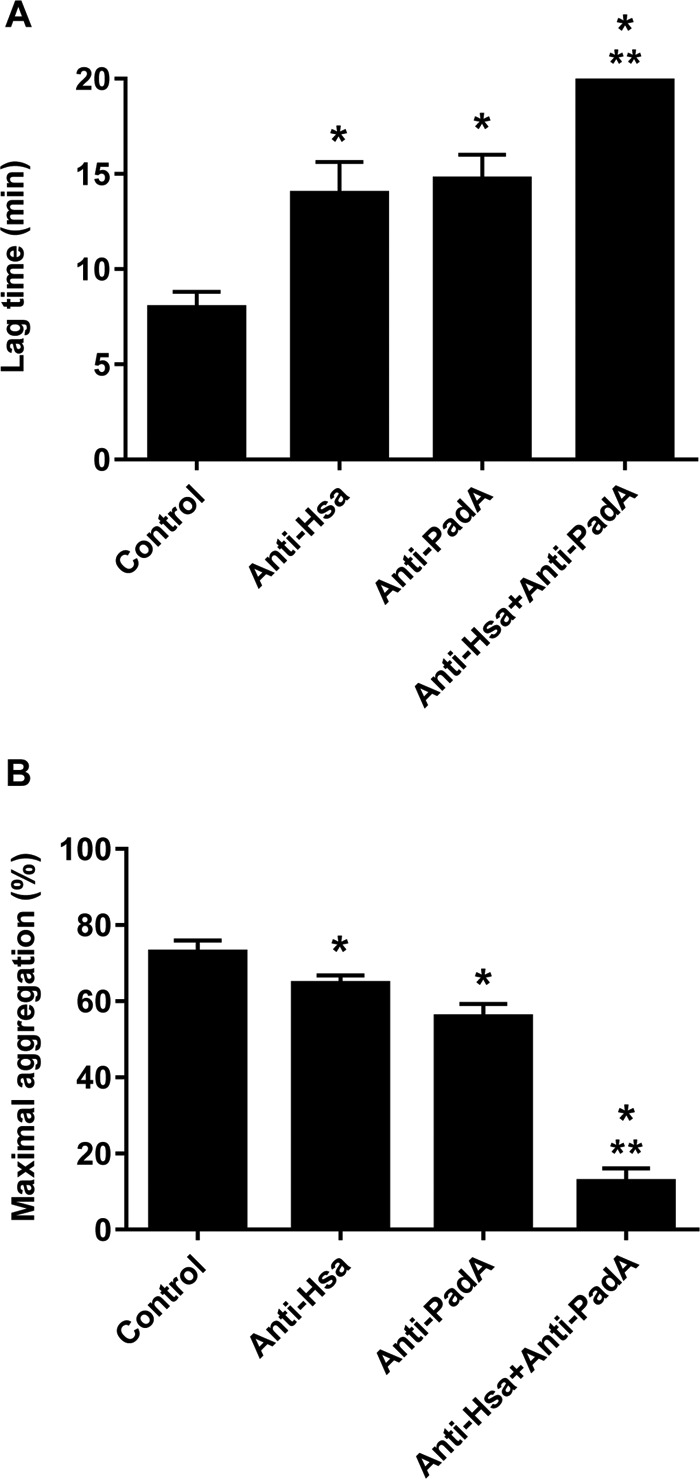
Effects of anti-Hsa, anti-PadA, and control sera on S. gordonii-induced platelet aggregation. Sera (10 μl) were preincubated with 108 CFU of S. gordonii (100 μl), and then platelet aggregability was assessed. Lag time (A) indicates the time between the addition of bacterium-serum mixtures to the PRP suspensions and the onset of aggregation response, and maximal aggregation (B) represents the total number (percent) of aggregated platelets. Results are expressed as the mean ± standard deviation from four samples from four individual rats per group, which were obtained in two independent assays. Lag times of 20 min indicate that there was no aggregation in the time window used to monitor platelet aggregation. *, P < 0.005 compared to controls (Student's t test); **, P < 0.001 compared to anti-Hsa and anti-PadA groups (Student's t test).
Vaccination with L. lactis Hsa-LysA2/PadA-LysA2 successfully protected rats against S. gordonii-induced experimental IE.
As shown in Fig. 5, while 14/15 (93%) sham-immunized rats developed S. gordonii IE, only 5/11 (45%) animals immunized with L. lactis Hsa-LysA2, 5/11 (45%) animals immunized with L. lactis PadA-LysA2, and 1/12 (9%) animals coimmunized with L. lactis Hsa-LysA2 and L. lactis PadA-LysA2 showed infected vegetations (P < 0.05). Interestingly, significantly higher levels of protection against S. gordonii IE were obtained with coimmunization than in single-immunized groups (P < 0.05).
FIG 5.
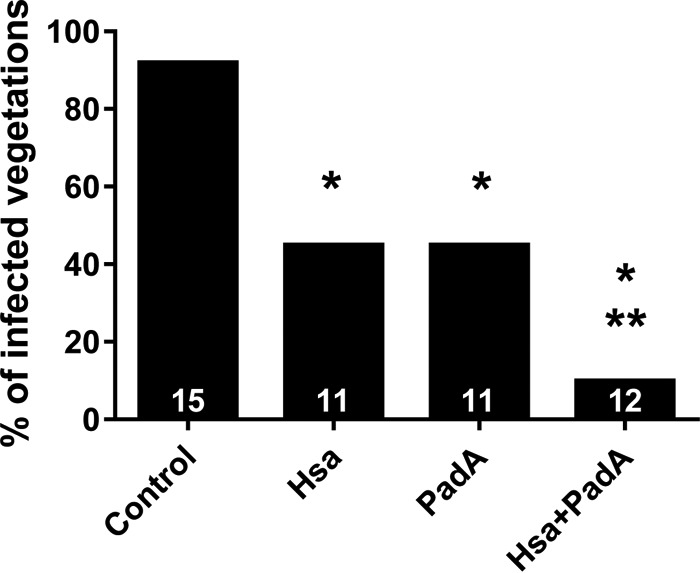
Protective effects of immunization with either L. lactis (control), L. lactis Hsa-LysA2 (Hsa), L. lactis PadA-LysA2 (PadA), or the combination of the two bacteria (Hsa+PadA) against experimental endocarditis induced by S. gordonii. Values on the bottom of each column are numbers of animals in each group. *, P < 0.05 by chi-square test compared to controls; **, P < 0.05 by chi-square test compared to Hsa and PadA groups.
Bacterial counts in vegetations that remained infected were not significantly different between immunized and control rats (6.38 to 7.09 log10 CFU/g).
DISCUSSION
In the present study, we investigated the potential of L. lactis displaying S. gordonii Hsa or PadA as a vaccine candidate against S. gordonii infection in a rat model of IE.
We used for the immunization a recently developed novel display system that allows highly efficient immobilization of antigens on the surface of lactic acid bacteria, such as L. lactis (23). This system is based on non-genetically modified L. lactis used as a vehicle for antigens bound on the surface by means of a high-affinity cell-binding domain (CBD) from L. casei A2 phage lysine LysA2 (24). Such a system offers some advantages over a previously used immunization method based on recombinant L. lactis expressing bacterial antigens, such as the Staphylococcus aureus clumping factor A (ClfA) or the fibronectin binding protein A (FnbpA) (26). First, it allows use of a non-genetically modified antigen-presenting system. Second, it permits the clustering of antigens on the L. lactis cell wall, a feature that increases antigen stimulation of the immune system. Consistent with this notion, while in a pilot study anti-Hsa antibody levels were barely detectable in rat plasma after immunization with recombinant L. lactis expressing Hsa (data not shown), vaccination with L. lactis with cell wall-saturating amounts of Hsa-LysA2 or PadA-LysA2 fusion proteins triggered production of antibodies against either immunogen with levels approximately 10 to 15 times higher than those found in the sham-immunized rats. Also, whether this strategy is more effective than the vaccination with proteins alone remains to be investigated.
Why then Hsa and PadA? Hsa and PadA were chosen as antigens for two main reasons. First, Hsa is a known virulence factor of S. gordonii associated with IE (30). Indeed, deletion of hsa was shown to significantly reduce the ability of S. gordonii to infect rat aortic valves (30). The role of PadA in IE has not been hitherto analyzed. Second, Hsa and PadA appear to be major factors in triggering S. gordonii platelet adhesion and aggregation, which are crucial steps in the pathogenesis of IE (15).
Consistent with this notion, we here showed that S. gordonii-induced platelet aggregation was delayed by anti-Hsa and anti-PadA and, most importantly, completely abolished by the combination of the two. These observations, however, are in contrast with previous results, which showed that concomitant genomic inactivation of hsa and padA did not have any effect on S. gordonii-induced platelet aggregation (21). These contradictory results might be attributed to the cross-reactivity of the anti-Hsa and anti-PadA antibodies with other S. gordonii (DL1) adhesins that interact with platelets, such as SspA and SspB (19). In support of this assumption, SspA and SspB sequences exhibit 20% identity to Hsa (see Fig. S1 in the supplemental material). Clearly, this hypothesis requires experimental validation.
The results of the vaccination studies also identified Hsa and PadA as factors primarily responsible for the early steps of IE induced by S. gordonii. In fact, rats immunized with Hsa or PadA were significantly protected from S. gordonii IE. Most importantly, coimmunization with the two proteins conferred full protection, suggesting that Hsa and PadA act synergistically to promote IE.
Thus, Hsa and PadA represent potential targets for vaccination against S. gordonii IE. To be ideal vaccinogens, however, S. gordonii Hsa and PadA should afford cross-protection against other VGS. PadA appears to be conserved among viridans streptococci, including Streptococcus sanguinis (99% identity), Streptococcus mitis (98% identity), Streptococcus parasanguinis (68% identity), and Streptococcus oralis (66% identity) (see Fig. S2 in the supplemental material). Although less well conserved, S. gordonii Hsa homologues can be found in S. mitis (83% identity), Streptococcus sanguis (named SrpA; 63% identity) (31), S. gordonii M99 (named GspB; 40% identity) (17, 32), and S. oralis (26% identity) (see Fig. S3). Whether the anti-Hsa and anti-PadA antibodies also cross-react with and block the function of the Hsa- and PadA-like proteins of other VGS is under investigation in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
We thank Juan Evaristo Suarez (University of Oviedo, Spain) for providing the pLysA2 vector and Christiane Gerschheimer (Service and Central Laboratory of Hematology, Lausanne University Hospital) for help with aggregometry.
None of the authors has any conflict of interests relevant to this study.
This work was funded by the Swiss National Science Foundation (310030-143799).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00810-16.
REFERENCES
- 1.Que YA, Moreillon P. 2011. Infective endocarditis. Nat Rev Cardiol 8:322–336. doi: 10.1038/nrcardio.2011.43. [DOI] [PubMed] [Google Scholar]
- 2.Roberts GJ. 1999. Dentists are innocent! “Everyday” bacteremia is the real culprit: a review and assessment of the evidence that dental surgical procedures are a principal cause of bacterial endocarditis in children. Pediatr Cardiol 20:317–325. doi: 10.1007/s002469900477. [DOI] [PubMed] [Google Scholar]
- 3.Forner L, Larsen T, Kilian M, Holmstrup P. 2006. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol 33:401–407. doi: 10.1111/j.1600-051X.2006.00924.x. [DOI] [PubMed] [Google Scholar]
- 4.Crasta K, Daly CG, Mitchell D, Curtis B, Stewart D, Heitz-Mayfield LJ. 2009. Bacteraemia due to dental flossing. J Clin Periodontol 36:323–332. doi: 10.1111/j.1600-051X.2008.01372.x. [DOI] [PubMed] [Google Scholar]
- 5.Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, Levison M, Bolger A, Cabell CH, Takahashi M, Baltimore RS, Newburger JW, Strom BL, Tani LY, Gerber M, Bonow RO, Pallasch T, Shulman ST, Rowley AH, Burns JC, Ferrieri P, Gardner T, Goff D, Durack DT. 2007. Prevention of infective endocarditis. Guidelines from the American Heart Association. A guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation 116:1736–1754. [DOI] [PubMed] [Google Scholar]
- 6.Habib G, Hoen B, Tornos P, Thuny F, Prendergast B, Vilacosta I, Moreillon P, de Jesus Antunes M, Thilen U, Lekakis J, Lengyel M, Muller L, Naber CK, Nihoyannopoulos P, Moritz A, Zamorano JL, Vahanian A, Auricchio A, Bax J, Ceconi C, Dean V, Filippatos G, Funck-Brentano C, Hobbs R, Kearney P, McDonagh T, McGregor K, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Vardas P, Widimsky P. 2009. Guidelines on the prevention, diagnosis, and treatment of infective endocarditis (new version 2009): the Task Force on the Prevention, Diagnosis, and Treatment of Infective Endocarditis of the European Society of Cardiology (ESC). Eur Heart J 30:2369–2413. doi: 10.1093/eurheartj/ehp285. [DOI] [PubMed] [Google Scholar]
- 7.Richey R, Wray D, Stokes T, Guideline Development Group . 2008. Prophylaxis against infective endocarditis: summary of NICE guidance. BMJ 336:770–771. doi: 10.1136/bmj.39510.423148.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pant S, Patel NJ, Deshmukh A, Golwala H, Patel N, Badheka A, Hirsch GA, Mehta JL. 2015. Trends in infective endocarditis incidence, microbiology, and valve replacement in the United States from 2000 to 2011. J Am Coll Cardiol 65:2070–2076. doi: 10.1016/j.jacc.2015.03.518. [DOI] [PubMed] [Google Scholar]
- 9.Durack DT, Gilliland BC, Petersdorf RG. 1978. Effect of immunization on susceptibility to experimental Streptococcus mutans and Streptococcus sanguis endocarditis. Infect Immun 22:52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheld WM, Thomas JH, Sande MA. 1979. Influence of preformed antibody on experimental Streptococcus sanguis endocarditis. Infect Immun 25:781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van de Rijn I. 1985. Role of culture conditions and immunization in experimental nutritionally variant streptococcal endocarditis. Infect Immun 50:641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Viscount HB, Munro CL, Burnette-Curley D, Peterson DL, Macrina FL. 1997. Immunization with FimA protects against Streptococcus parasanguis endocarditis in rats. Infect Immun 65:994–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitten T, Munro CL, Wang A, Macrina FL. 2002. Vaccination with FimA from Streptococcus parasanguis protects rats from endocarditis caused by other viridans streptococci. Infect Immun 70:422–425. doi: 10.1128/IAI.70.1.422-425.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douglas CW, Heath J, Hampton KK, Preston FE. 1993. Identity of viridans streptococci isolated from cases of infective endocarditis. J Med Microbiol 39:179–182. doi: 10.1099/00222615-39-3-179. [DOI] [PubMed] [Google Scholar]
- 15.Moreillon P, Que YA, Bayer AS. 2002. Pathogenesis of streptococcal and staphylococcal endocarditis. Infect Dis Clin North Am 16:297–318. doi: 10.1016/S0891-5520(01)00009-5. [DOI] [PubMed] [Google Scholar]
- 16.Kerrigan SW, Cox D. 2010. Platelet-bacterial interactions. Cell Mol Life Sci 67:513–523. doi: 10.1007/s00018-009-0207-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bensing BA, Lopez JA, Sullam PM. 2004. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibalpha. Infect Immun 72:6528–6537. doi: 10.1128/IAI.72.11.6528-6537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi Y, Yajima A, Cisar JO, Konishi K. 2004. Functional analysis of the Streptococcus gordonii DL1 sialic acid-binding adhesin and its essential role in bacterial binding to platelets. Infect Immun 72:3876–3882. doi: 10.1128/IAI.72.7.3876-3882.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jakubovics NS, Kerrigan SW, Nobbs AH, Stromberg N, van Dolleweerd CJ, Cox DM, Kelly CG, Jenkinson HF. 2005. Functions of cell surface-anchored antigen I/II family and Hsa polypeptides in interactions of Streptococcus gordonii with host receptors. Infect Immun 73:6629–6638. doi: 10.1128/IAI.73.10.6629-6638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerrigan SW, Jakubovics NS, Keane C, Maguire P, Wynne K, Jenkinson HF, Cox D. 2007. Role of Streptococcus gordonii surface proteins SspA/SspB and Hsa in platelet function. Infect Immun 75:5740–5747. doi: 10.1128/IAI.00909-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petersen HJ, Keane C, Jenkinson HF, Vickerman MM, Jesionowski A, Waterhouse JC, Cox D, Kerrigan SW. 2010. Human platelets recognize a novel surface protein, PadA, on Streptococcus gordonii through a unique interaction involving fibrinogen receptor GPIIbIIIa. Infect Immun 78:413–422. doi: 10.1128/IAI.00664-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keane C, Petersen HJ, Tilley DO, Haworth J, Cox D, Jenkinson HF, Kerrigan SW. 2013. Multiple sites on Streptococcus gordonii surface protein PadA bind to platelet GPIIbIIIa. Thromb Haemost 110:1278–1287. doi: 10.1160/TH13-07-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ribelles P, Benbouziane B, Langella P, Suarez JE, Bermudez-Humaran LG. 2013. Protection against human papillomavirus type 16-induced tumors in mice using non-genetically modified lactic acid bacteria displaying E7 antigen at its surface. Appl Microbiol Biotechnol 97:1231–1239. doi: 10.1007/s00253-012-4575-1. [DOI] [PubMed] [Google Scholar]
- 24.Ribelles P, Rodriguez I, Suarez JE. 2012. LysA2, the Lactobacillus casei bacteriophage A2 lysin is an endopeptidase active on a wide spectrum of lactic acid bacteria. Appl Microbiol Biotechnol 94:101–110. doi: 10.1007/s00253-011-3588-5. [DOI] [PubMed] [Google Scholar]
- 25.Que YA, Haefliger JA, Francioli P, Moreillon P. 2000. Expression of Staphylococcus aureus clumping factor A in Lactococcus lactis subsp. cremoris using a new shuttle vector. Infect Immun 68:3516–3522. doi: 10.1128/IAI.68.6.3516-3522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veloso TR, Mancini S, Giddey M, Vouillamoz J, Que YA, Moreillon P, Entenza JM. 2015. Vaccination against Staphylococcus aureus experimental endocarditis using recombinant Lactococcus lactis expressing ClfA or FnbpA. Vaccine 33:3512–3517. doi: 10.1016/j.vaccine.2015.05.060. [DOI] [PubMed] [Google Scholar]
- 27.Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veloso TR, Amiguet M, Rousson V, Giddey M, Vouillamoz J, Moreillon P, Entenza JM. 2011. Induction of experimental endocarditis by continuous low-grade bacteremia mimicking spontaneous bacteremia in humans. Infect Immun 79:2006–2011. doi: 10.1128/IAI.01208-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veloso TR, Que YA, Chaouch A, Giddey M, Vouillamoz J, Rousson V, Moreillon P, Entenza JM. 2015. Prophylaxis of experimental endocarditis with antiplatelet and antithrombin agents: a role for long-term prevention of infective endocarditis in humans? J Infect Dis 211:72–79. doi: 10.1093/infdis/jiu426. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi Y, Takashima E, Shimazu K, Yagishita H, Aoba T, Konishi K. 2006. Contribution of sialic acid-binding adhesin to pathogenesis of experimental endocarditis caused by Streptococcus gordonii DL1. Infect Immun 74:740–743. doi: 10.1128/IAI.74.1.740-743.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Ian Douglas CW. 2005. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol 129:101–109. doi: 10.1111/j.1365-2141.2005.05421.x. [DOI] [PubMed] [Google Scholar]
- 32.Xiong YQ, Bensing BA, Bayer AS, Chambers HF, Sullam PM. 2008. Role of the serine-rich surface glycoprotein GspB of Streptococcus gordonii in the pathogenesis of infective endocarditis. Microb Pathog 45:297–301. doi: 10.1016/j.micpath.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.