

Abstract
At present, there is no vaccine for enterotoxigenic Escherichia coli (ETEC), an important cause of diarrheal illness. Nevertheless, recent microbial pathogenesis studies have identified a number of molecules produced by ETEC that contribute to its virulence and are novel antigenic targets to complement canonical vaccine approaches. EtpA is a secreted two-partner adhesin that is conserved within the ETEC pathovar. EtpA interacts with the tips of ETEC flagella to promote bacterial adhesion, toxin delivery, and intestinal colonization by forming molecular bridges between the bacteria and the epithelial surface. However, the nature of EtpA interactions with the intestinal epithelium remains poorly defined. Here, we demonstrate that EtpA interacts with glycans presented by transmembrane and secreted intestinal mucins at epithelial surfaces to facilitate pathogen-host interactions that culminate in toxin delivery. Moreover, we found that a major effector molecule of ETEC, the heat-labile enterotoxin (LT), may enhance these interactions by stimulating the production of the gel-forming mucin MUC2. Our studies suggest, however, that EtpA participates in complex and dynamic interactions between ETEC and the gastrointestinal mucosae in which host glycoproteins promote bacterial attachment while simultaneously limiting the epithelial engagement required for effective toxin delivery. Collectively, these data provide additional insight into the intricate nature of ETEC interactions with the intestinal epithelium that have potential implications for rational approaches to vaccine design.
INTRODUCTION
Enterotoxigenic Escherichia coli (ETEC) is a major cause of infectious diarrhea in the developing world (1), where this organism causes millions of infections and hundreds of thousands of deaths, particularly in young children (2). This organism contributes substantially to the large global burden of diarrheal illness that has been associated with a number of important but poorly understood sequelae, including “environmental enteropathy” and compromised growth and cognitive development (3).
ETEC was discovered more than 40 years ago in cases of diarrheal illness clinically indistinguishable from cholera (4) and is defined by the production of plasmid-encoded heat-labile enterotoxin (LT) and/or heat-stable enterotoxin (ST). LT and ST must successfully engage cognate receptors (GM-1 ganglioside and guanylyl cyclase C, respectively) on the surface of epithelial cells to activate the production of cyclic nucleotides. The respective increases in intracellular cyclic AMP (cAMP) and cGMP trigger signaling events culminating in altered salt and water absorption in the small intestine accompanied by voluminous watery diarrhea (5).
Because of their substantial impact on global health, these pathogens are a principal target for vaccine development. Unfortunately, there is currently no vaccine that affords significant broad-based protection against ETEC (6). Despite decades of research, the current understanding of the pathogenesis of this complex and highly varied pathogen remains incomplete. This is especially true of ETEC interactions with the intestinal epithelium that are critical for efficient delivery of enterotoxins. While the majority of ETEC pathogenesis investigations, and consequently vaccine development efforts, have focused on plasmid-encoded fimbrial colonization factors (6, 7), recent studies have suggested that the interactions of ETEC with the intestinal mucosa and with host epithelial cells are highly complex and involve the orchestrated deployment of virulence factors unique to this pathovar, as well as highly conserved chromosomally encoded molecules (8, 9).
Elucidation of ETEC molecular interactions with the intestinal epithelium can identify new potential interdiction targets and potentially inform novel approaches to vaccine development (10). Recent studies have identified several molecules that engage intestinal epithelial cells (11) or work to degrade intestinal mucins (12, 13) to permit ETEC to gain access to the intestinal epithelium and enhance toxin delivery.
One potential novel vaccine candidate, EtpA, is a secreted adhesin (14) that appears to act as a molecular bridge between ETEC and the host epithelium (15), thereby promoting intestinal colonization and toxin delivery. Although EtpA binds avidly to the tips of ETEC flagella (15), permitting the bacteria to use these long peritrichous structures to engage the intestinal surface, the targets of EtpA-mediated adhesion and intestinal colonization remain undefined. Here we demonstrate that this high-molecular-weight extracellular adhesin interacts with glycans present on intestinal mucins to promote bacterial adhesion and toxin delivery.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
ETEC strain H10407 was obtained from the good manufacturing practices lots of bacteria produced at the Walter Reed Army Institute of Research, USA. The etpA mutant (jf1668) and complemented (jf1697) strains of H10407 were generated in a prior study (15). ETEC strains were routinely grown in Luria broth (LB) or on Luria agar plates at 37°C from glycerol stocks preserved at −80°C. The bacterial strains and plasmids used in this study are described in Table 1.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference(s) or source |
|---|---|---|
| Strains | ||
| H10407 | Wild-type ETEC strain O78:H11 CFA/1 LT+/ST+ EtpA+ | 45, 46 |
| jf576 | LT− (eltAB::Kmr) mutant of H10407 | 47 |
| jf876 | lacZYA::Kmr | 47 |
| jf1668 | etpA::Cmr mutant of H10407 | 15 |
| jf1696 | TOP10(pJL017, pJL030) Ampr Cmr | 16 |
| TOP10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| Plasmids | ||
| pJL017 | etpBA cloned into pBAD/Myc-His A with etpA in frame with myc and 6His coding regions, Ampr | 20 |
| pJL030 | etpC gene cloned into pACYC184, Cmr | 15 |
Kmr, kanamycin resistance; Cmr, chloramphenicol resistance; Ampr, ampicillin resistance.
Protein expression and purification.
Recombinant EtpA (rEtpA) glycoprotein (rEtpAmycHis6) was purified from culture supernatants of E. coli TOP10 carrying plasmids pJL017 and pJL030 as previously described (16). Briefly, E. coli TOP10 carrying pJL017 and pJL030 (Table 1) was grown overnight from frozen glycerol stocks and then diluted 1:100 into fresh Luria broth containing ampicillin (100 μg/ml) and chloramphenicol (15 μg/ml) at 37°C and 230 rpm until the optical density at 600 nm reached ∼0.5 to 0.6. Recombinant protein expression was then induced with 0.0002% arabinose for 6 h at 37°C. Culture supernatants containing secreted protein were then concentrated with a filter with a 100-kDa cutoff (Millipore), and rEtpAmycHis6 protein was purified by immobilized metal affinity chromatography (16).
Gel-forming MUC2 mucin was purified as described previously (12). Briefly, the tissue culture supernatant of LS 174T cells was concentrated with a filter with a 100-kDa cutoff and size exclusion chromatography was performed with Sepharose CL-2B resin. MUC2-containing fractions were collected in the void column volume, pooled, and preserved at −80°C.
Tissue culture and RNA interference (RNAi).
All of the human intestinal epithelial cell lines used (Table 2), including LS 174T (ATCC CL-188), Caco-2 (ATCC HTB-37), CC2BBe1 (ATCC CRL-2102), and HT-29 (ATCC HTB-38), were purchased from the American Type Culture Collection. LS 174T and Caco-2 cells were cultured in Eagle's minimum essential medium with 10 or 20% (final concentration) fetal bovine serum (FBS), respectively, at 37°C in 5% CO2. CC2BBe1 (brush border-expressing derivative of Caco-2) cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS and human transferrin (10 μg/ml). HT-29 cells were routinely cultured in McCoy's 5A medium supplemented with 10% FBS (McCoy's 5A-10) at 37°C in 5% CO2.
TABLE 2.
Human intestinal cell lines and siRNAs used in this study
| Cell line or siRNA | Description or target description | Gene ID | Source (no.) | Reference |
|---|---|---|---|---|
| Cell lines | ||||
| Caco-2 | Epithelial (enterocyte), colonic adenocarcinoma | ATCC HTB-37 | ||
| CC2BBe1 | Enterocyte, polarizes into monolayers with an apical brush border, derived from Caco-2 | ATCC CRL-2102 | ||
| HT-29 | Epithelial (enterocyte), colonic adenocarcinoma | ATCC HTB-38 | ||
| LS 174T | Colonic adenocarcinoma, MUC2 producing | ATCC CL-188 | ||
| siRNAs | ||||
| MUC2 | Oligomeric, gel-forming mucin | 4583 | TFa (9070) | |
| MUC3A/B | Cell surface membrane-bound mucin | 4584 | TF (s195330) | 48 |
TF, Thermo Fisher Scientific (Ambion).
Mucin gene silencing in human intestinal cell lines was carried out by RNAi. Reverse transfection was performed with Lipofectamine RNAiMAX (Invitrogen) and predesigned/prevalidated MUC3A/B- and MUC2-specific small interfering RNAs (siRNAs; Thermo Fisher Scientific) (Table 2) at a final concentration of 20 nM in 96-well tissue culture plates. Appropriate negative-control siRNA molecules were used at the same concentration. For cultures grown for several days postconfluence, fresh growth medium containing siRNA (20 nM) was replaced on alternate days. Mucin knockdown was confirmed by immunoblotting with specific antibodies as described above.
Protein interaction studies.
To examine EtpA interaction with MUC2 mucin by far-Western analysis, 10 μg of purified MUC2 was resolved by SDS-PAGE on NuPAGE Novex 3 to 8% Tris-acetate gel (Life Technologies) and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat milk in Tris-buffered saline–0.01% Tween 20 for 1 h and then incubated with 50 μg/ml purified rEtpA overnight at 4°C. Proteins were detected by immunoblotting with rabbit anti-MUC2 polyclonal antibody (H-300 sc15334; Santa Cruz) or affinity-purified anti-EtpA rabbit polyclonal antibody (15).
To test whether carbohydrate moieties are involved in EtpA-mucin interactions, 100 μg of purified glycoprotein rEtpA or MUC2 was treated with 10 mM (final concentration) sodium metaperiodate (NaIO4; Sigma S1878) in phosphate-buffered saline (PBS) for 1 h at 4°C in the dark. NaIO4-treated proteins were then dialyzed overnight against PBS and used in interaction studies.
To examine the interaction of EtpA with MUC3, cell lysate prepared from Caco-2 cells grown as described above for 8 days after they attained confluence was used as the source of MUC3. Briefly, Caco-2 cell monolayers cultured in 100- by 15-mm tissue culture plates were washed three times with PBS, scraped off, transferred to a 1.5-ml microcentrifuge tube, and lysed by five cycles of freezing (dry ice) and thawing (37°C water bath) in 500 μl of lysing buffer (50 mM sodium phosphate [pH 7.4], 250 mM NaCl, 5 mM EDTA, 0.1% Triton X-100, 0.1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail [EDTA-free complete protease cocktail; Roche]). Cell suspensions were sheared several times with a syringe and needle and then centrifuged at 10,000 × g, for 10 min at 4°C. A total of 10 mg of protein was recovered from the supernatant fraction of the cell lysate, diluted to a 1-ml final volume with PBS, and preclarified with 50 μl of rProtein A resin (Repligen). The clarified lysate was rotation mixed with 100 μg of purified rEtpA, 50 μl of protein A beads (50% slurry in PBS), and 5 μl of mouse anti-human MUC3 (IgG2a) monoclonal antibody (MA1-35702; Pierce) overnight at 4°C. The following day, protein A resin was collected by centrifugation, washed four times with PBS–0.01% Tween 20, and boiled in 50 μl of Laemmli sample buffer for 10 min. The proteins were resolved by SDS-PAGE as described above, and immunoblotting was performed with affinity-purified rabbit anti-EtpA polyclonal antibody or goat anti-MUC3A/B polyclonal antibody (sc-13314; Santa Cruz).
BLI.
Bio-Layer Interferometry (BLI) was used to determine the affinity of EtpA for MUC2 with OctetRed96 (Pall fortéBIO Corporation, USA). Briefly, purified MUC2 was biotin labeled with the protein biotinylation reagent EZ-Link Sulfo-NHS-LC-Biotin (Thermo Scientific) in accordance with the manufacturer's protocol and immobilized on streptavidin (SA) biosensors (18-5019; Pall fortéBIO Corporation). Twofold serial dilutions (1,000, 500, 250, 125, 62.5, and 31.25 nM) of purified rEtpA were prepared in 1× PBS and used as the analyte. Experiments were performed in triplicate, and affinity constants (KD values) were obtained from a global fit of real-time kinetic measurements from the titration series calculated as 1:1 binding with Octet software version 8.1 (Pall fortéBIO Corporation).
Real-time quantitative PCR (qPCR).
Following overnight treatment of HT-29 cells with LT (0.1 μg/ml), cells were washed once with PBS and then the contents of 3 wells of a 96-well plate were combined and resuspended in 500 μl of TRIzol reagent. Following RNA extraction with phenol-chloroform for 3 min on ice, samples were centrifuged at 14,000 × g for 15 min. A 200-μl volume of the aqueous phase was recovered, combined with isopropanol for 90 min at −80°C, and then centrifuged at 14,000 × g for 20 min. The pellet was washed once with 75% ethanol, air dried, and resuspended in DNase I (Invitrogen) for 30 min at room temperature, and the remaining RNA was purified (RNeasy; Qiagen). The total RNA was quantified by measurement of A260 (Eon Take3; BioTek, VT, USA).
cDNA was generated with the SuperScript VILO cDNA synthesis kit (Invitrogen) by using 0.5 μg of RNA with or without reverse transcriptase. qPCR was performed with SYBER Green master mix (Thermo Fischer) and 10 ng of cDNA. Glyceraldehyde 3-phosphate dehydrogenase was used for standardization with forward primer 5′-ACCCACTCCTCCACCTTTGA and reverse primer 5′-CTGTTGCTGTAGCCAAATTCG. MUC2 was detected with forward primer 5′-GCTCATTGAGAACGATG and reverse primer 5′-CTTAGTGTCCAGCTCCAGCA. All reactions were performed in triplicate on an Applied Biosystems 7500 real-time PCR system and analyzed with 7500 System Software v1.40. Fold change was calculated by ΔΔCT with Excel software.
ETEC adhesion and toxin delivery assays.
In vitro assays of ETEC adhesion and delivery of LT were performed with control and MUC3A/B siRNA-treated Caco-2 cells in 96-well plates. Caco-2 cells were cultured for up to 8 days after cell confluence to permit cell differentiation and MUC3 protein expression to become detectable by immunoblotting. Bacteria to be tested were grown overnight in 2 ml of LB with appropriate antibiotics, diluted 1:100 into fresh medium on the morning of the experiment, and grown for an additional 90 min to mid-logarithmic growth phase. A 1-μl sample of bacterial culture was then added to each well of the 96-well Caco-2 tissue culture plate in quadruplicate. After 1 h, the monolayers were washed three times with tissue culture medium and then treated with 0.1% Triton X-100 in PBS for 5 min. Triton X-100 lysates containing total cell-associated bacteria were then diluted 1:10 in PBS and plated onto Luria agar with appropriate antibiotics. Bacterial adherence was calculated as the percentage of organisms recovered per CFU of inoculum.
To examine effective delivery of LT, cultures of bacteria were grown overnight and Caco-2 monolayers were infected as described above. After incubation at 37°C in 5% CO2 for 2 h, monolayers were washed three times with prewarmed tissue culture medium. After the medium was replaced, the plates were returned to the tissue culture incubator for an additional 2.5 h. The efficiency of toxin delivery was determined by measuring the cellular cAMP levels induced in monolayers following ETEC infection relative to those in cells infected with LT mutant jf576 (Table 1) with a commercial enzyme-linked immunosorbent assay kit (Arbor Assays, Ann Arbor, MI).
Effect of LT on in vitro MUC2 expression and ETEC adhesion.
The effect of LT or the closely related cholera toxin (CT, catalog no. 110B; List Biological Laboratories, Inc.) on MUC2 expression was studied in HT-29 cells. Briefly, confluent HT-29 cell monolayers were cultured in 96-well tissue culture plates or on coverslips in 24-well plates as mentioned above and toxin treatment was performed by replacing the culture medium with fresh medium containing a 0.1-μg/ml final concentration of LT or CT for the times indicated. Confocal immunofluorescence microscopy was performed to detect HT-29 cell surface-associated MUC2. To detect cell-associated MUC2, HT-29 cell monolayers were lysed in Laemmli sample buffer and samples were boiled, resolved by SDS-PAGE, and transferred to nitrocellulose membranes prior to immunoblotting.
Periodate oxidation of cell monolayers.
To investigate the role of mucin glycans expressed on the surface of HT29 cells in bacterial adhesion, ETEC adhesion assays were performed as described above with untreated cells or cells fixed with 2% paraformaldehyde for 30 min at 37°C in 5% CO2. NaIO4 (2 mM final concentration) oxidation of fixed cell monolayers was performed for 30 min, followed by the addition of 1% glycerol and incubation for 10 min to quench excess periodate. Cells were washed with PBS, and an adherence assay was performed as described above. Control cells were treated in an identical manner, except for the addition of periodate.
ETEC murine colonization.
Five- to 8-week-old female mice (C57BL/6) were purchased from Charles River Laboratories International Inc., USA. Mice were pretreated with streptomycin (5 g/liter) in drinking water for 24 h to inhibit gut microbes, followed by the provision of drinking water without antibiotics for 12 h as previously described (17). A 125-μl volume of famotidine (20-mg/ml stock) was injected intraperitoneally per mouse to reduce gastric acidity 1 h before the mice were challenged by oral gavage with ∼106 CFU of the lacZYA::Kmr jf876 or etpA::Cmr jf1668 mutant strain. Mice were sacrificed 24 h after infection, the small intestine was collected and lysed in 5% saponin as previously described (17), and lysates were plated onto Luria agar plates containing kanamycin (25 μg/ml) or chloramphenicol (15 μg/ml).
To examine the role of etpA in mediation of the colonization of C57BL/6 parental mice, competition experiments were performed as previously described (15). Briefly, mice were prepared as described above and then challenged with ∼1 × 106 CFU of jf876 (Table 1) mixed with the etpA mutant jf1668 (etpA::Cmr) in a final volume of 400 μl. After 24 h, the intestinal lysates were plated separately onto Luria agar medium containing either chloramphenicol (15 μg/ml) or kanamycin (25 μg/ml). The competitive index (CI) was then determined for individual mice as follows: CI = [mutant (Cmr)/wild-type (Kmr) output CFU]/(mutant/wild-type input CFU), where the input fraction was determined directly by colony counting prior to final preparation of the inoculum.
The duration of intestinal colonization of C57BL/6 mice was first examined by assaying the number of CFU of bacteria shed over time following an intestinal challenge with 1 × 105 CFU of either strain jf876 (lacZYA::Kmr) or etpA mutant (etpA::Cmr) strain jf1668 bacteria. Starting 1 day after the challenge, six fecal pellets per mouse were collected every 24 h for 7 days and resuspended in PBS. Dilutions in PBS were then plated onto Luria agar plates containing kanamycin or chloramphenicol.
Immunohistology and confocal microscopy.
Sections of mouse small intestine were collected and preserved in Carnoy's fixation solution (chloroform-ethanol-acetic acid, 3:6:1) to preserve mucins and then paraffin embedded. For mucin immunostaining, slides were deparaffinized by sequential washing in xylene and ethanol. Briefly, slides were immersed in fresh xylene twice for 10 min each and then transferred to 100% ethanol for 2 min, followed by sequential washing for 1 min each in 95, 70, 50, and 30% ethanol. Slides were then transferred to 0.85% NaCl solution for 2 min and immersed in 1× PBS for 2 min. Deparaffinized and rehydrated slides were then labeled with anti-Muc2 (MUC2) rabbit or anti-Muc3 (MUC3) goat polyclonal primary antibody as described above, followed by Alexa Fluor 488-conjugated goat anti-rabbit or donkey anti-goat antibody (Life Technologies). To study EtpA colocalization with mucins, mouse intestinal sections fixed in Carnoy's solution were deparaffinized, blocked with 1% bovine serum albumin in PBS, and incubated with biotinylated rEtpA (50-μg/ml final concentration) for 2 h at 37°C or overnight at 4°C. After washing with PBS three times, EtpA was detected with Qdot 605 SA conjugate (Life Technologies). In experiments involving EtpA colocalization with Dolichos biflorus agglutinin (DBA) lectin, unlabeled rEtpA (50-μg/ml final concentration) and biotinylated DBA lectin (1:100; Vector Laboratories catalog no. B-1035) were added to mouse intestinal sections fixed in Carnoy's solution as described above. EtpA was detected with rabbit anti-EtpA polyclonal antibody (1:200), followed by Alexa Fluor 488-conjugated goat anti-rabbit antibody (1:400) (Life Technologies), and SA-conjugated Qdot 605 (1:400) was used to detect biotinylated DBA lectin (Life Technologies). Confocal microscopy images were captured with a Zeiss LSM 510 Meta confocal laser scanning microscope (CLSM). Data were then analyzed with Volocity three-dimensional image analysis software (version 6.2; PerkinElmer, Inc.) as previously described (13).
RESULTS
EtpA interacts with the gel-forming mucin MUC2.
In the intestinal lumen, bacteria encounter mucin glycoproteins, including MUC2, which is secreted by goblet cells within the mucosa. In the colon, MUC2 forms a thick complex barrier between the heavy burden of commensal organisms and the epithelium (18) and MUC2 adjacent to the colonic epithelial surface is densely packed and firmly attached, thereby effectively excluding bacteria (19).
ETEC preferentially colonizes the small intestine, where the organism effectively delivers its toxin payload (5). In contrast to the mucin layer in the colon, the MUC2 layer in the small intestine is relatively thin and loosely attached to the epithelial surface (19). We therefore questioned whether, in addition to deploying recently described mucin-degrading enzymes (12, 13), this pathogen might be equipped to engage MUC2 to establish intestinal colonization. Because the EtpA adhesin promotes effective small intestinal colonization (20) and early studies demonstrated binding to goblet cells (15), we first examined the interaction of this secreted protein with MUC2.
In far-Western analyses, rEtpA interacted with immobilized MUC2 purified from goblet cell-like line LS 174T (Fig. 1a). To confirm these interactions, we examined EtpA binding to MUC2 by BLI. Here, rEtpA bound to MUC2 with submicromolar affinity (KD, ∼10−7) (Fig. 1b; see Data Set S1 in the supplemental material). Likewise, we found that rEtpA colocalized with Muc2 in the murine small intestinal mucosa (Fig. 1c). Following infection and mouse small intestinal colonization with ETEC, we found that EtpA secreted by the bacteria also colocalized with Muc2 and that the bacteria appeared to be coated with Muc2 (Fig. 1d). Collectively, these data suggested that the previously noted EtpA-mediated colonization of the small intestine (20) occurs in part through the interactions of this secreted adhesin with Muc2 in the intestinal lumen.
FIG 1.
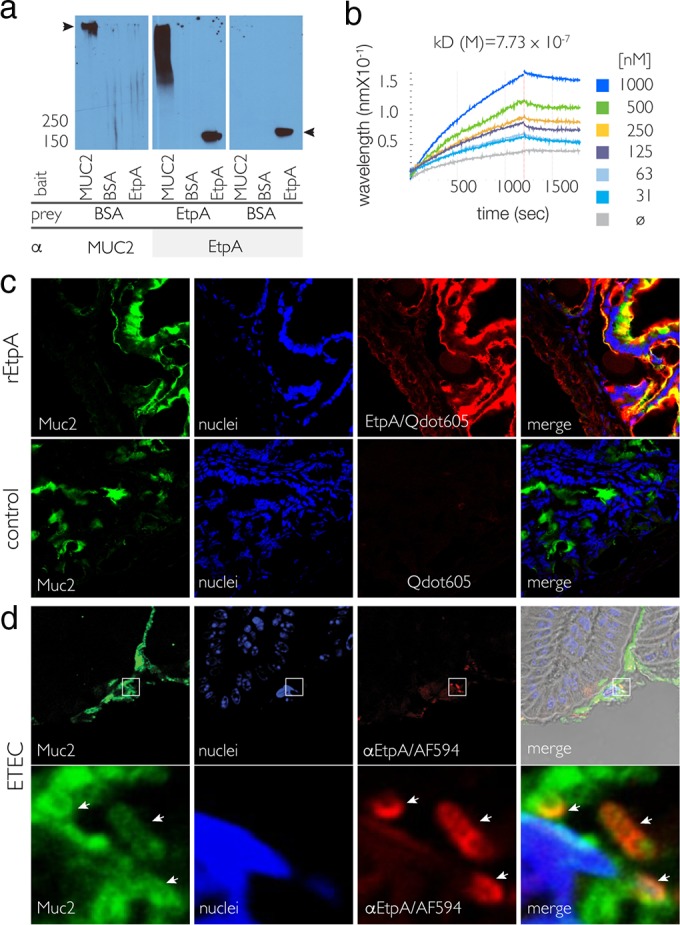
EtpA interacts with the major secreted intestinal gel-forming mucin MUC2. (a) Far-Western immunoblot analysis demonstrating binding of full-length rEtpA glycoprotein to purified human MUC2 (middle). On the left and right are negative controls. The arrowheads at the left and right represent the migration of MUC2 and EtpA, respectively. BSA, bovine serum albumin. (b) BLI studies with rEtpA glycoprotein and purified MUC2. The dissociation constant (KD) determined from a global fit of real-time kinetics from the rEtpA titration series shown in the key at the right is shown above the binding curves. The gray line (ø) represents MUC2 alone. (c) EtpA colocalizes with Muc2 in the murine small intestine. Shown are CLSM images of Muc2 (green), bound biotinylated rEtpA detected with SA-coated nanodots (red, Qdot 605), and epithelial cell nuclei (blue). EtpA-negative controls are shown in the bottom row. (d) During ETEC infection of murine small intestinal mucosal surfaces, Muc2 (green) colocalizes with EtpA (red) on the surface of ETEC. The images in the bottom row represent magnified, deconvolved regions enclosed at lower magnifications in the top row. Locations of individual bacteria are indicated by arrows.
EtpA interacts with intestinal glycocalyx transmembrane mucin MUC3.
ETEC has long been known to interact with the glycocalyx on the apical surface of enterocytes (21–23). This glycoprotein-rich layer coating the microvilli of the small intestine is thought to be formed by a number of transmembrane mucins (24), the most abundant and best studied being MUC3 (25, 26). Because we had previously shown that EtpA also interacts with the surface of intestinal epithelial cells (15), we examined whether this adhesin could engage MUC3. Incubation of EtpA with sections of small intestine from mice demonstrated that EtpA adhesin bound to the surface intestinal villi at sites of Muc3 expression (Fig. 2a). In addition, we demonstrated that following incubation with polarized human Caco-2 BBe enterocytes, which make apical brush borders (27), EtpA colocalized with MUC3 (Fig. 2b). Likewise, coimmunoprecipitation of lysates from these cells with antibodies against MUC3 yielded EtpA (Fig. 2c). The results suggest that EtpA can engage both secreted gel-forming mucins and transmembrane molecules. To examine the functional significance of EtpA interactions with MUC3, we depleted target epithelial cells of MUC3 by RNAi (Fig. 2d). Cells treated with MUC3 siRNA exhibited significantly (P < 0.0001) diminished binding of EtpA (Fig. 2e). Likewise, EtpA-expressing ETEC adhered less efficiently to cells depleted of MUC3 (Fig. 2f), resulting in decreased functional delivery of LT, as determined by cAMP activation (Fig. 2g).
FIG 2.
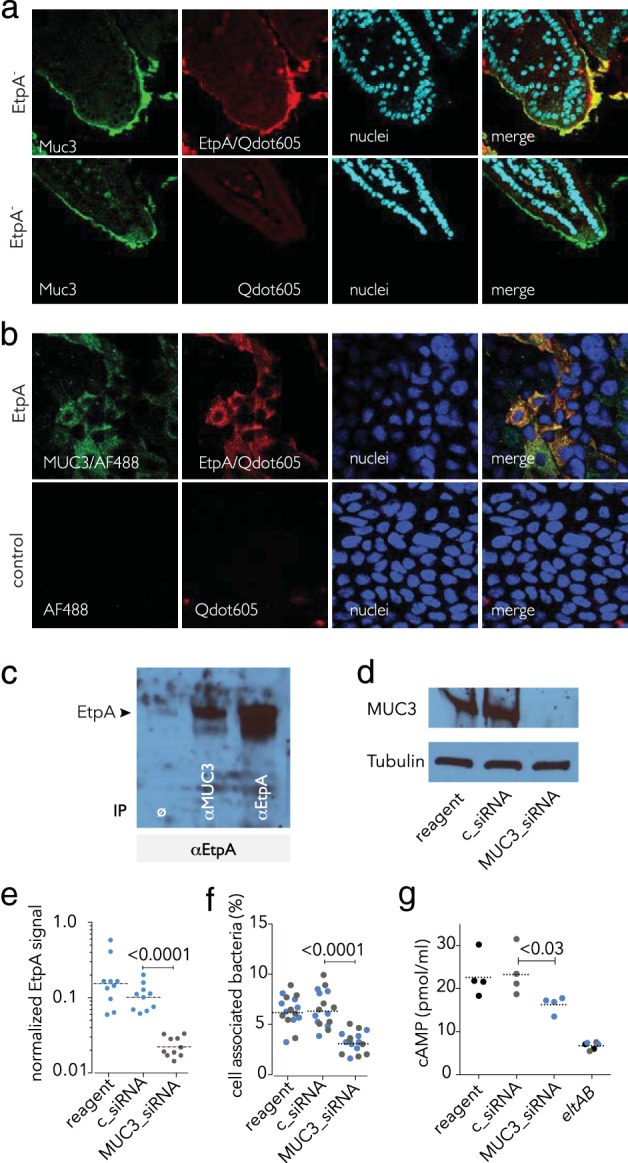
EtpA interaction with cell surface MUC3 is required for optimal ETEC epithelial cell interaction and toxin delivery in vitro. (a) EtpA interacts with Muc3 in the murine small intestine. The top row shows rEtpA (biotinylated) binding to the surface of ileal sections detected with Qdot 605. Negative controls lack rEtpA (row 2). (b) EtpA colocalizes with MUC3 on the apical surface of intestinal enterocytes. The top row shows biotinylated EtpA binding (detected with SA-Qdot 605) to Caco-2 BBe cells and colocalization with MUC3 (green); negative controls (no EtpA, no anti-MUC3 primary antibody) are shown in the bottom row. (c) EtpA immunoblot assay showing EtpA coimmunoprecipitation studies. Lanes: 1, no-antibody negative control (ø); 2, coimmunoprecipitation with anti-MUC3 antibody; 3, anti-EtpA antibody (+immunoprecipitation [IP] control). (d) siRNA-mediated depletion of MUC3 from the surface of Caco-2 epithelial cells. Shown in the immunoblot assay at the top is MUC3 produced by Caco-2 epithelial cells following treatment with transfection reagent alone (lane 1), control siRNA (lane 2), or MUC3 siRNA (lane 3). Tubulin was used as a loading control. (e) Depletion of cell surface MUC3 leads to diminished EtpA interaction with Caco-2 cells. Data represent EtpA immunofluorescence signals quantitated with Volocity imaging analysis software in MUC3 siRNA-treated cells and controls. (f) Optimal ETEC interaction with epithelial cells requires MUC3. Shown are bacteria adherent to MUC3 siRNA-treated cells and controls. (Colors represent results from two independent experiments performed on different days). (g) Optimal delivery of LT by ETEC requires MUC3. The LT-negative (eltAB) mutant was included as a negative control. Dashed horizontal lines represent geometric mean values. Statistical calculations were performed by Mann-Whitney two-tailed nonparametric testing.
Glycans expressed on mucins are important for recognition by EtpA.
Gastrointestinal mucins are heavily glycosylated proteins in which more than 80% of the structure consists of carbohydrates (24). Because EtpA interacts with multiple mucins, we investigated the contribution of glycans expressed on these proteins to EtpA binding. As shown in Fig. 3a, interruption of the glycan structure with NaIO4 drastically reduced EtpA binding to MUC2, suggesting that one or more sugars decorating the mucin protein backbone are important for EtpA binding. Mucin molecules are O glycosylated at serine or threonine residues, where N-acetylgalactosamine (GalNAc) is the first sugar added by GalNAc transferase enzymes in the Golgi apparatus, followed by “core” enzymes that add additional sugars to GalNAc. The “core 3” enzyme β1,3-N-acetylglucosaminyltransferase (C3GnT), which is most abundant in the intestinal tract, extends the GalNAc O-glycan structure by adding GlcNAc (28). The resulting core 3-derived O glycans can then be extensively modified by additional glycosyltransferases and other enzymes, resulting in complex mucin glycoprotein structures. We found that EtpA bound relatively poorly to small intestinal sections of mice that were deficient in either Muc2 expression (Muc2−/−) or C3GnT (C3GnT−/−) (Fig. 3b), again suggesting that EtpA interacts primarily with one or more glycans on mucins rather than the protein backbone.
FIG 3.

EtpA interaction with MUC2 is glycan dependent. (a) Far-Western immunoblot assay of EtpA binding to MUC2 before and after treatment with NaIO4 to open the saccharide ring structures. The control blot on the left shows MUC2 protein detected before and after NaIO4 treatment. (b) EtpA interactions with murine small intestinal Muc2 are dependent on core glycan synthesis. Confocal immunofluorescence images of EtpA interaction with Muc2 in sections of small intestine from wild-type (wt) (C57BL/6) mice (row 2). A no-EtpA control (−) is shown in row 1, and interaction with a Muc2−/− homozygous deletion mutant is shown in row 3. The bottom row demonstrates the limited interaction of EtpA in sections from mice deficient in C3GnT.
EtpA interactions with GalNAc direct binding to epithelium.
Mucins in the intestine are O glycosylated by the addition of GalNAc to Ser and Thr moieties of the respective proteins, and further substitution of these GalNAc residues yields a diverse repertoire of glycan structures (29) that may be differentially glycosylated, dependent on their relative location within the intestine (30). Expression of blood group-related antigens that terminate in GalNAc are among glycans that exhibit varied expression in the gastrointestinal tract (31). Interestingly, we found that in the murine small intestine, EtpA colocalized with the DBA lectin, which binds to terminal GalNAc residues (Fig. 4a). Similarly, we were able to completely block EtpA interactions with small intestinal Muc2 by using exogenous GalNAc, but not N-acetylglucosamine (GlcNAc) (Fig. 4b). Similarly, the addition of exogenous GalNAc, but not GlcNAc, significantly impaired bacterial adhesion (Fig. 4c) and EtpA-dependent toxin delivery (Fig. 4d), suggesting that EtpA may exert its function as an adhesin by acting as a GalNAc lectin, similar to other previously described microbial adhesins that target intestinal mucins (32, 33).
FIG 4.
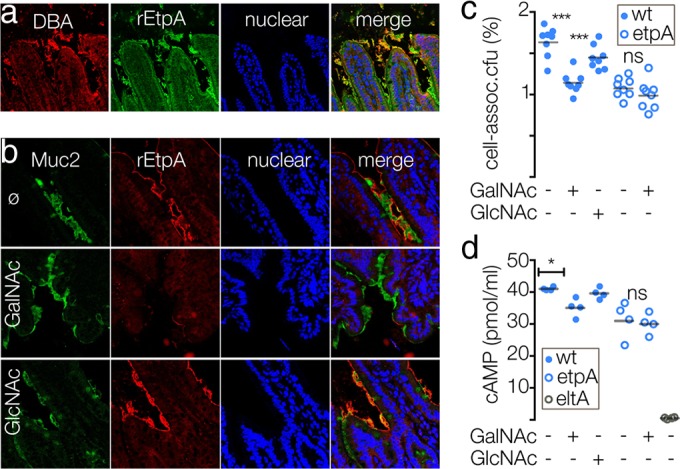
EtpA interactions with glycans containing GalNAc. (a) EtpA localizes to epithelial surface structures that bind the DBA GalNAc lectin. (b) Addition of exogenous GalNAc (middle row) prevents EtpA binding to Muc2 on the epithelial surface. The top row shows an EtpA binding control in the absence of exogenous sugars (ø), and the bottom row shows a GlcNAc control. (c) Addition of exogenous GalNAc inhibits EtpA-mediated bacterial adhesion. (d) GalNAc impairs LT-mediated activation of cAMP to target epithelial cells by ETEC expressing EtpA. Solid blue circles represent the EtpA-expressing wild-type (wt) H10407 strain, open blue circles represent the isogenic etpA deletion mutant, and open gray circles represent the LT-negative (eltA) mutant. Statistical comparisons were performed by two-tailed Mann-Whitney nonparametric testing (*, P < 0.05; ***, P < 0.001; ns, not significant).
LT alters mucin expression, favoring bacterial adhesion.
We have previously shown that LT is required for efficient intestinal colonization (17) and that it enhances bacterial adhesion (34); however, the mechanisms underlying these effects are as yet unknown. CT has previously been shown to promote mucin secretion in the intestinal lumen (35). Therefore, we questioned whether LT, which is a structural and functional homologue of CT, could enhance mucin expression on the surface of the intestinal epithelium and promote bacterial adhesion through EtpA. Interestingly, we found that exposure of enterocytes to exogenous LT promoted the expression of MUC2 mucin (Fig. 5a and b) and promoted the binding of rEtpA to the surface of target epithelial cells (Fig. 5c). Adhesion by wild-type ETEC bacteria, as well as the LT-negative isogenic eltA mutant, was also significantly enhanced following treatment of target host cells with LT; however, these changes were largely abrogated in the etpA mutant strain (Fig. 5d). In addition, pretreatment of cells with siRNA targeting MUC2 expression (Fig. 5e) or with NaIO4 (Fig. 5f) significantly impaired the adhesion of EtpA-producing wild-type bacteria. Collectively, these findings suggest that LT is able to induce changes in target epithelial cells that include enhanced display of surface proteoglycans that promote interactions with ETEC through one or more adhesin molecules, including EtpA.
FIG 5.

LT enhances mucin expression and EtpA-dependent ETEC adhesion. (a) Production of MUC2 is enhanced by LT. Shown in immunoblot images are cell lysates probed with anti-MUC2 antibody following exposure to LT at a final concentration of 0.1 μg/ml for the times indicated. (b) MUC2 gene expression is enhanced by LT (n = 4 replicates, P = 0.03 by Mann-Whitney nonparametric testing). (c) EtpA binding to the surface of intestinal epithelial cells is enhanced following LT exposure. Shown are CLSM images of EtpA binding to intestinal epithelial cells with and without LT treatment. (d) Pretreatment with LT enhances adhesion of ETEC to target epithelial cells. Shown are the percentages of bacteria associated with HT29 cells 1 h after infection of LT-pretreated cells (+) or control untreated cells (−). Dashed horizontal lines represent geometric mean values (n = 8 replicates). ***, P < 0.001 by Mann-Whitney nonparametric testing. (e) LT-induced enhancement of adhesion is dependent on MUC2. (n = 7 replicates per group). (f) LT-mediated increase in adhesion requires intact glycan targets. Shown are LT-associated increases in adhesion to epithelial cells with and without NaIO4. wt, wild type.
EtpA, mucin, and intestinal glycans direct ETEC colonization.
Earlier studies with outbred mice suggested that EtpA plays an important role in establishing intestinal colonization and that immunization with this secreted ETEC antigen impairs the ability of ETEC to infect the small intestine (20, 36, 37). To first establish the role of EtpA in inbred (C57BL/6) mice, we performed competition assays with wild-type ETEC and the etpA isogenic mutant. As anticipated, the etpA mutant colonized the small intestines of these mice poorly relative to the wild-type strain (Fig. 6a). Likewise, in fecal shedding studies, we found that infection with the etpA mutant was eliminated more rapidly than wild-type infection, with only one of nine etpA mutant-infected mice remaining colonized at the end of 1 week, compared to eight of nine mice infected with the wild-type strain (Fig. 6b).
FIG 6.

EtpA and intestinal glycoproteins impact intestinal colonization. (a) Competition experiments demonstrating impaired colonization of etpA mutant strain jf1668 relative to wild-type ETEC strain jf876 in parental C57BL/6 mice. (b) etpA mutants (open blue symbols) exhibit accelerated fecal shedding relative to wild-type (wt, solid gray symbols) H10407 ETEC. Symbols depict geometric mean colonization values, and superscript values adjacent to the symbols indicate the numbers of mice (out of nine in each group) that have completely cleared the infection at each time point. (c) Alteration of intestinal glycoprotein production alters intestinal colonization by ETEC. Shown are parental C57BL/6 mice (solid symbols), Muc2 mutant mice (open blue symbols), and C3GnT mutant mice (open gray symbols) infected with wild-type ETEC jf876. Dashed horizontal lines represent geometric mean values throughout panels a, c, d, and e. *, P < 0.05; **, P < 0.01 by Mann-Whitney nonparametric testing. (d) Colonization of Muc2−/− mice by wild-type ETEC or etpA mutant jf1668. (e) Colonization of C3GnT−/− mice by wild-type ETEC or etpA mutant jf1668. (f) The image at the top left shows a single wild-type ETEC bacterium (identified by anti-O78 antibody, green) bound to the epithelial surface of a small intestinal villus following infection of C57BL/6 mice. The higher-magnification image at the bottom left is an averaged z-stack projection. The graph at the right depicts the number of O78+ bacteria attached to the surface of enterocytes in sections of proximal small intestine from wild-type C57BL/6, C3GnT−/−, and Muc2−/− mice. Blue symbols represent duodenum, and gray symbols represent jejunum. Horizontal bars represent geometric mean numbers of bacteria per villus. *, P < 0.05 by Mann-Whitney two-tailed nonparametric comparisons.
We next examined the impact of intestinal mucin glycoprotein expression on intestinal colonization by ETEC. Interestingly, in Muc2−/− or C3GnT−/− mice, infection with wild-type ETEC resulted in an unanticipated increase in overall colonization of the proximal small intestine relative to that in parental C57BL/6 mice (Fig. 6c). Colonization of Muc2−/− mice by the etpA mutant remained somewhat diminished relative to that by wild-type ETEC (Fig. 6d), potentially reflecting the ability of EtpA to engage multiple intestinal glycoproteins in addition to Muc2. On the basis of the diminished capacity of EtpA to interact with mucosa from mice lacking C3GnT, we anticipated that the impact of the etpA mutation on colonization would be diminished in these mice. Indeed, the etpA mutant colonized C3GnT−/− mice at least as efficiently as the ETEC wild-type strain (Fig. 6e), potentially reflecting the multiplicity of ETEC adhesins that can direct colonization in the absence of EtpA.
These data suggested a complex relationship between intestinal colonization and eukaryotic glycoprotein expression. We speculated that ETEC may engage multiple intestinal glycoproteins with a variety of different adhesins, including EtpA, and that while mucin could serve as a point of attachment for ETEC in the lumen, these glycoproteins could also preclude efficient access of the bacteria to the epithelial surface. Accordingly, we observed a significantly greater number of ETEC bacteria attached to the epithelial surface of the proximal small intestine in Muc2−/− or C3GnT−/− mice than in C57BL/6 mice (Fig. 6f). These findings extend earlier observations made with other E. coli pathovars that preferentially colonize the colon (18) and demonstrate that the relatively thin layer of secreted mucin in the small intestine likewise limits the migration of ETEC to enterocytes, where toxin delivery occurs.
DISCUSSION
Collectively, the findings described here suggest that ETEC has evolved to exploit and manipulate a number of glycoproteins expressed by the intestinal epithelium, including secreted and transmembrane mucins. Interestingly, the data suggest that these bacteria are equipped with at least one protein, the secreted EtpA adhesin, that can engage multiple mucin glycoproteins to facilitate bacterial attachment and ultimately delivery of their enterotoxin payloads. Previously, EtpA has been shown to bind to the ends of ETEC flagella, where it is thought to permit the use of these long peritrichous structures to initiate adhesion (15). The studies described here suggest that EtpA may also accumulate at the surface of ETEC bacteria as they migrate through the mucin layer in the small intestine, potentially permitting these organisms to transiently engage the mucosa en route to the apical surface of enterocytes, where toxin delivery occurs.
ETEC may be equipped to exploit the thin layer of MUC2 (38) coating the small intestine in initial colonization events mediated by EtpA. Further enzymatic degradation of this layer by recently described MUC2-degrading proteases (12, 13) could then permit ETEC direct access to the fine glycocalyx layer on the apical surface of small intestinal enterocytes likely formed by transmembrane mucins, including MUC3 (24). A potential limitation of murine colonization studies is that because of sequence divergence between mucin orthologues, it is not clear whether the EatA serine protease, which degrades human MUC2 (12), is capable of degrading murine Muc2 mucin and an inability to effectively negotiate the barrier formed by this secreted mucin could alter the dynamics of intestinal colonization. While the in vitro data presented here suggest that interaction with secreted glycoproteins, including MUC2, and transmembrane mucins, including MUC3, may ultimately be required for efficient toxin delivery at the epithelial surface, our data may also suggest that engaging these intestinal glycoproteins also permits the elimination of these organisms, as wild-type bacteria more effectively colonized the glycoprotein-deficient mice. Our findings are therefore in keeping with earlier studies that demonstrated that MUC2 is involved in keeping both pathogens and commensal strains from effectively interacting with epithelial cells (18), particularly in the colon, where a thicker layer of mucin serves as an effective barrier to a large burden of organisms (19).
The interaction of ETEC with the intestinal mucosa, similar to that of other enteric pathogens, is likely to be highly dynamic, involving the interaction of a number of adhesins, including the canonical colonization factor antigens, as well as mucin-modifying enzymes (39). Interestingly, a principal ETEC effector, LT, has previously been shown to enhance intestinal colonization (17) and bacterial adhesion (34). While this phenotype could relate to a number of alterations in the host epithelium, the data presented here indicate that ETEC may utilize LT to augment the display of one or more intestinal glycoprotein targets, including MUC2, which the bacteria can then exploit to promote effective bacterial adhesion. LT and the structurally and functionally similar CT increase cAMP in the target intestinal epithelium, leading to protein kinase A-mediated activation of the cystic fibrosis transmembrane regulator (CFTR) and ultimately net efflux of salt and water into the intestinal lumen. Interestingly, both CT- and forskolin-induced increases in cAMP (40) have been shown to stimulate the production and secretion of mucin in the intestine (35, 41), and bicarbonate secretion via the CFTR has been shown to be required for effective mucin secretion (42). Therefore, enterotoxin production of LT by ETEC could theoretically alter the glycoprotein landscape in a number of ways that transiently favor the bacterium.
Intriguingly, recent studies have demonstrated that during the peak of the severe cholera-like illness caused by ETEC, this pathogen emerges in diarrheal stool in a predominant clonal population that coincides with transient displacement of the commensal microbiota (43, 44). However, within days, ETEC bacteria are supplanted as the commensal microbiota re-emerges and the infection resolves. Studies aimed at further defining the complex nature of pathogen-host interactions that permit ETEC to gain a transient foothold in the small intestine could inform novel interdiction strategies and vaccine development approaches.
Supplementary Material
ACKNOWLEDGMENTS
LT was kindly provided by John Clements of Tulane University.
This work was supported by funding from the Department of Veterans Affairs (5I01BX001469-04), grants 2R01AI89894 and 1R01AI126887-01 (J.M.F.), as well as funding from the Canadian Institute of Health Research (CIHR) and Natural Sciences and Engineering Research Council (NSERC) to B.A.V. Funding was also received from the Washington University DDRCC (NIDDK P30 DK052574) from the National Institutes of Health. K.B. was funded by a Vanier Canada Graduate Scholarship, and B.A.V. is the Children with Intestinal and Liver Disorders (CHILD) Foundation Research Chair in Pediatric Gastroenterology.
The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of the NIAID, NIH, NIDDK, or VA.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00692-16.
REFERENCES
- 1.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 2.Qadri F, Saha A, Ahmed T, Al Tarique A, Begum YA, Svennerholm AM. 2007. Disease burden due to enterotoxigenic Escherichia coli in the first 2 years of life in an urban community in Bangladesh. Infect Immun 75:3961–3968. doi: 10.1128/IAI.00459-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Platts-Mills JA, Babji S, Bodhidatta L, Gratz J, Haque R, Havt A, McCormick BJ, McGrath M, Olortegui MP, Samie A, Shakoor S, Mondal D, Lima IF, Hariraju D, Rayamajhi BB, Qureshi S, Kabir F, Yori PP, Mufamadi B, Amour C, Carreon JD, Richard SA, Lang D, Bessong P, Mduma E, Ahmed T, Lima AA, Mason CJ, Zaidi AK, Bhutta ZA, Kosek M, Guerrant RL, Gottlieb M, Miller M, Kang G, Houpt ER, MAL-ED Network Investigators. 2015. Pathogen-specific burdens of community diarrhoea in developing countries: a multisite birth cohort study (MAL-ED). Lancet Glob Health 3:e564–575. doi: 10.1016/S2214-109X(15)00151-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sack RB. 2011. The discovery of cholera-like enterotoxins produced by Escherichia coli causing secretory diarrhoea in humans. Indian J Med Res 133:171–180. [PMC free article] [PubMed] [Google Scholar]
- 5.Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, Steinsland H. 2010. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect 12:89–98. doi: 10.1016/j.micinf.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Svennerholm AM, Lundgren A. 2012. Recent progress toward an enterotoxigenic Escherichia coli vaccine. Expert Rev Vaccines 11:495–507. doi: 10.1586/erv.12.12. [DOI] [PubMed] [Google Scholar]
- 7.Zhang W, Sack DA. 2012. Progress and hurdles in the development of vaccines against enterotoxigenic Escherichia coli in humans. Expert Rev Vaccines 11:677–694. doi: 10.1586/erv.12.37. [DOI] [PubMed] [Google Scholar]
- 8.Kansal R, Rasko DA, Sahl JW, Munson GP, Roy K, Luo Q, Sheikh A, Kuhne KJ, Fleckenstein JM. 2013. Transcriptional modulation of enterotoxigenic Escherichia coli virulence genes in response to epithelial cell interactions. Infect Immun 81:259–270. doi: 10.1128/IAI.00919-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleckenstein JM, Munson GM, Rasko DA. 2013. Enterotoxigenic Escherichia coli: orchestrated host engagement. Gut Microbes 4:392–396. doi: 10.4161/gmic.25861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleckenstein J, Sheikh A, Qadri F. 2014. Novel antigens for enterotoxigenic Escherichia coli vaccines. Expert Rev Vaccines 13:631–639. doi: 10.1586/14760584.2014.905745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheikh A, Luo Q, Roy K, Shabaan S, Kumar P, Qadri F, Fleckenstein JM. 2014. Contribution of the highly conserved EaeH surface protein to enterotoxigenic Escherichia coli pathogenesis. Infect Immun 82:3657–3666. doi: 10.1128/IAI.01890-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar P, Luo Q, Vickers TJ, Sheikh A, Lewis WG, Fleckenstein JM. 2014. EatA, an immunogenic protective antigen of enterotoxigenic Escherichia coli, degrades intestinal mucin. Infect Immun 82:500–508. doi: 10.1128/IAI.01078-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo Q, Kumar P, Vickers TJ, Sheikh A, Lewis WG, Rasko DA, Sistrunk J, Fleckenstein JM. 2014. Enterotoxigenic Escherichia coli secretes a highly conserved mucin-degrading metalloprotease to effectively engage intestinal epithelial cells. Infect Immun 82:509–521. doi: 10.1128/IAI.01106-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleckenstein JM, Roy K, Fischer JF, Burkitt M. 2006. Identification of a two-partner secretion locus of enterotoxigenic Escherichia coli. Infect Immun 74:2245–2258. doi: 10.1128/IAI.74.4.2245-2258.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roy K, Hilliard GM, Hamilton DJ, Luo J, Ostmann MM, Fleckenstein JM. 2009. Enterotoxigenic Escherichia coli EtpA mediates adhesion between flagella and host cells. Nature 457:594–598. doi: 10.1038/nature07568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleckenstein JM, Roy K. 2009. Purification of recombinant high molecular weight two-partner secretion proteins from Escherichia coli. Nat Protoc 4:1083–1092. doi: 10.1038/nprot.2009.87. [DOI] [PubMed] [Google Scholar]
- 17.Allen KP, Randolph MM, Fleckenstein JM. 2006. Importance of heat-labile enterotoxin in colonization of the adult mouse small intestine by human enterotoxigenic Escherichia coli strains. Infect Immun 74:869–875. doi: 10.1128/IAI.74.2.869-875.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. 2010. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog 6:e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. 2008. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A 105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy K, Hamilton D, Allen KP, Randolph MP, Fleckenstein JM. 2008. The EtpA exoprotein of enterotoxigenic Escherichia coli promotes intestinal colonization and is a protective antigen in an experimental model of murine infection. Infect Immun 76:2106–2112. doi: 10.1128/IAI.01304-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knutton S, Lloyd DR, Candy DC, McNeish AS. 1984. In vitro adhesion of enterotoxigenic Escherichia coli to human intestinal epithelial cells from mucosal biopsies. Infect Immun 44:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knutton S, McConnell M, Rowe B, McNeish A. 1989. Adhesion and ultrastructural properties of human enterotoxigenic Escherichia coli producing colonization factor antigens III and IV. Infect Immun 57:3364–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knutton S, Lloyd DR, Candy DC, McNeish AS. 1985. Adhesion of enterotoxigenic Escherichia coli to human small intestinal enterocytes. Infect Immun 48:824–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansson ME, Sjovall H, Hansson GC. 2013. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol 10:352–361. doi: 10.1038/nrgastro.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiss AA, Babyatsky MW, Ogata S, Chen A, Itzkowitz SH. 1996. Expression of MUC2 and MUC3 mRNA in human normal, malignant, and inflammatory intestinal tissues. J Histochem Cytochem 44:1161–1166. doi: 10.1177/44.10.8813081. [DOI] [PubMed] [Google Scholar]
- 26.Van Klinken BJ, Tytgat KM, Buller HA, Einerhand AW, Dekker J. 1995. Biosynthesis of intestinal mucins: MUC1, MUC2, MUC3 and more. Biochem Soc Trans 23:814–818. doi: 10.1042/bst0230814. [DOI] [PubMed] [Google Scholar]
- 27.Peterson MD, Mooseker MS. 1992. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci 102(Pt 3):581–600. [DOI] [PubMed] [Google Scholar]
- 28.Bergstrom KS, Xia L. 2013. Mucin-type O-glycans and their roles in intestinal homeostasis. Glycobiology 23:1026–1037. doi: 10.1093/glycob/cwt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsson JM, Karlsson H, Sjovall H, Hansson GC. 2009. A complex, but uniform O-glycosylation of the human MUC2 mucin from colonic biopsies analyzed by nanoLC/MSn. Glycobiology 19:756–766. doi: 10.1093/glycob/cwp048. [DOI] [PubMed] [Google Scholar]
- 30.Karlsson NG, Herrmann A, Karlsson H, Johansson ME, Carlstedt I, Hansson GC. 1997. The glycosylation of rat intestinal Muc2 mucin varies between rat strains and the small and large intestine. A study of O-linked oligosaccharides by a mass spectrometric approach. J Biol Chem 272:27025–27034. [DOI] [PubMed] [Google Scholar]
- 31.Rausch P, Steck N, Suwandi A, Seidel JA, Kunzel S, Bhullar K, Basic M, Bleich A, Johnsen JM, Vallance BA, Baines JF, Grassl GA. 2015. Expression of the blood-group-related gene B4galnt2 alters susceptibility to Salmonella infection. PLoS Pathog 11:e1005008. doi: 10.1371/journal.ppat.1005008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petri WA Jr, Chapman MD, Snodgrass T, Mann BJ, Broman J, Ravdin JI. 1989. Subunit structure of the galactose and N-acetyl-d-galactosamine-inhibitable adherence lectin of Entamoeba histolytica. J Biol Chem 264:3007–3012. [PubMed] [Google Scholar]
- 33.Houpt E, Barroso L, Lockhart L, Wright R, Cramer C, Lyerly D, Petri WA. 2004. Prevention of intestinal amebiasis by vaccination with the Entamoeba histolytica Gal/GalNac lectin. Vaccine 22:611–617. doi: 10.1016/j.vaccine.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 34.Johnson AM, Kaushik RS, Francis DH, Fleckenstein JM, Hardwidge PR. 2009. Heat-labile enterotoxin promotes Escherichia coli adherence to intestinal epithelial cells. J Bacteriol 191:178–186. doi: 10.1128/JB.00822-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forstner JF, Roomi NW, Fahim RE, Forstner GG. 1981. Cholera toxin stimulates secretion of immunoreactive intestinal mucin. Am J Physiol 240:G10–16. [DOI] [PubMed] [Google Scholar]
- 36.Roy K, Hamilton D, Ostmann MM, Fleckenstein JM. 2009. Vaccination with EtpA glycoprotein or flagellin protects against colonization with enterotoxigenic Escherichia coli in a murine model. Vaccine 27:4601–4608. doi: 10.1016/j.vaccine.2009.05.076. [DOI] [PubMed] [Google Scholar]
- 37.Luo Q, Vickers TJ, Fleckenstein JM. 2016. Immunogenicity and protective efficacy against ETEC colonization following intradermal, sublingual or oral vaccination with EtpA adhesin. Clin Vaccine Immunol 23:628–637. doi: 10.1128/CVI.00248-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ermund A, Schutte A, Johansson ME, Gustafsson JK, Hansson GC. 2013. Studies of mucus in mouse stomach, small intestine, and colon. I. Gastrointestinal mucus layers have different properties depending on location as well as over the Peyer's patches. Am J Physiol Gastrointest Liver Physiol 305:G341–G347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGuckin MA, Linden SK, Sutton P, Florin TH. 2011. Mucin dynamics and enteric pathogens. Nat Rev Microbiol 9:265–278. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 40.Bradbury NA. 2000. Protein kinase-A-mediated secretion of mucin from human colonic epithelial cells. J Cell Physiol 185:408–415. doi: 10.1002/1097-4652(200012)185:3<408::AID-JCP11>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 41.Epple HJ, Kreusel KM, Hanski C, Schulzke JD, Riecken EO, Fromm M. 1997. Differential stimulation of intestinal mucin secretion by cholera toxin and carbachol. Pflugers Arch 433:638–647. [DOI] [PubMed] [Google Scholar]
- 42.Gustafsson JK, Ermund A, Ambort D, Johansson ME, Nilsson HE, Thorell K, Hebert H, Sjovall H, Hansson GC. 2012. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J Exp Med 209:1263–1272. doi: 10.1084/jem.20120562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahl JW, Sistrunk JR, Fraser CM, Hine E, Baby N, Begum Y, Luo Q, Sheikh A, Qadri F, Fleckenstein JM, Rasko DA. 2015. Examination of the enterotoxigenic Escherichia coli population structure during human infection. mBio 6:e00501-15. doi: 10.1128/mBio.00501-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.David LA, Weil A, Ryan ET, Calderwood SB, Harris JB, Chowdhury F, Begum Y, Qadri F, LaRocque RC, Turnbaugh PJ. 2015. Gut microbial succession follows acute secretory diarrhea in humans. mBio 6:e00381-15. doi: 10.1128/mBio.00381-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans DG, Silver RP, Evans DJ Jr, Chase DG, Gorbach SL. 1975. Plasmid-controlled colonization factor associated with virulence in Escherichia coli enterotoxigenic for humans. Infect Immun 12:656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans DJ Jr, Evans DG. 1973. Three characteristics associated with enterotoxigenic Escherichia coli isolated from man. Infect Immun 8:322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dorsey FC, Fischer JF, Fleckenstein JM. 2006. Directed delivery of heat-labile enterotoxin by enterotoxigenic Escherichia coli. Cell Microbiol 8:1516–1527. doi: 10.1111/j.1462-5822.2006.00736.x. [DOI] [PubMed] [Google Scholar]
- 48.Pratt WS, Crawley S, Hicks J, Ho J, Nash M, Kim YS, Gum JR, Swallow DM. 2000. Multiple transcripts of MUC3: evidence for two genes, MUC3A and MUC3B. Biochem Biophys Res Commun 275:916–923. doi: 10.1006/bbrc.2000.3406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.