

Abstract
Enoyl-acyl carrier protein reductase catalyzes the last step in each elongation cycle of type II bacterial fatty acid synthesis and is a key regulatory protein in bacterial fatty acid synthesis. Genes of the facultative intracellular pathogen Listeria monocytogenes encode two functional enoyl-acyl carrier protein isoforms based on their ability to complement the temperature-sensitive growth phenotype of Escherichia coli strain JP1111 [fabI(Ts)]. The FabI isoform was inactivated by the FabI selective inhibitor AFN-1252, but the FabK isoform was not affected by the drug, as expected. Inhibition of FabI by AFN-1252 decreased endogenous fatty acid synthesis by 80% and lowered the growth rate of L. monocytogenes in laboratory medium. Robust exogenous fatty acid incorporation was not detected in L. monocytogenes unless the pathway was partially inactivated by AFN-1252 treatment. However, supplementation with exogenous fatty acids did not restore normal growth in the presence of AFN-1252. FabI inactivation prevented the intracellular growth of L. monocytogenes, showing that neither FabK nor the incorporation of host cellular fatty acids was sufficient to support the intracellular growth of L. monocytogenes. Our results show that FabI is the primary enoyl-acyl carrier protein reductase of type II bacterial fatty acid synthesis and is essential for the intracellular growth of L. monocytogenes.
INTRODUCTION
Listeria monocytogenes is a facultative intracellular Gram-positive bacterium that is the primary cause of the disease listeriosis (1). The environmentally hardy and ubiquitous L. monocytogenes is ingested through contaminated food products and can cause noninvasive diseases, such as gastroenteritis, or invade through the intestinal epithelium to cause central nervous system infections or bacteremia. L. monocytogenes poses a major medical threat for pregnant women and immunocompromised patients. Yearly outbreaks of L. monocytogenes in a variety of food products from fruits to ice cream underscore the need to understand the requirements for L. monocytogenes infection and discover new methods to inhibit the growth of L. monocytogenes.
The phospholipids of L. monocytogenes are composed of branched-chain fatty acids synthesized by the L. monocytogenes type II fatty acid synthesis system. Decreasing the synthesis of branched-chain fatty acids by the genetic deletion of the branched-chain α-keto acid dehydrogenase compromises the environmental survival and intracellular pathogenesis of L. monocytogenes (2–6). The pathways for fatty acid synthesis, phospholipid synthesis, and exogenous fatty acid incorporation of L. monocytogenes are predicted to be the same as those for the phylogenetically related and well-characterized Staphylococcus aureus system (Fig. 1). Fatty acids are synthesized using a prototypical type II bacterial fatty acid synthesis, with 2-methylbutyryl-coenzyme A (CoA) as the primer, to make branch-chain fatty acids (3, 5, 7–9). An acyl-acyl carrier protein (ACP) of the appropriate length is used as the acyl donor by the PlsX/Y/C system to make phosphatidic acid from glycerol-3-phosphate (10, 11). Exogenous fatty acids are incorporated via the fatty acid kinase system (12–14). An unusual feature of L. monocytogenes fatty acid synthesis is the presence of four genes that may encode enoyl-acyl carrier protein reductases, including a FabI, a FabL, and two FabK isoforms, rather than the single FabI found in S. aureus and a variety of other bacterial pathogens. The FabL isoform was discovered in the phylogenetically related Bacillus, which also has a FabI (15). Both proteins function in fatty acid synthesis in Bacillus. The FabK isoform is a flavoprotein unrelated to FabI that was originally discovered in Streptococcus pneumoniae (16). The Enterococcus faecalis genome encodes both FabI and FabK, but FabI is responsible for the bulk of fatty acid synthesis while FabK plays a minor role in supporting fatty acid synthesis (17).
FIG 1.

Fatty acid synthesis, phospholipid synthesis, and exogenous fatty acid incorporation system of L. monocytogenes. The locus tags for the genes in L. monocytogenes strain EGD-e are annotated below the enzyme symbols. The L. monocytogenes genome encodes the same basic type II bacterial fatty acid synthesis, acyltransferase, and exogenous fatty acid incorporation system as the characterized S. aureus system. The unique aspect of L. monocytogenes is that the L. monocytogenes genome has four putative enoyl-acyl carrier protein reductase genes of three different isoforms. The gene LMO0970 is predicted to encode the FabI isoform (53% identity; E value of 1e−89 versus FabI from S. aureus). The gene LMO1688 is predicted to encode the FabL isoform (69% identity; E value of 1e−120 versus FabL from B. subtilis). The gene LMO0814 is predicted to encode the FabK isoform (50% identity; E value of 1e−100 versus FabK from S. pneumoniae). The gene LMO2170 is also predicted to encode the FabK isoform (51% identity; E value of 7e−99 versus FabK from S. pneumoniae).
The goal of this study was to identify which of the putative L. monocytogenes reductase genes code for functional enoyl-acyl carrier protein reductases, assess their contribution to supporting type II bacterial fatty acid synthesis, and determine their role in planktonic and intracellular growth. Complete inhibition of FabI using the FabI selective inhibitor AFN-1252 reduced the fatty acid synthesis of L. monocytogenes strain 10403S by 80% and reduced the growth rate of L. monocytogenes in laboratory medium. In contrast, FabI inactivation through AFN-1252 stopped the intracellular growth of L. monocytogenes. These results establish that FabI is the major enoyl acyl carrier protein reductase of type II fatty acid synthesis and that FabI-dependent type II fatty acid synthesis is essential for the intracellular growth of L. monocytogenes and validates FabI as an effective drug target against intracellular L. monocytogenes.
MATERIALS AND METHODS
Materials.
Chemicals were from Fisher Scientific or Sigma-Aldrich. Bacterial medium supplies were from BD Medical Technology, while human cell culture supplies were from Life Technologies. Radioactive chemicals were from American Radiolabeled Chemicals, and [7,7,8,8-D4]palmitic acid (D4-16:0) was from Cambridge Isotope Laboratories.
Bacterial growth.
L. monocytogenes strain 10403S was grown in brain heart infusion medium for routine growth and maintenance. L. monocytogenes was grown in tryptic soy broth for growth inhibition, pathway labeling, and exogenous fatty acid supplementation experiments because tryptic soy broth lacked the exogenous fatty acids in brain heart infusion medium. L. monocytogenes was grown at 37°C with shaking at 200 rpm unless otherwise indicated.
Molecular biology.
The predicted L. monocytogenes FabI (LmFabI; LMO0970), LmFabL (LMO1688), LmFabK1 (LMO0814), and LmFabK2 (LMO2170) proteins were optimized for expression in Escherichia coli through GeneArt gene synthesis technology (Life Technologies). An NdeI restriction site was engineered at the 5′ end of the gene with the start codon in the NdeI restriction site, while a stop codon and an EcoRI restriction site were engineered at the 3′ end of the gene. The genes were cloned into the plasmid pPJ131. The pPJ131 plasmid is a pBluescript plasmid (Stratagene) containing the multiple-cloning site from pPET28a for protein expression in E. coli (18, 19). The resulting expressed protein has an N-terminal histidine tag from the pET28a multiple-cloning site. The polyhistidine tag allows for the detection and purification of the expressed protein. Although tags sometimes affect protein function, there are no examples of histidine tags interfering with FabI or FabK activity. The protein encoded by the plasmid is constitutively expressed because the pPJ131 plasmid is a high-copy-number plasmid, and the endogenous lacI is titrated by copy number.
Complementation.
The expression constructs containing the four putative L. monocytogenes enoyl-acyl carrier protein reductases (LMO0814, LMO0970, LMO1688, and LMO2170) were transformed into the fabI temperature-sensitive E. coli strain JP1111 [fabI(Ts)] to determine complementation. The pPJ131 parent plasmid was used as a negative complementation control, and the pPJ131 plasmid expressing FabI from S. aureus was used as the positive control (20). Strain JP1111 is able to grow at 30°C but was nonviable at 42°C without FabI complementation.
Western blotting.
JP1111 cells harboring plasmids containing LMO0814, LMO0970, LMO1688, and LMO2170 were grown at 30°C in lysogeny broth to an A600 of 1. The cells were washed twice with phosphate-buffered saline and harvested via centrifugation. The cells were resuspended in B-PER bacterial protein extraction reagent (1 ml per 25-ml culture; Thermo Scientific) and shaken for 30 min to lyse the cells. The resulting mixture was centrifuged at 20,000 × g to separate the supernatant from the cellular debris. Equal volumes of the supernatant from each expression strain were electrophoresed on a NuPAGE Novex 10% Bis-Tris protein gel (Life Technologies), and protein expression was visualized via Western blot analysis using a primary rabbit polyclonal IgG His-probe antibody (Santa Cruz Biotechnology) (21) and a secondary goat alkaline phosphatase-linked anti-rabbit IgG antibody (Sigma-Aldrich) (20). The blot was visualized using ECF substrate (GE Healthcare) on a Typhoon FLA 9500 imager in the fluorescence detection mode.
Susceptibility of L. monocytogenes enoyl-acyl carrier protein reductase to AFN-1252.
The L. monocytogenes enoyl-acyl carrier protein reductase genes that complemented the growth of JP1111 were transformed into E. coli strain ANS1 (ΔtolC), an efflux pump-deficient E. coli strain with increased susceptibility to AFN-1252 (20). The AFN-1252 MIC against ANS1 expressing the parent pPJ131 plasmid, the pPJ131 plasmid expressing the S. aureus fabI gene, and the pPJ131 plasmid expressing the functional L. monocytogenes enoyl-acyl carrier protein reductase genes was determined to evaluate the susceptibility of the L. monocytogenes enoyl-acyl carrier protein reductases to AFN-1252.
Growth experiments.
L. monocytogenes was cultured overnight in tryptic soy broth medium. The overnight culture was back diluted to an A600 of 0.02 in tryptic soy broth medium, and the A600 was monitored over 5 h for AFN-1252 in growth rate experiments. The overnight culture was back diluted to an A600 of 0.02 in tryptic soy broth medium with 0.1% Brij-58 in fatty acid growth complementation experiments. Fatty acids are known to have inhibitory effects on the growth of L. monocytogenes (22), but delivering fatty acids in the detergent Brij-58 (0.1%) overcomes the inhibitory effects (23, 24). The culture was grown until an A600 of ≈0.05 and split into six 14-ml aliquots: dimethyl sulfoxide (DMSO) only added, DMSO with 100 μM D4-16:0 and 100 μM 18:1, DMSO with 100 μM anteiso-15:0 and 100 μM anteiso-17:0, 50 μM AFN-1252, 50 μM AFN-1252 with 100 μM D4-16:0 and 100 μM 18:1, and 50 μM AFN-1252 with 100 μM anteiso-15:0 and 100 μM anteiso-17:0. Growth was monitored over 5 h or until the A600 becomes greater than 1.5. After 5 h or an A600 of >1.5 was reached, 2.5 ml of the culture was harvested, washed with phosphate-buffered saline, extracted for lipids via the Bligh and Dyer method (25), and analyzed via molecular species analysis to determine the phospholipid species (24, 26). Growth curves and molecular species analysis experiments were conducted on two independent biological replicates, and representative curves and spectra are shown in the figures.
Acetate labeling experiments.
L. monocytogenes was cultured overnight in tryptic soy broth medium. The overnight culture was back diluted to an A600 of 0.1 in tryptic soy broth medium and grown to an A600 of 0.4. The culture was split into 5-ml aliquots, and the aliquots were incubated with a final concentration of 0, 0.04, 0.15, 0.6, 2.5, 10, and 50 μM AFN-1252 for 15 min. [14C]acetate (5 μCi) was added to each aliquot and incubated for 45 min. The cells were pelleted via centrifugation and washed twice with phosphate-buffered saline, and the lipids were isolated (25). The amount of [14C]acetate incorporated into the phospholipids was measured via liquid scintillation counting (Tri-Carb 2910 TR; PerkinElmer Life Sciences). Data were plotted as the fraction of incorporation compared to that of the untreated cells and fitted via a 50% inhibitory concentration (IC50) equation with partial inhibition in GraphPad Prism 5.
Pathway labeling experiments.
L. monocytogenes was cultured overnight in tryptic soy broth medium. The overnight culture was back diluted to an A600 of 0.1 in tryptic soy broth medium and grown to an A600 of 0.4. The culture was split into 10-ml aliquots, and the aliquots were incubated with a final concentration of 0.5% DMSO or 50 μM AFN-1252 for 15 min. The aliquots then were labeled for 45 min with 10 μCi of an 3H-labeled l-amino acid mixture, [3H]thymidine, or [3H]uracil to measure protein, DNA, and RNA synthesis, respectively. The cells were harvested via filtration and washed with 3 ml of phosphate-buffered saline 3 times. The filters were incubated with the liquid scintillation solution for 2 h, and the radioactive incorporation into the cells was measured via liquid scintillation counting of the filter. Data are plotted as fractional incorporation relative to the untreated cells normalized to the final A600 of the cultures. Plotted data were derived from two biological replicates. The statistical significance of whether each pathway was inhibited by AFN-1252 was determined using Student's t test.
Intracellular infection.
L. monocytogenes strain 10403S was used to infected HeLa cells as previously described (27). Briefly, L. monocytogenes was grown overnight in brain heart infusion medium at 37°C with shaking at 200 rpm. The culture was back diluted to an A600 of 0.25 in brain heart infusion medium and grown for 1 h. L. monocytogenes was washed with phosphate-buffered saline and resuspended in DMEM (Dulbecco's modified Eagle medium with 10% fetal bovine serum) at the appropriate concentration such that 2 ml of medium per well gave the desired multiplicity of infection (MOI). HeLa cell monolayers grown to 80% confluence in 6-well plates (ca. 1 × 106 HeLa cells per well) were washed twice with Hanks' balanced salt solution (HBSS). L. monocytogenes resuspended in DMEM was added to the HeLa cells. The plates were centrifuged at 900 × g for 15 min at room temperature and then incubated for 105 min at 37°C in 5% CO2. Cells next were washed 2 times with HBSS and incubated in 2 ml of DMEM and 40 μg/liter gentamicin for 60 min. The cells were washed 2 more times with HBSS and incubated in DMEM and 40 μg/liter gentamicin (and DMSO or AFN-1252 where appropriate) at 37°C in 5% CO2 until ready for use. For microscopy experiments, glass coverslips were placed at the bottom of the well, and the HeLa cells were seeded onto the coverslips. All other infection and growth procedures remained the same. Growth curve and mass spectrometry experiments were conducted at an MOI of 1, while microscopy experiments were conducted at an MOI of 15.
The number of L. monocytogenes organisms per well was determined by lysing the HeLa cells and observing the number of CFU of L. monocytogenes. Briefly, the infected HeLa cells were washed with HBSS twice and resuspended in HBSS. The cells were resuspended via cell scraping, collected into microcentrifuge tubes, and resuspended in 0.1% Triton X-100 on ice. The solution was alternately vortexed thoroughly and incubated on ice for 20 min to lyse the HeLa cells to free the intracellular L. monocytogenes. The solution was serially diluted in brain heart infusion medium and plated on brain heart infusion medium agar plates to determine the CFU per well. For intracellular growth curve experiments, the infected HeLa cells were lysed immediately after infection (0 h), 3.5 h after infection, 20 h after infection, and 44 h after infection. The cells were collected via scraping and then washed twice with phosphate-buffered saline before lipid extraction and analysis for mass spectrometry experiments.
Molecular species analysis.
Phospholipid molecular species fingerprints were determined using direct infusion electrospray ionization-mass spectrometry technology. Mass spectrometry analysis was performed using a QTrap 4500 (Sciex, Framingham, MA) equipped with a Turbo V ion source. Lipid extracts were resuspended in 50:50 (vol/vol) chloroform-methanol plus 1% formic acid. The instrument was operated in the negative ion mode for phosphatidylglycerol (PG), phosphatidylinositol (PI), and fatty acid scan analysis. The ion source parameters were the following: ion spray voltage, −4,500 V; curtain gas, 15 lb/in2; temperature, 270°C; collision gas, medium; ion source gas 1, 15 lb/in2; and ion source gas 2, 25 lb/in2. Parameters for PG analysis were the following: scan range, 600 to 900 m/z; declustering potential, −30 V; collision energy, −45 V; peak width, Q1 and Q3 0.7 FWHM (full width at half maximum). For PI and 15:0 fatty acid analysis, lipid classes were separated using a Discovery DSC-NH2 solid-phase extraction column (Supelco, Bellefonte, PA). In brief, the column was conditioned with 8 ml of hexane and lipid extract was added. Nonpolar lipids were eluted with 6 ml of 2:1 (vol/vol) chloroform-isopropyl alcohol, fatty acids were eluted with 6 ml of ether plus 2% acetic acid, phosphatidylcholine and phosphatidylethanolamine were eluted with 6 ml of methanol, and phosphatidylglycerol and phosphatidylinositol were eluted with 6 ml of chloroform–methanol–0.8 M sodium acetate (60:30:4.5, vol/vol/vol). PI and 15:0 fatty acid parameters were the following: precursor ion, 241 m/z; scan range, 600 to 1,000 m/z; declustering potential, −35 V; collision energy, −40 V; peak width, Q1 and Q30.7 FWHM.
Microscopy.
The DMSO- or AFN-1252-treated infected HeLa cells were visualized via immunofluorescence microscopy. Glass coverslips in the 6-well plates were washed twice with phosphate-buffered saline and then fixed with 4% formaldehyde and 2% goat serum in phosphate-buffered saline. Hydration chambers were constructed from 150-mm petri dishes. Coverslips were washed with phosphate-buffered saline five times and placed in hydration chambers. Cells were permeabilized with 300 μl of 0.1% NP-40, 2% goat serum in phosphate-buffered saline for 10 min at room temperature and washed with phosphate-buffered saline five times. The samples were then blocked with 10% goat serum in phosphate-buffered saline for 30 min at room temperature. The blocking solution was aspirated and the samples were incubated with the primary rabbit anti-Listeria antibody (1:1,000 dilution of stock AB35132 from AbCam [28, 29]) in 2% goat serum, 0.1% NP-40, and phosphate-buffered saline for 1 h at room temperature. The sample was washed 5 times with phosphate-buffered saline and then incubated with anti-rabbit antibody conjugated to Alexa Fluor-488 (1:1,000 dilution of stock from Thermo Fisher) in 2% goat serum, 0.1% NP-40, and phosphate-buffered saline for 1 h at room temperature in the dark. In experiments with 4′,6-diamidino-2-phenylindole (DAPI), 1 μg/ml of dye was added with secondary antibody. Samples were washed five times with phosphate-buffered saline. In experiments with Alexa Fluor 594-conjugated phalloidin (Thermo Fisher), 200 μl of 100 nM phalloidin was added and incubated in the dark at room temperature for 15 min, and then samples were washed five times with phosphate-buffered saline. Samples were mounted in FluorSave (Calbiochem), dried overnight, and sealed with nail polish. High-resolution images looking at the intracellular shape of the bacteria were taken using a Zeiss LSM 780 microscope and Zeiss Zen software. Other images were taken using an Olympus BX41 using SPOT imaging software. The number of Listeria monocytogenes cells per HeLa nuclei from 3 representative fields containing at least 100 HeLa nuclei was determined for AFN-1252- and DMSO-treated Listeria monocytogenes-infected HeLa cells. The statistical significance between the two treatment groups was determined using Student's t test.
RESULTS
The genome of L. monocytogenes encodes two enoyl-acyl carrier protein reductase isoforms.
Enoyl-acyl carrier protein reductase catalyzes the reduction of trans-2-enoyl-ACP to acyl-ACP using the reducing potential of NAD(P)H in the last, rate-determining reaction of the elongation cycle (30, 31). Several isoforms of enoyl-acyl carrier protein reductase are known in bacteria. FabI is the first discovered isoform and is a member of the short-chain dehydrogenase/reductase protein family (32). The FabL isoform also belongs to the short-chain dehydrogenase/reductase protein family but is structurally divergent from FabI (15). FabL was discovered in Bacillus, which also expresses FabI. The FabK isoform is a flavoprotein unrelated to FabI and FabL. It was originally discovered in S. pneumoniae (16) and is found predominantly in other Gram-positive bacteria. The FabV isoform is found only in select Gram-negative bacteria (33, 34).
FabI from S. aureus, FabL from Bacillus subtilis, FabK from S. pneumoniae, and FabV from Vibrio cholerae were used to search the L. monocytogenes genome for related proteins. The L. monocytogenes genome encoded four enoyl-acyl carrier protein reductase gene homologues (Fig. 1 and NCBI Microbial Genome Resources). The LMO0970 gene encodes a putative FabI, the LMO1688 gene encodes a putative FabL, and the LMO0814 and LMO2170 genes encode putative FabKs. None of these genes are neighbored by another fatty acid metabolism gene. These bioinformatics predictions were validated experimentally because S. aureus and Bacillus subtilis encode FabK homologues that do not possess enoyl-acyl carrier protein reductase function (16). Whether these genes can function as enoyl-acyl carrier protein reductases was tested by assessing if the expression of these genes, using a constitutive promoter on a multicopy plasmid, complemented the growth of the temperature-sensitive E. coli strain JP1111 [fabI(Ts)] (35). LmFabI (LMO0970) and LmFabK1 (LMO0814) complemented the growth of JP1111 at the nonpermissive temperature, while LmFabL (LMO1688) weakly complemented growth at the nonpermissive temperature (Fig. 2A and B). LmFabK2 (LMO2170) expression did not restore the growth of the temperature-sensitive strain. The expression of the four predicted L. monocytogenes enoyl-acyl carrier protein reductases in the JP1111 cells was verified to ensure that the inability to complement was due to the lack of enoyl-acyl carrier protein reductase activity rather than lack of protein expression. All four proteins were expressed in JP1111 cells (Fig. 2C), showing that that LmFabI and LmFabK1 were the two most active enoyl-acyl carrier protein reductases encoded by L. monocytogenes genes.
FIG 2.
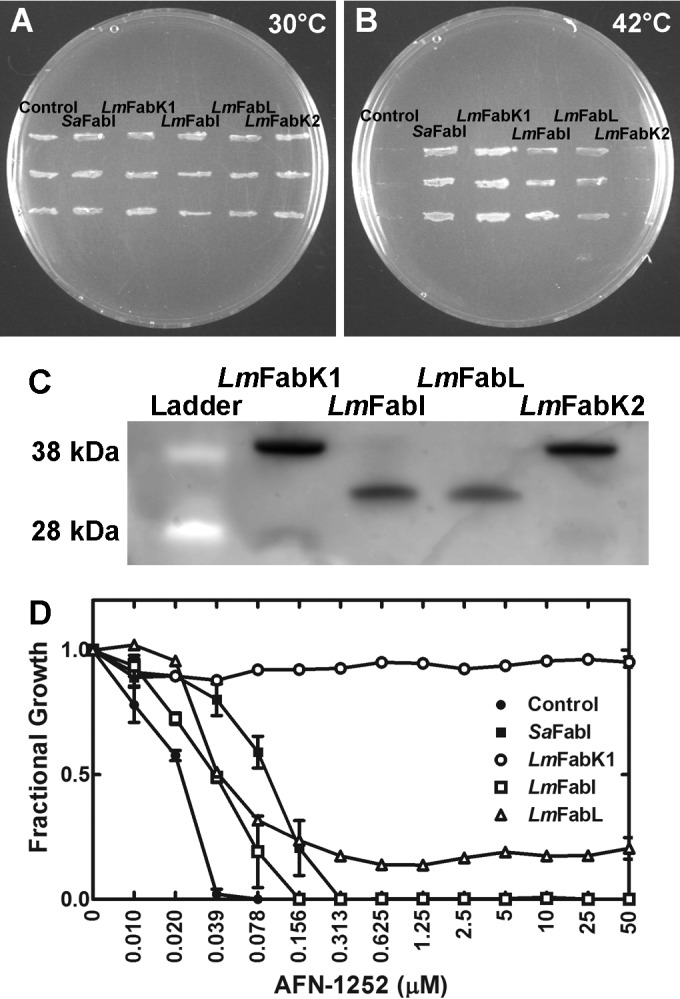
L. monocytogenes encodes two functional enoyl-acyl carrier protein reductase isoforms. (A) Growth of the temperature-sensitive E. coli strain JP1111 [fabI(Ts)] transformed with the empty plasmid (Control) or plasmids expressing either S. aureus FabI (SaFabI), predicted L. monocytogenes FabK1 (LmFabK1; LMO0814), predicted L. monocytogenes FabI (LmFabI; LMO0970), predicted L. monocytogenes FabL (LmFabL; LMO1688), or predicted L. monocytogenes FabK2 (LmFabK2; LMO2170) under the permissive growth condition (30°C). (B) Growth of the same strains at the nonpermissive temperature (42°C). (C) The expression of the predicted L. monocytogenes enoyl-acyl carrier protein reductases in JP1111 was verified via Western blot analysis. JP1111 cells expressing the enoyl-acyl carrier protein reductases were grown at 30°C to an A600 of 1. The cells were lysed, and the supernatant was separated from the cell debris. Western blot analysis against the supernatant fraction using an antihistidine tag was performed to ensure that the predicted enoyl-acyl carrier protein reductase was expressed. All four predicted enoyl-acyl carrier protein reductases (LmFabK1, 32.4 kDa; LmFabI, 28.3 kDa; LmFabL, 27.2 kDa; LmFabK2, 32.5 kDa; each with 2.1 kDa added from the N-terminal histidine tag from pPJ131 plasmid) were expressed in JP1111. Data shown as a representative blot are from 2 biological replicates. (D) MIC of AFN-1252 against the E. coli strain ANS1 (ΔtolC) expressing empty plasmid (control), S. aureus FabI (SaFabI), L. monocytogenes FabK (LmFabK1; LMO0814), L. monocytogenes FabI (LmFabI; LMO0970), and L. monocytogenes FabL (LmFabL; LMO1688).
Validation of AFN-1252.
AFN-1252 is a potent and selective inhibitor of FabI that has no known off-target effects (20, 24, 36, 37). AFN-1252 inhibits the growth of bacteria with a single essential FabI, including intracellular pathogens (18, 20, 24, 38), but is ineffective against bacterial genes encoding the other enoyl-acyl carrier protein reductase isoforms (39). These data, coupled with the observation that S. aureus accD mutants are refractory to AFN-1252 (14), show there is little or no off-target toxicity of this drug. Therefore, AFN-1252 was used as a tool to determine the contribution of LmFabI versus LmFabK1 to fatty acid synthesis and growth of L. monocytogenes. AFN-1252 was tested against E. coli strain ANS1 (ΔtolC), expressing LmFabI, LmFabK1, or LmFabL. Strain ANS1, harboring the empty plasmid (control), had an AFN-1252 MIC of 0.039 μM, whereas the MIC in the strain expressing S. aureus FabI (SaFabI) increased by 8-fold to 0.313 μM (Fig. 2D). Strain ANS1, expressing LmFabI, had an AFN-1252 MIC of 0.156 μM, which was a 4-fold increase compared to that of the control. This result showed that LmFabI, like SaFabI, was completely inhibited by AFN-1252. In contrast, strain ANS1 expressing the LmFabK1 gene was unaffected by AFN-1252 up to 50 μM, demonstrating that this FabK was able to fully support growth and was resistant to AFN-1252, as expected. The cells expressing LmFabL grew poorly in the presence of greater than 0.039 μM AFN-1252, showing that while resistant to AFN-1252, LmFabL was not sufficient to support a normal rate of E. coli growth. Therefore, FabL cannot substitute for the enoyl-acyl carrier protein reductase of type II bacterial fatty acid synthesis. Together, these experiments showed that the L. monocytogenes genome encoded an LmFabI that was completely inhibited by AFN-1252 as well as an LmFabK1 that was resistant to AFN-1252.
AFN-1252 inhibited L. monocytogenes growth and fatty acid synthesis.
The effect of increasing concentrations of AFN-1252 on the growth rate of L. monocytogenes in planktonic culture was measured to determine the contribution of LmFabI versus LmFabK1 to fatty acid synthesis and growth. AFN-1252 caused the cessation of cell growth after 1 to 2 cellular doublings in bacterial genes encoding a single, essential FabI (24, 38). In contrast, increasing concentrations of AFN-1252 slowed but did not stop the growth of L. monocytogenes (Fig. 3A). Pathway labeling experiments showed that AFN-1252 selectively inhibited lipid synthesis, with minimal perturbation to protein, DNA, and RNA synthesis (Fig. 3B). Therefore, the reduction of growth rate by AFN-1252 was attributed to on-target inhibition of LmFabI and fatty acid synthesis.
FIG 3.

AFN-1252 causes on-target, partial inhibition of phospholipid synthesis of L. monocytogenes. (A) The effect of increasing concentrations of AFN-1252 on the growth of L. monocytogenes in tryptic soy broth medium. AFN-1252 was added to cells at the time indicated by the arrow. The growth curve shown is representative of duplicate biological replicates. (B) The effect of AFN-1252 on the major biosynthetic pathways was measured by treating growing L. monocytogenes cells with 50 μM AFN-1252 and comparing them to untreated cells. Metabolic labeling with [14C]acetate was used for lipid biosynthesis, an 3H-labeled l-amino acid mixture was used to measure protein biosynthesis, [3H]thymidine incorporation measured DNA biosynthesis, and [3H]uracil measured stable RNA biosynthesis, as described in Materials and Methods. Data shown are averages with standard deviations from two biological replicates. Lipid biosynthesis was the only pathway inhibited by AFN-1252. Significance was determined using Student's t test. ***, P < 0.001. (C) AFN-1252 inhibition of [14C]acetate incorporation into the phospholipids of L. monocytogenes in tryptic soy broth medium. The fractional [14C]acetate incorporation was compared to that of the no-inhibitor control. Data shown are averages with standard deviations from two biological replicates. The data were fit to an IC50 equation with partial inhibition. The IC50 is the concentration of AFN-1252 needed to inhibit 50% of the sensitive fatty acid synthesis activity. Vmin is the fraction of residual fatty acid synthesis activity that could not be inhibited by AFN-1252. (D) The effect of branched-chain fatty acid (Brc; 100 μM anteiso-15:0 and 100 μM anteiso-17:0) and straight-chain fatty acid (Str; 100 μM D4-16:0 and 100 μM 18:1) supplementation on AFN-1252 inhibition of L. monocytogenes growth in tryptic soy broth medium.
The incorporation of [14C]acetate into the phospholipid fraction at increasing concentrations of AFN-1252 was measured to determine the fraction of fatty acid synthesis that was inhibited by AFN-1252. Increasing concentrations of AFN-1252 caused a dose-dependent inhibition of [14C]acetate incorporation, but there was a residual 20% [14C]acetate incorporation that could not be eliminated by AFN-1252 (Fig. 3C). This result showed that in the absence of LmFabI, LmFabK1 was only able to support 20% of the normal fatty acid synthesis rate during planktonic growth. Together, these experiments point to LmFabI as the enoyl-acyl carrier protein reductase dedicated to type II bacterial fatty acid synthesis, but the LmFabK1 was expressed and active in L. monocytogenes.
Exogenous fatty acid metabolism in L. monocytogenes.
L. monocytogenes is predicted to encode a fatty kinase system as in S. aureus for the incorporation of exogenous fatty acids (Fig. 1) (13, 14). In this system, exogenous fatty acids are phosphorylated by FakA/FakB to make acyl-phosphates (12), which can then enter phospholipid synthesis (10). The acyl-phosphate can be used by the glycerol-3-phosphate acyltransferase (PlsY) to make lysophosphatidic acid. The acyl-phosphate can also be converted into acyl-ACP by PlsX. The resulting acyl-ACP can undergo additional elongation cycles or be used by the 1-acyl-sn-glycerol-3-phosphate acyltransferase PlsC to make phosphatidic acid, the common precursor to phospholipid synthesis in bacteria. Genes encoding alternative pathways of exogenous fatty acid incorporation, such as acyl-ACP or acyl-CoA synthetase, were not found in the L. monocytogenes genome. β-Oxidation genes for breaking down fatty acids also were not found in the L. monocytogenes genome, so the only known fate of exogenous fatty acids is incorporation into the phospholipids.
Untreated and AFN-1252-treated L. monocytogenes organisms were supplemented with two combinations of fatty acids to determine if exogenous fatty acids bypassed the effect of AFN-1252 inhibition. The straight-chain combination contained 100 μM D4-16:0 and 100 μM 18:1, which corresponded to two abundant fatty acid species found in mammalian serum and cellular phospholipids. This combination of fatty acids allowed the determination of whether host fatty acids can bypass FabI inhibition in L. monocytogenes. The deuterium-labeled D4-16:0 fatty acids have a greater mass than L. monocytogenes-synthesized 16:0 fatty acid, so the uptake of D4-16:0 can be monitored using mass spectrometry. The 18:1 fatty acid can be monitored without isotopic labeling because L. monocytogenes cannot synthesize unsaturated fatty acids (3, 5, 8, 40), so all of the incorporated 18:1 must be exogenous in origin. The branched-chain combination contained 100 μM anteiso-15:0 and 100 μM anteiso-17:0. Branched-chain fatty acids are not found in mammalian hosts but are the normal acyl chains found in L. monocytogenes phospholipids. L. monocytogenes requires branched-chain fatty acids for optimum growth (5). The branched-chain combination was intended to test if the fatty acids found in L. monocytogenes phospholipids can overcome FabI inhibition. Neither fatty acid combination altered the growth rate of untreated L. monocytogenes (Fig. 3D). Both fatty acid combinations increased the growth rate of AFN-1252-treated L. monocytogenes but did not restore wild-type growth rates. These data suggested that the reduction in growth rate by AFN-1252 could be partially overcome if L. monocytogenes was able to access host fatty acids.
The acyl chain composition of L. monocytogenes phospholipids was determined to verify that the exogenous fatty acids affected growth through incorporation into L. monocytogenes phospholipids. Molecular species analysis was performed on the phosphatidylglycerol (PG) of L. monocytogenes. Only the straight-chain combination was analyzed, because the anteiso-17:0 and anteiso-15:0 supplementation produced the exact same molecular species normally found in L. monocytogenes. The PG major molecular species in untreated L. monocytogenes consisted of 17:0/15:0 with smaller amounts of 15:0/15:0 and 16:0/15:0 (Fig. 4A). Only small amounts of exogenous fatty acids were incorporated into the phospholipids of untreated L. monocytogenes, illustrating the inefficient uptake of fatty acids by L. monocytogenes (Fig. 4B). Both the D4-16:0 and 18:1 species were paired with the endogenously synthesized anteiso-15:0 to give D4-16:0/15:0 and 18:1/15:0 PG molecular species. These mass spectra showed that L. monocytogenes genes encoded the ability to incorporate exogenous fatty acids, but this pathway made a minor contribution to phospholipid synthesis when type II bacterial fatty acid synthesis was active. The acyl chain composition of AFN-1252-treated L. monocytogenes was similar to that of untreated L. monocytogenes, with a low-abundance, new molecular species of 16:0/16:1 appearing (Fig. 4C). The 16:1 fatty acid was a component of the complex medium used in these experiments, because the L. monocytogenes genome does not encode the ability to synthesize unsaturated fatty acids (3, 5, 8, 40). The D4-16:0/15:0 and 18:1/15:0 combinations became major molecular species in fatty acid-supplemented, AFN-1252-treated L. monocytogenes, while the 17:0/15:0, 15:0/15:0, and 16:0/15:0 combinations from endogenous synthesis were reduced (Fig. 4D). Bacteria synthesizing branched-chain fatty acids have been characterized as having high selectivity for anteiso-15:0 fatty acids in the 2-position of their phospholipids and low acyl chain selectivity for the 1-position (38, 41, 42). The mass spectra showed that serum fatty acid species replaced the fatty acids at the 1-position of L. monocytogenes phospholipids, but the 2-position has retained high selectivity for endogenously synthesized anteiso-15:0. Together, these experiments showed that L. monocytogenes did not incorporate significant amounts of exogenous fatty acids when type II bacterial fatty acid synthesis was operational. Exogenous fatty acid incorporation was more pronounced when endogenous fatty acid synthesis was inhibited.
FIG 4.

Exogenous fatty acid metabolism in L. monocytogenes. Growing cultures of untreated or AFN-1252 (50 μM)-treated L. monocytogenes in tryptic soy broth medium were not supplemented with exogenous fatty acids or were supplemented with straight-chain fatty acid (100 μM D4-16:0 and 100 μM 18:1) and grown to an A600 of ≈1.5. The lipids were harvested from the cells and the PG molecular species were determined. The identities of the major molecular species are annotated in the figure, and the new molecular species arising from the incorporation of the fatty acid supplements are highlighted in red. New molecular species (16:0/16:1) arising from incorporation of fatty acids present in tryptic soy broth are highlighted in blue. Spectra shown are representative examples from two biological replicates. (A) PG molecular species profile of L. monocytogenes with no drug treatment and no fatty acid supplementation. (B) PG molecular species profile of L. monocytogenes with no drug treatment and straight-chain fatty acid supplementation. (C) PG molecular species profile of L. monocytogenes with AFN-1252 treatment and no fatty acid supplementation. (D) PG molecular species profile of L. monocytogenes with AFN-1252 treatment and straight-chain fatty acid supplementation.
The ability to use exogenous fatty acids to replace endogenously synthesized fatty acids varies by bacterial species (12). Streptococcus and Neisseria both use straight-chain and unsaturated fatty acids, which are abundant in mammalian hosts, to make their phospholipids. Genes of both species also encode the ability to incorporate exogenous fatty acids. Streptococcus shuts down endogenous fatty acid synthesis in the presence of exogenous fatty acids and therefore can overcome type II bacterial fatty acid synthesis inhibition (38, 43). In contrast, Neisseria cannot overcome type II bacterial fatty acid synthesis inhibition despite its genes encoding the ability to incorporate exogenous fatty acids (24). The low levels of exogenous fatty acid incorporation when type II bacterial fatty acid synthesis was active, coupled with the inability of exogenous fatty acids to fully restore the growth rate of L. monocytogenes when FabI is inhibited, suggests that L. monocytogenes cannot efficiently use exogenous fatty acids to support growth. These results raised the question of whether the fatty acids found in the host cell cytosol are able to relieve FabI inhibition for intracellular L. monocytogenes growth.
AFN-1252 inhibited the intracellular growth of L. monocytogenes.
The effect of AFN-1252 on the intracellular growth of L. monocytogenes was determined in a HeLa cell infection model (44, 45). AFN-1252 has been used previously in the intracellular infection model of Chlamydia trachomatis and showed no toxic effects against HeLa cells (18). The total number of intracellular L. monocytogenes organisms in HeLa cells was determined by lysing the infected HeLa cells and plating the lysate in serial dilutions. The number of L. monocytogenes CFU increased exponentially in untreated HeLa cells, with 1,300 times the initial number of CFU at 44 h (Fig. 5A). In contrast, the number of L. monocytogenes CFU increased initially for 2 to 3 doublings in AFN-1252-treated HeLa cells before decreasing over time (Fig. 5A). The reduction in L. monocytogenes titer under AFN-1252 treatment was corroborated using immunofluorescence microscopy (Fig. 5B to D) and lipid mass spectrometry (Fig. 5E and F). Untreated and AFN-1252-treated samples at 20 h after infection were stained with anti-L. monocytogenes antibody and DAPI for the host cell nuclei. AFN-1252 treatment caused a 26-fold reduction in the number of L. monocytogenes cells (green) observed per HeLa cell (DAPI staining, blue) (Fig. 5B to D). The decrease in L. monocytogenes cells caused by AFN-1252 inhibition observed via microscopy corroborated the decrease observed through plating the infected HeLa cell lysate, demonstrating that AFN-1252 inhibited the intracellular replication of L. monocytogenes.
FIG 5.

AFN-1252 inhibits the intracellular growth of L. monocytogenes. (A) Growth curve of untreated or AFN-1252 (50 μM)-treated intracellular L. monocytogenes in HeLa cells. (B) L. monocytogenes-infected HeLa cells were stained with DAPI and anti-Listeria antibody. Shown are the average numbers of L. monocytogenes organisms per HeLa cell counted from three sets of representative fields containing at least 100 HeLa cells of untreated and AFN-1252-treated L. monocytogenes. Significance was determined using Student's t test. ***, P < 0.001. (C) Immunofluorescence image of untreated HeLa cells infected with L. monocytogenes at 20 h postinfection. DAPI (blue) stain was used for the HeLa cell nucleus. Anti-Listeria antibody (green) stains for L. monocytogenes. (D) Immunofluorescence image of AFN-1252-treated HeLa cells infected with L. monocytogenes at 20 h postinfection. (E) Molecular species profile of lipids containing a 15:0 fatty acid or the inositol head group (loss of 241) for the untreated L. monocytogenes-infected HeLa cells. (F) Molecular species profile of lipids containing a 15:0 fatty acid or the inositol head group (loss of 241) for the AFN-1252-treated L. monocytogenes-infected HeLa cells.
Molecular species analysis of the PG was also performed on untreated and AFN-1252-treated samples at 20 h after infection. A loss scan of 241 molecular weight was conducted to identify phospholipid species containing 15:0 fatty acids or inositol head groups. This method allowed the detection of only the L. monocytogenes-synthesized phospholipids, along with host cell phosphatidylinositol for comparison, and ignored the most abundant host phospholipid species present in the sample. The 17:0/15:0, 15:0/15:0, and 16:0/15:0 combinations were the major PG species in L. monocytogenes-infected HeLa cells (Fig. 5E), showing that the acyl chain composition of L. monocytogenes remained the same in intracellular and planktonic growth, and that intracellular L. monocytogenes did not incorporate significant amounts of exogenous fatty acids like in normal planktonic growth. These molecular species peaks were significantly reduced relative to those of the host phosphatidylinositol species in the AFN-1252-treated infected HeLa cells (Fig. 5F), demonstrating that AFN-1252 inhibited intracellular L. monocytogenes fatty acid and phospholipid synthesis.
The effect of AFN-1252 on the L. monocytogenes invasion cycle was characterized by immunofluorescence microscopy through staining for both L. monocytogenes (green) and cellular actin (red). In a normal invasion cycle, extracellular L. monocytogenes is initially phagocytized into the lysosome (1). Listeriolysin O produced by L. monocytogenes dissolves the lysosome and allows L. monocytogenes to enter the cytosol, where it recruits actin and replicates (46). Intracellular L. monocytogenes can use the polymerization of host actin to move intracellularly or spread extracellularly. L. monocytogenes cells colocalized with actin represent cytosolic bacteria, while L. monocytogenes cells not colocalized with actin represent extracellular or lysosomal L. monocytogenes. L. monocytogenes organisms appeared as short green rods in untreated L. monocytogenes-infected HeLa cells at 20 h postinfection (Fig. 6A). Some of the rods colocalized with actin (red stain), consistent with these bacteria residing in the cytosol. Many of the cytosolic L. monocytogenes cells appeared as longer, diploid-like rods, consistent with these bacteria undergoing cell division in the cytosol. Abnormally long rods colocalizing with actin were observed in AFN-1252-treated, L. monocytogenes-infected HeLa cells at 20 h postinfection (Fig. 6B). The lengths of the bacteria under both conditions were quantified using ImageJ software (Fig. 7). Listeria cells under normal intracellular growth conditions were 3.1 ± 1.3 μm long, and in the presence of AFN-1252 they were 4.6 ± 3.4 μm. The large standard error was due to the bimodal distribution of cell lengths in the AFN-1252-treated cells, with many cells being considerably longer and shorter than normal. This phenotype suggested that these bacteria were unable to properly divide due to the on-target inhibition of fatty acid synthesis.
FIG 6.
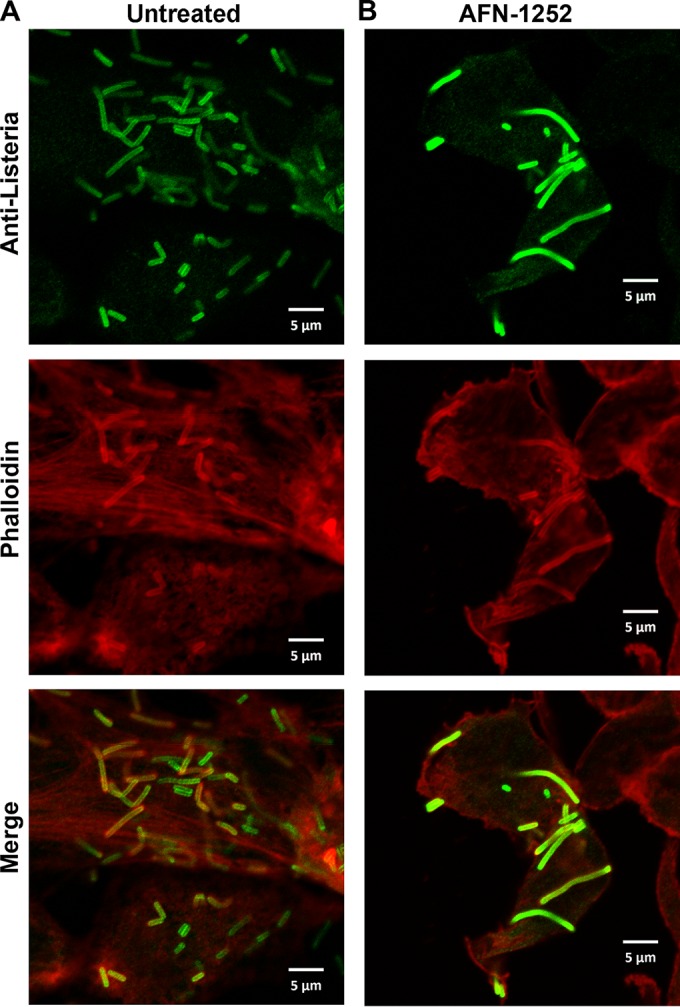
AFN-1252 arrested the division of intracellular L. monocytogenes. L. monocytogenes-infected HeLa cells were labeled with anti-Listeria-specific rabbit IgG antibodies, Alexa Fluor 488 (green)-conjugated goat anti-rabbit IgG, and Alexa Fluor 594 (red)-conjugated phalloidin. (Top) L. monocytogenes staining (green). (Middle) Cellular actin staining (red). (Bottom) Merges of the two images from the top and middle. (A) Untreated HeLa cells infected with L. monocytogenes for 20 h. (B) AFN-1252 (50 μM)-treated HeLa cells infected with L. monocytogenes for 20 h.
FIG 7.
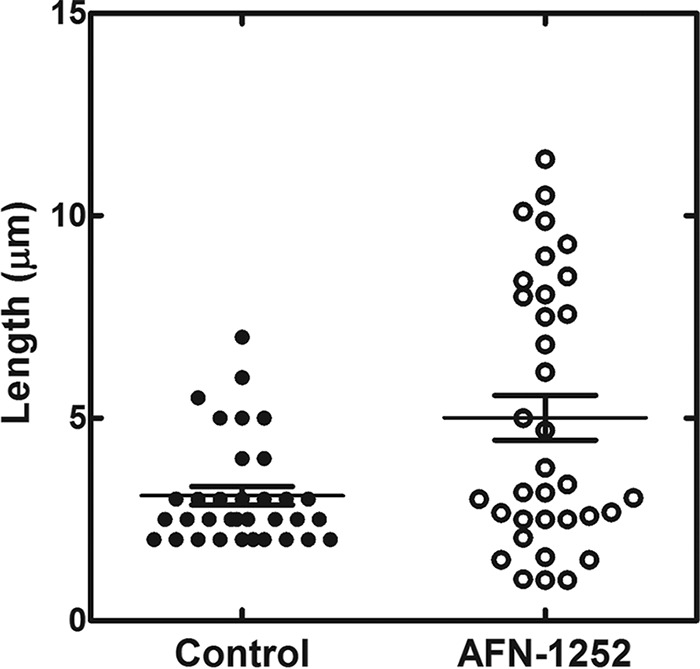
Length distribution of untreated and AFN-1252-treated L. monocytogenes in the intracellular infection model. The lengths of the intracellular L. monocytogenes cells were determined using the analyze/measure function in ImageJ software.
AFN-1252 caused the cessation of cellular growth after 1 to 2 cellular doublings in bacterial genomes encoding a single, essential FabI, such as those of Neisseria and Staphylococcus (24, 38). A similar phenotype was observed in intracellular L. monocytogenes, where L. monocytogenes is able to grow for 2 to 3 doublings before cell growth stopped and the number of CFU slowly decreased over time (Fig. 5A). This result showed that LmFabI was essential for the proper intracellular replication and division of L. monocytogenes, and that LmFabK1 and exogenous fatty acid incorporation were unable to support intracellular growth. The slow decrease of L. monocytogenes titer from 20 h to 44 h in AFN-1252-treated cells suggested that arrested cells eventually became nonviable. Whether this is through the direct effect of the inhibitor on the viability of the bacteria or the clearing of the improperly dividing bacteria with lipid-deficient membrane by the host cell remains to be determined.
DISCUSSION
This study shows that FabI is the principal enoyl-ACP reductase of L. monocytogenes FASII and is required for the intracellular growth of this facultative intracellular pathogen. Inactivation of LmFabI caused an 80% decrease in fatty acid synthesis. Although LmFabK1 is capable of complementing FASII when expressed in the heterologous E. coli system, the physiological level of LmFabK1 expression in L. monocytogenes can only support 20% of FASII activity in the absence of FabI. This leads to a reduced growth rate of planktonic cultures treated with AFN-1252 to inactivate FabI. Exogenous fatty acids can partially offset the growth-inhibitory effects of AFN-1252 in planktonic culture and are incorporated into membrane phospholipids, showing that the growth defect is due to reduced FASII activity. However, FabI inactivation with AFN-1252 had a more drastic effect on the intracellular growth of L. monocytogenes. The bacterial cells became unusually elongated, failed to divide, and eventually were cleared from the cells. In this physiological context, neither LmFabK1 expression nor the acquisition of host cell fatty acids could support L. monocytogenes proliferation when LmFabI is inactivated. These results show that inhibitors of FabI, or any other enzyme in FASII (Fig. 1), would be effective in preventing intracellular L. monocytogenes replication.
L. monocytogenes does not actively incorporate exogenous fatty acids into its membrane phospholipids. The L. monocytogenes genome encodes the fatty acid kinase system, which is the first step in exogenous fatty acid incorporation (Fig. 1). However, the primary purpose of this system does not appear to be the incorporation of exogenous fatty acids. Exogenous fatty acids incorporated via the fatty acid kinase system constitute half the total membrane phospholipid fatty acids in S. aureus (14), but exogenous fatty acids only contribute a few percent to the membrane of L. monocytogenes during planktonic growth. Incorporation of exogenous fatty acids became significant only when fatty acid synthesis is blocked by AFN-1252. However, this enhanced exogenous fatty acid incorporation did not completely rescue the growth defect caused by FabI inhibition during planktonic growth. Incorporation of the host cell fatty acids was not able to overcome FASII inhibition in the context of intracellular growth, demonstrating that the L. monocytogenes fatty acid kinase system is not able to use host fatty acids to support growth. The correct acyl chain composition has been clearly demonstrated to play a key role in the environmental and intracellular survival of L. monocytogenes, and increasing the straight-chain composition reduces the fitness of the organism (2–6). Furthermore, fatty acids, particularly branched chains, are inhibitors of L. monocytogenes growth (22). In S. aureus, the fatty acid kinase system is used to sense the environment and acts as the master regulator of virulence, in addition to its role in the incorporation of exogenous fatty acids (14). Further work is needed to determine if the fatty acid kinase system functions in virulence signaling in L. monocytogenes.
ACKNOWLEDGMENTS
We thank the Hartwell Center DNA sequencing shared resources for DNA sequencing and Yannan Ouyang and the St. Jude Cell and Tissue Imaging Center for microscopy assistance. Listeria monocytogenes strain 10403S was a gift from Thirumala-Devi Kanneganti.
REFERENCES
- 1.Ireton K. 2013. Molecular mechanisms of cell-cell spread of intracellular bacterial pathogens. Open Biol 3:130079. doi: 10.1098/rsob.130079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun Y, Wilkinson BJ, Standiford TJ, Akinbi HT, O'Riordan MXD. 2012. Fatty acids regulate stress resistance and virulence factor production for Listeria monocytogenes. J Bacteriol 194:5274–5284. doi: 10.1128/JB.00045-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu K, Ding X, Julotok M, Wilkinson BJ. 2005. Exogenous isoleucine and fatty acid shortening ensure the high content of anteiso-C15:0 fatty acid required for low-temperature growth of Listeria monocytogenes. Appl Environ Microbiol 71:8002–8007. doi: 10.1128/AEM.71.12.8002-8007.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun Y, O'Riordan MXD. 2010. Branched-chain fatty acids promote Listeria monocytogenes intracellular infection and virulence. Infect Immun 78:4667–4673. doi: 10.1128/IAI.00546-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu K, Bayles DO, Xiong A, Jayaswal RK, Wilkinson BJ. 2005. Precursor and temperature modulation of fatty acid composition and growth of Listeria monocytogenes cold-sensitive mutants with transposon-interrupted branched-chain a-keto acid dehydrogenase. Microbiology 151:615–623. doi: 10.1099/mic.0.27634-0. [DOI] [PubMed] [Google Scholar]
- 6.Lucic D, Huang ZH, Gu D, Subbaiah PV, Mazzone T. 2007. Cellular sphingolipids regulate macrophage apolipoprotein E secretion. Biochemistry 46:11196–11204. doi: 10.1021/bi701106v. [DOI] [PubMed] [Google Scholar]
- 7.Rock CO, Jackowski S. 2002. Forty years of fatty acid biosynthesis. Biochem Biophys Res Commun 292:1155–1166. doi: 10.1006/bbrc.2001.2022. [DOI] [PubMed] [Google Scholar]
- 8.Singh VK, Hattangady DS, Giotis ES, Singh AK, Chamberlain NR, Stuart MK, Wilkinson BJ. 2008. Insertional inactivation of branched-chain a-keto acid dehydrogenase in Staphylococcus aureus leads to decreased branched-chain membrane fatty acid content and increased susceptibility to certain stresses. Appl Environ Microbiol 74:5882–5890. doi: 10.1128/AEM.00882-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh AK, Zhang Y-M, Zhu K, Subramanian C, Li Z, Jayaswal RK, Gatto C, Rock CO, Wilkinson BJ. 2009. FabH selectivity for anteiso branched-chain fatty acid precursors in low temperature adaptation in Listeria monocytogenes. FEMS Microbiol Lett 301:188–192. doi: 10.1111/j.1574-6968.2009.01814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao J, Rock CO. 2013. Phosphatidic acid synthesis in bacteria. Biochim Biophys Acta 1831:495–502. doi: 10.1016/j.bbalip.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu Y-J, Zhang Y-M, Grimes KD, Qi J, Lee RE, Rock CO. 2006. Acyl-phosphates initiate membrane phospholipid synthesis in gram-positive pathogens. Mol Cell 23:765–772. doi: 10.1016/j.molcel.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 12.Yao J, Rock CO. 2015. How bacterial pathogens eat host lipids: implications for the development of fatty acid synthesis therapeutics. J Biol Chem 290:5940–5946. doi: 10.1074/jbc.R114.636241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsons JB, Frank MW, Jackson P, Subramanian C, Rock CO. 2014. Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol Microbiol 92:234–245. doi: 10.1111/mmi.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons JB, Broussard TC, Bose JL, Rosch JW, Jackson P, Subramanian C, Rock CO. 2014. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc Natl Acad Sci U S A 111:10532–10537. doi: 10.1073/pnas.1408797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heath RJ, Su N, Murphy CK, Rock CO. 2000. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J Biol Chem 275:40128–40133. doi: 10.1074/jbc.M005611200. [DOI] [PubMed] [Google Scholar]
- 16.Marrakchi H, DeWolf WE Jr, Quinn C, West J, Polizzi BJ, So CY, Holmes DJ, Reed SL, Heath RJ, Payne DJ, Rock CO, Wallis NG. 2003. Characterization of Streptococcus pneumoniae enoyl-[acyl carrier protein] reductase (FabK). Biochem J 370:1055–1062. doi: 10.1042/bj20021699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu L, Bi H, Ma J, Hu Z, Zhang W, Cronan JE, Wang H. 2013. The two functional enoyl-acyl carrier protein reductases of Enterococcus faecalis do not mediate triclosan resistance. mBio 4:e00613. doi: 10.1128/mBio.00613-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao J, Abdelrahman YM, Robertson RM, Cox JV, Belland RJ, White SW, Rock CO. 2014. Type II fatty acid synthesis is essential for the replication of Chlamydia trachomatis. J Biol Chem 289:22365–22376. doi: 10.1074/jbc.M114.584185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paoletti L, Lu Y-J, Schujman GE, de Mendoza D, Rock CO. 2007. Coupling of fatty acid and phospholipid synthesis in Bacillus subtilis. J Bacteriol 189:5816–5824. doi: 10.1128/JB.00602-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao J, Maxwell JB, Rock CO. 2013. Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI). J Biol Chem 288:36261–36271. doi: 10.1074/jbc.M113.512905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morosky S, Lennemann NJ, Coyne CB. 2016. BPIFB6 regulates secretory pathway trafficking and enterovirus replication. J Virol 90:5098–5107. doi: 10.1128/JVI.00170-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang LL, Johnson EA. 1992. Inhibition of Listeria monocytogenes by fatty acids and monoglycerides. Appl Environ Microbiol 58:624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons JB, Yao J, Frank MW, Jackson P, Rock CO. 2012. Membrane disruption by antimicrobial fatty acids releases low molecular weight proteins from Staphylococcus aureus. J Bacteriol 194:5294–5304. doi: 10.1128/JB.00743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao J, Bruhn DF, Frank MW, Lee RE, Rock CO. 2016. Activation of exogenous fatty acids to acyl-acyl carrier protein cannot bypass FabI inhibition in Neisseria. J Biol Chem 291:171–181. doi: 10.1074/jbc.M115.699462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 26.Yao J, Dodson VJ, Frank MW, Rock CO. 2015. Chlamydia trachomatis scavenges host fatty acids for phospholipid synthesis via an acyl-acyl carrier protein synthetase. J Biol Chem 290:22173. doi: 10.1074/jbc.M115.671008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Francis M, Thomas CJ. 1996. Effect of multiplicity of infection on Listeria monocytogenes pathogenicity for HeLa and Caco-2 cell lines. J Med Microbiol 45:323–330. doi: 10.1099/00222615-45-5-323. [DOI] [PubMed] [Google Scholar]
- 28.Köprülü AD, Kastner R, Wienerroither S, Lassnig C, Putz EM, Majer O, Reutterer B, Sexl V, Kuchler K, Mnller M, Decker T, Ellmeier W. 2013. The tyrosine kinase Btk regulates the macrophage response to Listeria monocytogenes infection. PLoS One 8:e60476. doi: 10.1371/journal.pone.0060476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kastner R, Dussurget O, Archambaud C, Kernbauer E, Soulat D, Cossart P, Decker T. 2011. LipA, a tyrosine and lipid phosphatase involved in the virulence of Listeria monocytogenes. Infect Immun 79:2489–2498. doi: 10.1128/IAI.05073-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergler H, Fuchsbichler S, Högenauer G, Turnowsky F. 1996. The enoyl-[acyl-carrier-protein] reductase (FabI) of Escherichia coli, which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Eur J Biochem 242:689–694. doi: 10.1111/j.1432-1033.1996.0689r.x. [DOI] [PubMed] [Google Scholar]
- 31.Heath RJ, Rock CO. 1995. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem 270:26538–26542. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 32.Bergler H, Wallner P, Ebeling A, Leitinger B, Fuchsbichler S, Aschauer H, Kollenz G, Högenauer G, Turnowsky F. 1994. Protein envM is the NADH-dependent enoyl-ACP reductase fabI of Escherichia coli. J Biol Chem 269:5493–5496. [PubMed] [Google Scholar]
- 33.Massengo-Tiasse RP, Cronan JE. 2008. Vibrio cholerae fabV defines a new class of enoyl acyl-carrier-protein reductase. J Biol Chem 283:1308–1316. doi: 10.1074/jbc.M708171200. [DOI] [PubMed] [Google Scholar]
- 34.Hirschbeck MW, Kuper J, Lu H, Liu N, Neckles C, Shah S, Wagner S, Sotriffer CA, Tonge PJ, Kisker C. 2012. Structure of the Yersinia pestis FabV enoyl-ACP reductase and its interaction with two 2-pyridone inhibitors. Structure 20:89–100. doi: 10.1016/j.str.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bergler H, Högenauer G, Turnowsky F. 1992. Sequences of the envM gene and two mutated alleles in Escherichia coli. J Gen Microbiol 138:2093–2100. doi: 10.1099/00221287-138-10-2093. [DOI] [PubMed] [Google Scholar]
- 36.Kaplan N, Albert M, Awrey D, Bardouniotis E, Berman J, Clarke T, Dorsey M, Hafkin B, Ramnauth J, Romanov V, Schmid MB, Thalakada R, Yethon J, Pauls HW. 2012. Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob Agents Chemother 56:5865–5874. doi: 10.1128/AAC.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaplan N, Awrey D, Bardouniotis E, Berman J, Yethon J, Pauls HW, Hafkin B. 2013. In vitro activity (MICs and rate of kill) of AFN-1252, a novel FabI inhibitor, in the presence of serum and in combination with other antibiotics. J Chemother 25:18–25. doi: 10.1179/1973947812Y.0000000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. 2011. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc Natl Acad Sci U S A 108:15378–15383. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karlowsky JA, Kaplan N, Hafkin B, Hoban DJ, Zhanel GG. 2009. AFN-1252, a FabI inhibitor, demonstrates a Staphylococcus-specific spectrum of activity. Antimicrob Agents Chemother 53:3544–3548. doi: 10.1128/AAC.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giotis ES, McDowell DA, Blair IS, Wilkinson BJ. 2007. Role of branched-chain fatty acids in pH stress tolerance in Listeria monocytogenes. Appl Environ Microbiol 73:997–1001. doi: 10.1128/AEM.00865-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaneda T. 1991. Iso- and anteiso-fatty acids in bacteria: biosynthesis, function, and taxonomic significance. Microbiol Rev 55:288–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao J, Cherian PT, Frank MW, Rock CO. 2015. Chlamydia trachomatis relies on autonomous phospholipid synthesis for membrane biogenesis. J Biol Chem 290:18874–18888. doi: 10.1074/jbc.M115.657148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. 2009. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 458:83–86. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- 44.Tang P, Sutherland CL, Gold MR, Finlay BB. 1998. Listeria monocytogenes invasion of epithelial cells requires the MEK-1/ERK-2 mitogen-activated protein kinase pathway. Infect Immun 66:1106–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weiglein I, Goebel W, Troppmair J, Rapp UR, Demuth A, Kuhn M. 1997. Listeria monocytogenes infection of HeLa cells results in listeriolysin O-mediated transient activation of the Raf-MEK-MAP kinase pathway. FEMS Microbiol Lett 148:189–195. doi: 10.1111/j.1574-6968.1997.tb10287.x. [DOI] [PubMed] [Google Scholar]
- 46.Hamon MA, Ribet D, Stavru F, Cossart P. 2012. Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol 20:360–368. doi: 10.1016/j.tim.2012.04.006. [DOI] [PubMed] [Google Scholar]