

Abstract
Our recent study demonstrated that virulent Coxiella burnetii Nine Mile phase I (NMI) is capable of infecting and replicating within peritoneal B1a cells and that B1a cells play an important role in host defense against C. burnetii infection in mice. However, it remains unknown if avirulent Nine Mile phase II (NMII) can infect and replicate in B1a cells and whether NMI and NMII can differentially interact with B1a cells. In this study, we examined if NMI and NMII can differentially modulate host cell apoptotic signaling in B1a cells. The results showed that NMII induced dose-dependent cell death in murine peritoneal B1a cells but NMI did not, suggesting that NMI and NMII may differentially activate host cell apoptotic signaling in B1a cells. Western blotting indicated that NMII-induced B1a cell death was not dependent on either caspase-3 or PARP-1 cleavage, but cleavage of caspase-1 was detected in NMII-infected B1a cells. In addition, inhibition or deficiency of caspase-1 activity blocked NMII-induced B1a cell death. These results suggest that NMII induces a caspase-1-dependent pyroptosis in murine peritoneal B1a cells. We also found that heat-killed NMII and type 4 secretion system (T4SS) mutant NMII were unable to induce B1a cell death and that NMII infection did not induce cell death in peritoneal B1a cells from Toll-like receptor 2 (TLR-2)- or NLRP3 inflammasome-deficient mice. These data suggest that NMII-induced caspase-1-dependent pyroptosis may require its T4SS and activation of the TLR-2 and NLRP3 signaling pathways.
INTRODUCTION
Coxiella burnetii is an obligate intracellular Gram-negative bacterial pathogen that causes acute and chronic Q fever in humans. Acute Q fever manifests as a flu-like febrile illness, atypical pneumonia, or hepatitis that is usually self-limiting or effectively treated by antibiotics (1), while chronic Q fever is a severe, sometimes fatal disease (2–4). A recent outbreak in the Netherlands from 2007 to 2010 resulted in more than 3,500 reported clinical Q fever cases (5), which highlights that this worldwide zoonotic pathogen remains a significant threat to public health. Antibiotic treatment for acute Q fever is most effective when it is initiated within the first 3 days of illness, but accurate early diagnosis of Q fever is difficult and often overlooked due to nondescript flu-like symptoms. However, there is no alternative strategy for treatment of more advanced infections in cases where the disease is neglected or incorrectly treated due to misdiagnosis. Additionally, chronic Q fever is much more difficult to treat effectively and often requires treatment with multiple antibiotics for several years (6). Therefore, it is necessary to discover novel drugs and alternative strategies for controlling C. burnetii infections.
The C. burnetii Nine Mile strain undergoes a lipopolysaccharide (LPS) phase variation in which its virulent smooth LPS phase I (NMI) converts to an avirulent rough LPS phase II (NMII) upon serial passage in eggs and tissue cultures (7). NMI is able to replicate in wild-type (WT) animals and cause disease, while NMII can be rapidly cleared in animals and does not cause disease (8). It has been shown that both NMI and NMII can infect several cell types and can slowly replicate in a Coxiella-containing vacuole (CCV) in a low-pH (∼4.5) environment within cells (9–12). Thus, the mechanisms for C. burnetii intracellular survival and the establishment of a persistent infection may be related to its ability to modulate host responses and subvert the microbicidal functions of phagocytes.
Cell death in eukaryotic cells can happen in a programmed fashion in various distinct forms with either a noninflammatory or inflammatory nature, and this dictates important physiological outcomes. Apoptosis is the noninflammatory form of cell death first well studied in eukaryotic cells. It is characterized by cleavage of caspases 3, 6, and 8, DNA fragmentation, chromatin condensation, and packaging of cellular contents into small bodies with intact plasma membranes that are released and phagocytosed (13). Other cell death programs include autophagy, oncosis, and caspase-1-dependent programmed cell death. Caspase-1-dependent programmed cell death is also known as pyroptosis. This is a more recently identified form of programmed cell death induced by a variety of microbes, including Shigella, Salmonella, Francisella, Legionella, and Listeria (14–17). Pyroptosis is characterized by caspase-1 cleavage, DNA fragmentation, cellular swelling, and rupture of the plasma membrane and the release of proinflammatory contents and cytokines, such as interleukin-1β (IL-1β), IL-18, and tumor necrosis factor alpha (TNF-α) (13). It has been shown that pyroptosis is critical for the clearance of some intracellular pathogens, such as Legionella pneumophila and Burkholderia thailandensis, which is independent of the production of IL-1β and IL-18 (16). However, some intracellular pathogens have evolved mechanisms to block pyroptosis induction. Listeria monocytogenes avoids initiating pyroptosis by suppressing flagellin expression at the host temperature (18). Thus, pyroptosis is a critical form of cell death that is involved in inflammation, development of the immune response, and elimination of bacterial pathogens (17).
Previous studies have shown that C. burnetii is capable of inducing or inhibiting host cell apoptosis, depending on its interaction with host cells under different conditions. Voth and Heinzen showed that both NMI and NMII bacteria were able to partially prevent exogenously induced apoptosis in differentiated THP-1 cells as well as in primary monkey alveolar macrophages (19, 20). Similarly, Lührmann and Roy demonstrated that NMII inhibited exogenously induced apoptosis in Chinese hamster ovary and HeLa cells at a late stage of infection (21). These observations suggest that C. burnetii-infected cells possess an antiapoptotic ability in the presence of exogenously applied apoptotic stimuli that may be important for C. burnetii to establish a persistent infection in vitro. In contrast, one previous study showed that NMII induces apoptosis in undifferentiated THP-1 cells during an early stage of infection through a caspase-3-independent pathway (22). These data suggest that the sensitivity of host cells to apoptosis in response to C. burnetii stimuli may be varied on the basis of the cell type, the state of cellular maturation, and differentially expressed cell surface receptors.
It has been shown that B cells can regulate immune functions by producing cytokines, acting as antigen-presenting cells (APCs), and potentially, killing phagocytosed bacteria and other immune cells (23–30). Naive B cells can be classified into three subsets: B1 cells, follicular B cells, and marginal zone (MZ) B cells (31). B1 cells, after origination, are destined to the peritoneal and pleural cavities (32). B1 cells can be further classified into B1a and B1b cells on the basis of the expression of CD5, with B1a cells being CD5+ and B1b cells being CD5−. B1a cells contribute to innate immunity-like immune responses and have demonstrated roles in the immune response to a variety of pathogens (33–36). B1b cells contribute to adaptive immunity. Our recent study demonstrated that peritoneal B1a cells were able to phagocytose virulent NMI and B1a cells play an important role in regulating the immune response and controlling C. burnetii replication in vivo (30). Further understanding of the mechanisms of cellular interaction between C. burnetii and phagocytic B1a cells may provide information useful for identifying novel therapeutic targets against C. burnetii infection.
In this study, we examined if virulent NMI and avirulent NMII can differentially activate host cell apoptotic signaling in B1a cells in vitro. The results suggest that NMII induces a caspase-1-dependent pyroptosis through activation of the Toll-like receptor 2 (TLR-2) and NLRP3 signaling pathways.
MATERIALS AND METHODS
Bacteria.
C. burnetii NMI clone 7 (RSA493) and NMII clone 4 (RSA439) were propagated in L929 cells and purified by density gradient centrifugation as described previously (37). The type 4 secretion system (T4SS)-deficient NMII (dotA mutant) (a kind gift from Paul Beare) was propagated in acidified citrate cysteine medium 2 (ACCM-2) (38). NMI was handled under biosafety level 3 (BSL3) conditions at the University of Missouri Laboratory for Infectious Disease Research. All infections were done at a multiplicity of infection (MOI) of 100 unless otherwise stated. For heat killing, NMII was treated in a boiling water bath for 10 min.
Animals.
Specific-pathogen-free, 6- to 8-week-old female BALB/c, C57BL/6, TLR-2−/−, and NLRP3−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Caspase-1−/− caspase-11−/− mice were kindly donated by Jerod Skyberg (University of Missouri, Columbia, MO). Animals were housed in microisolator cages at a conventional animal facility at the University of Missouri. All research involving animals was conducted in accordance with animal care and use guidelines, and all animal use protocols were approved by the Animal Care and Use Committee at the University of Missouri.
Isolation of peritoneal cells.
Peritoneal cells were isolated from 6- to 8-week-old female BALB/c, C57BL6, Caspase-1−/− caspase-11−/−, NLRP3−/−, and TLR-2−/− mice using peritoneal lavage as described previously (39). Briefly, mice were intraperitoneally (i.p.) injected with 5 to 10 ml of RPMI 1640 supplemented with 12 mM HEPES and 10% fetal bovine serum (FBS), and the fluid was then removed using a fresh 25-gauge needle (BD Biosciences, Franklin Lakes, NJ). Purification of total B cells was performed using magnetically activated cell sorting (MACS) CD19 microbeads (Miltenyi Biotec Inc., CA) and LS columns according to manufacturer specifications. B1a cells were purified using a MACS B1a cell isolation kit with LS and MS columns (Miltenyi Biotec Inc., CA) according to manufacturer specifications. Purities were confirmed using flow cytometry with a Beckman Coulter CyAN ADP analyzer. Data were analyzed using Summit (v5.3) software. The purity of total B cells following CD19 microbead treatment was >95%. The purity of purified B1a cells following separation was >90%.
Indirect immunofluorescence assay (IFA).
To stain both intracellular and extracellular markers in B cells, purified peritoneal B cells were plated at 5 × 105 cells/well and then allowed to adhere to poly-d-lysine-coated coverslips (Neuvitro) for 15 min at room temperature prior to infection. After infection with NMII at an MOI of 100 for 24 h, the cells were washed twice with RPMI 1640 containing 2.5% FBS to remove extracellular bacteria. At day 1 and day 3 postinfection, cells were blocked with mouse BD Fc block and stained extracellularly with CD19–R-phycoerythrin (eBiosciences) or CD5-Alexa Fluor 647 (BioLegend, San Diego, CA). Following staining, cells were fixed and permeabilized using a BD Cytofix/Cytoperm Plus kit (BD biosciences) according to manufacturer specifications. The cells were then stained intracellularly with rabbit anti-Coxiella polyclonal antibodies followed by incubation with goat anti-rabbit IgG (Southern Biotech). Nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI) for 5 min at 4°C. Coverslips were mounted in ProLong gold antifade reagent (Invitrogen) and allowed to cure overnight at room temperature. Microscopic analysis was performed using an Olympus IX70 inverted microscope.
Confocal microscopy.
Purified peritoneal B cells were plated at 5 × 105 cells and infected with NMII at an MOI of 100. At 1 and 3 days postinfection, cells were fixed with 2% paraformaldehyde and then permeabilized with −20°C methanol. The cells were blocked with 5% normal goat serum and then stained with rabbit anti-Coxiella polyclonal antibodies, followed by staining with CD107a-phycoerythrin (PE) (for detection of LAMP-1), goat anti-rabbit IgG-fluorescein isothiocyanate (FITC), and/or CD282-PE (for detection of TLR-2) (BioLegend, San Diego, CA). Coverslips were mounted as described above. Microscopic analysis was performed using a Leica TCP SP8 MP inverted spectral confocal microscope.
Flow cytometry.
After infection with NMII at an MOI of 100 for 1 or 3 days, 5 × 105 whole peritoneal cells were blocked with mouse BD Fc Block (BD Biosciences) and then stained with CD19 (eBioscience), CD5 (BD Biosciences), and CD11b (BD Biosciences) for 40 min at 4°C in fluorescence-activated cell sorting buffer (1× phosphate-buffered saline [PBS] supplemented with 0.5% bovine serum albumin, 2 mM EDTA, and 0.1% sodium azide). Coxiella was visualized using rabbit anti-Coxiella polyclonal antibodies and goat anti-rabbit IgG-FITC. Cells were then fixed using 2% paraformaldehyde in PBS. Flow cytometry was performed using a Beckman Coulter CyAN ADP analyzer, and the data were analyzed using Summit (v5.3) software.
Real-time PCR.
To measure NMII replication within cells, 5 × 105 purified B cells were infected with NMII at an MOI of 100, and excess bacteria were washed away at 1 day postinfection with two washes of RPMI 1640–5% FBS. The cells were scraped and lysed with 200 μl lysis buffer (1 M Tris, 0.5 M EDTA, 7 mg/ml glucose, 28 mg/ml lysozyme) and 10 μl proteinase K (20 mg/ml) and incubated overnight at 60°C. DNA was extracted using a High Pure PCR template preparation kit (Roche) according to manufacturer specifications. The com1 gene copy number was quantified using a standard curve with SYBR green (Applied Biosciences) on an Applied Biosystems 7300 real-time PCR system. Recombinant plasmid DNA (the com1 gene ligated into the pET23a vector) was used as a standard to quantify com1 gene copy numbers.
ELISA.
Supernatants from NMII-infected (MOI, 100) and uninfected control B cells were analyzed for tumor necrosis factor alpha (TNF-α) or interleukin-1β (IL-1β) production using a mouse TNF-α, IL-1β, or IL-18 enzyme-linked immunosorbent assay (ELISA; Ready-SET-Go! kit; eBioscience) according to manufacturer specifications. The absorbance was measured at 490 nm using a Molecular Devices SpectraMax plus plate reader and SoftMax software.
Western blotting.
B1a cells were isolated as described above. B1a cells (1 × 106) were allowed to attach in a 24-well plate for 1 h. Then, the cells were infected with NMII (MOI, 100) or left uninfected and incubated at 37°C in an incubator with 5% CO2 for 1 or 3 days postinfection. Proteins were extracted from the cells using the M-PER mammalian protein extraction reagent (Thermo Fisher Scientific, Grand Island, NY) mixed with Halt protease inhibitor cocktail according to manufacturer specifications. The protein concentration was measured by using a Pierce bicinchoninic acid protein assay kit (Thermo Scientific, Rockford, IL). Equal amounts of proteins (20 to 30 μg per sample) were separated on 8%, 12%, and 15% acrylamide gels and transferred to a nitrocellulose membrane. The membranes were blocked with 5% nonfat dry milk prepared in Tris-buffered saline–Tween 20 (TBST; 200 mM Tris [pH 7.5], 1.38 M NaCl, 0.1% Tween 20) and incubated overnight at 4°C with primary antibodies. The following antibodies were added in 5% nonfat dry milk made in TBST: PARP-1 antibody (Cell Signaling Technologies), which binds to the uncleaved form (116 kDa) and cleaved form (89 kDa) of PARP-1; caspase-3 antibody, which recognizes the 35-kDa proform and cleaved proteins of 19 kDa and 17 kDa of caspase-3 (Cell Signaling Technologies); caspase-1 antibody, which recognizes the p20 active form and the proform of 42 kDa of caspase-1 (eBioscience); and β-actin (Cell Signaling Technologies). Then, the membranes were washed and incubated with the corresponding secondary antibodies coupled with horseradish peroxidase for 2 h at room temperature or overnight at 4°C. Bands were visualized using an enhanced chemiluminescence Western blot detection kit (Thermo Scientific). The intensities of the protein bands were captured on autoradiography film. A representative blot from at least two experiments is shown in the figures.
MTS assay.
Cell death was measured indirectly by using a CellTiter 96 Aqueous One Solution cell proliferation assay kit (Promega, Madison, WI). This solution contains a tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)] and an electron-coupling reagent (phenazine ethosulfate [PES]). The MTS compound was bioreduced by the NADPH or NADH produced by dehydrogenase enzymes in metabolically active living cells to a soluble formazan product, which was quantified by taking the absorbance at 490 nm. The absorbance is directly proportional to the number of living cells in the culture. Purified B1a cells (1.5 × 105), isolated as described above from BALB/c, C57BL6, TLR-2−/−, caspase-1−/− caspase-11−/−, and NLRP3−/− mice, were allowed to attach for 1 h and infected with either NMI or NMII at MOIs of 5, 50, and 100 for 3 days or at an MOI of 100 for 1 day or 3 days in 96-well plates. The plates were incubated at 37°C in 5% CO2. At the end of each time point, the floating cells were removed and adherent cells were washed 2 times with 2% FBS–RPMI 1640. MTS solution in 100 μl of the same medium was added, and the cells were incubated at 37°C for 1 to 4 h for color development. The absorbance at 490 nm was measured using a Molecular Devices SpectraMax Plus plate reader and SoftMax software, and percent cell death was calculated. All these experiments were repeated three times, and the data are presented as percent cell death, calculated as follows: [(average OD for infected cells − average OD for control cells)/average OD for control cells] × 100, where OD represents optical density. The background absorbance at an OD of 630 nm was measured, and the value was subtracted from the sample ODs.
TNF-α neutralization.
To neutralize the TNF-α secreted by infected culture cells, neutralizing antibody (BioLegend, San Diego, CA) was used at 0.5 μg/ml. Briefly 1.5 × 105 B1a cells were allowed to attach for 1 h and pretreated for 1 h with neutralizing antibody in triplicate wells in a 96-well plate. The cells were then infected with NMII at an MOI of 100 and incubated at 37°C for 3 days. At the end of the day 3 time point, the MTS assay was performed as described above.
Caspase-1 inhibition.
To analyze the contribution of caspase-1 to NMII-induced cell death, we used the caspase-1 inhibitor acetyl-Tyr-Val-AlaAsp-chloromethylketone (Ac-YVAD-cmk), which is an irreversible inhibitor of caspase-1 (Sigma). B cells (1.5 × 105) were allowed to attach for 1 h in a 96-well culture plate, and the cells were pretreated for 1 h with 200 μM inhibitor or its vehicle control solution, dimethyl sulfoxide, in triplicate wells. The cells were infected with NMII at an MOI of 100 and incubated at 37°C for 3 days. At the end of the day 3 time point, the MTS assay was performed as described above.
Double staining of intracellular C. burnetii and apoptotic cell DNA.
Purified peritoneal B cells were allowed to adhere to coverslips for 15 min at room temperature and then infected with NMII at an MOI of 100. For inhibition of replication using rifampin, B1 cells were allowed to attach for 1 h and treated for 1 h with rifampin (Sigma) at 10 μg/ml prior to NMII infection. At 1 day and 3 days postinfection, the cells were fixed and permeabilized as described above. Intracellular C. burnetii staining was performed in the same manner described above. Staining by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) was performed using the in situ death detection kit TMR red (Roche) according to manufacturer specifications. Briefly, following washing, the TUNEL reaction mix was added to uninfected and NMII-infected B cells, and the cells were incubated for 1 h at 37°C in the dark. The cells were then washed with PBS and examined using a fluorescence microscope.
Statistical analysis.
Statistical analysis was performed using Prism (v5.0) software (GraphPad Software Inc., San Diego, CA). All experiments are performed at least three times. Results were compared using a two-sample Student's t test. Differences were considered significant if P was ≤0.05.
RESULTS
C. burnetii NMII can infect and replicate in peritoneal B cells.
Our recent work showed that peritoneal B cells were able to phagocytose virulent C. burnetii NMI bacteria (30). In this study, IFA was used to determine whether peritoneal B cells can take up avirulent NMII bacteria. Purified peritoneal B cells were infected with NMII at an MOI of 100 for 1 and 3 days and stained with anti-C. burnetii and anti-CD19 antibodies. As shown in Fig. 1A, colocalization between the B cell marker CD19 and C. burnetii was observed in NMII-infected peritoneal B cells at 3 days postinfection. To further determine whether NMII bacteria can proliferate within Coxiella-containing vacuoles (CCVs) of infected B cells, NMII-infected peritoneal B cells were intracellularly stained with anti-C. burnetii and anti-LAMP-1 antibodies and observed by confocal microscopy. As shown in Fig. 1B, colocalization between intracellular C. burnetii and LAMP-1 was observed in NMII-infected B cells at 3 days postinfection. In addition, flow cytometry was used to analyze the NMII infection rate in B cells at 1 and 3 days postinfection. Figure 1C shows flow cytometry scatter plots of uninfected or NMII-infected B cells double stained with anti-C. burnetii and anti-CD19 antibodies at 3 days postinfection. B cells stained with both anti-C. burnetii and anti-CD19 antibodies were considered infected. Compared to the infection rate at 1 day postinfection, the NMII infection rate was significantly increased at 3 days postinfection (Fig. 1D). Furthermore, to determine whether NMII bacteria can replicate within B cells, real-time quantitative PCR was used to measure the number of copies of the C. burnetii genome in NMII-infected peritoneal B cells at 1 and 3 days postinfection. As shown in Fig. 1E, the C. burnetii genome copy number was significantly increased in NMII-infected B cells at 3 days postinfection compared to that in NMII-infected B cells at 1 day postinfection, suggesting that C. burnetii was able to replicate within peritoneal B cells. Collectively, these results demonstrate that avirulent NMII bacteria are able to infect and replicate in CCVs inside peritoneal B cells.
FIG 1.

Primary peritoneal B cells take up NMII. Primary peritoneal B cells were harvested by lavage and purified using CD19 MACS beads. (A) Following infection with NMII, cells were stained with antibodies against CD19 and NMII and DNA was visualized with DAPI. (B) Following infection with NMII, cells were stained with antibodies against NMII and LAMP-1 and visualized by confocal microscopy. (C) Flow cytometry plots of NMII-infected B cells at day 3 postinfection. The value at top right indicates the percentage of stained cells. (D) Infection rates observed by IFA at 1 and 3 days postinfection. Two hundred cells per slide were counted. ***, P < 0.001. (E) Measurement of the com1 gene copy number in infected purified B cells by real-time PCR at day 1 and day 3 postinfection. *, P < 0.05. These data indicate that primary peritoneal B cells are infected by NMII and that the bacteria are taken up into LAMP-1-positive vacuoles. DPI, days postinfection.
NMII bacteria can infect both the B1a and B1b subsets of B cells, while a reduction of B1a cells was detected among NMII-infected peritoneal B cells.
To determine whether avirulent NMII bacteria can infect peritoneal subset B cells, purified peritoneal B cells were infected with NMII at an MOI of 100 and stained with anti-C. burnetii IgG, CD19, CD11b, and CD5 at 1 and 3 days postinfection. Flow cytometry was used to analyze the NMII infection rate for the B1a and B1b subsets of B cells at 1 and 3 days postinfection. Figure 2A shows flow cytometry scatter plots of uninfected or NMII-infected peritoneal B cells (gated on live, CD19+ CD11b+ B1 cells) double stained with anti-C. burnetii and anti-CD5 antibodies at 3 days postinfection. Since CD5+ and CD5− B cells are classified into the B1a and B1b subsets, B cells double stained with anti-C. burnetii and anti-CD5 antibodies were counted as NMII-infected B1a cells, while B cells stained with anti-C. burnetii antibody but not with anti-CD5 antibody were counted as NMII-infected B1b cells. As shown in Fig. 2B, both NMII-infected B1a and B1b cells were detected among NMII-infected peritoneal B cells at 1 and 3 days postinfection. However, although the infection rate was similar between B1a and B1b cells at 1 and 3 days postinfection, the infection rate for B1a and B1b cells at 3 days postinfection was significantly higher than their infection rate at 1 day postinfection. These results suggest that NMII bacteria are able to infect and proliferate in both B1a and B1b cells at similar rates. Figure 2C shows a comparison of the total number of B1a and B1b cells among uninfected control B cells and NMII-infected B cells at 1 and 3 days postinfection. Interestingly, although the total numbers of B1a cells among uninfected control B cells and NMII-infected B cells were similar at 1 day postinfection, the total number of B1a cells among NMII-infected B cells was significantly less than the total number of B1a cells among uninfected control B cells at 3 days postinfection. In contrast, the total number of B1b cells was not significantly different between uninfected control and NMII-infected B cells at both 1 and 3 days postinfection. These results suggest that NMII infection may induce the death of the B1a subset of B cells.
FIG 2.

NMII-infected B cells were visualized and examined using flow cytometry. Cells were purified using CD19 beads and infected with NMII. Cells were then stained with antibodies against CD19 and NMII. (A) Infected peritoneal B cells were visible using flow cytometry at day 3 postinfection. Gating was on live cells. Values indicate the percentages of stained cells. (B) The quantity of infected B cell subsets was measured at day 1 and day 3 postinfection. ***, P < 0.001. (C) The absolute number of B cell subsets at day 1 and day 3 postinfection. *, P < 0.05. These data indicate that NMII infection rates peak at day 3 postinfection.
Apoptotic DNA was detected in NMII-infected peritoneal B cells.
To determine if NMII infection can induce apoptosis in peritoneal B cells, we used staining by TUNEL to detect fragmented cell DNA in NMII-infected peritoneal B cells at different times postinfection. As shown in Fig. 3A, fragmented cell DNA was observed in NMII-infected B cells at 3 days postinfection. A significantly higher number of TUNEL-positive cells was detected among NMII-infected B cells than uninfected cells (Fig. 3B). These observations suggest that NMII infection may induce an apoptotic cell death in peritoneal B cells.
FIG 3.

NMII-infected B cells undergo DNA fragmentation. Purified peritoneal B cells were allowed to adhere to poly-d-lysine-coated coverslips and infected at an MOI of 100 with NMII, treated with PBS (Uninfected), or treated with staurosporine (positive control). At 3 days postinfection, uninfected and infected cells were fixed, permeabilized, and stained with antibodies against NMII and with the TUNEL reaction kit. (A) DNA fragmentation is visible within NMII-infected and staurosporine-treated cells at 3 days postinfection. (B) The number of cells with fragmented DNA was counted. Two hundred cells per coverslip on three separate slips were counted for each time point. ***, P < 0.001. These data indicate that NMII infection induces DNA fragmentation in peritoneal B cells.
Avirulent NMII bacteria induce B1a subset B cell death in a dose-dependent manner, but virulent NMI bacteria do not.
When culturing purified peritoneal B cells, we noted that there were two different cell populations present in the cell culture plate; one cell population strongly adhered to the cell culture plate, while another cell population floated in the supernatant. To determine which cell population undergoes cell death, the MTS assay was used to measure the percent cell death for adhered and floating cells infected with different doses of NMII bacteria. As shown in Fig. 4A, cell death was detected only for adhered cells that were infected with NMII bacteria at an MOI of 50 or 100, and the percentage of dead cells among adhered cells infected with NMII at an MOI of 100 was significantly higher than that among adhered cells infected with NMII at an MOI of 50 at 3 days postinfection. These results indicate that NMII infection can induce adhered B cell death in a dose-dependent manner. To identify which subsets of B cells were present among adhered cells, adhered cells were scraped after 24 h of infection; stained with anti-CD19, anti-CD11b, and anti-CD5 antibodies; and analyzed by flow cytometry. As shown in Fig. 4B, approximately 90% of the adherent B cells were stained with anti-CD19, anti-CD11b, and anti-CD5 antibodies. This result indicates that the major population of adhered B cells is B1a subset B cells. This finding is consistent with data from our flow cytometry analysis that showed a reduction in the number of B1a cells among NMII-infected peritoneal B cells (Fig. 2C). Staining by TUNEL was also used to confirm this observation. Purified B cells were infected with NMII for 3 days and stained by use of the TUNEL reaction and with antibodies against NMII and CD5, a B1a cell marker. Figure 5 demonstrates that NMII-infected CD5+ B cells were TUNEL positive (indicated by white arrows). These results suggest that NMII infection can induce dose-dependent B1a cell death. However, it was also noted that several cells not stained with anti-C. burnetii antibodies were TUNEL positive, while several cells stained with anti-C. burnetii antibodies were TUNEL negative. In addition, the MTS assay was used to investigate if virulent NMI and avirulent NMII bacteria differentially induce cell death in B1a cells. Figure 4C shows a comparison of the percent cell death between NMI- and NMII-infected B1a cells at 3 days postinfection. Interestingly, the percentage of dead cells among NMII-infected cells was significantly higher than the percentage of dead cells among NMI-infected cells at 3 days postinfection. Additionally, although a low percentage of dead cells was detected among NMI-infected cells, it was not statistically significantly different from the percentage of dead cells among uninfected control cells. These results indicate that virulent NMI infection did not induce significant B1a cell death. Collectively, these data suggest that avirulent NMII bacteria induce B1a subset B cell death in a dose-dependent manner but virulent NMI bacteria do not.
FIG 4.
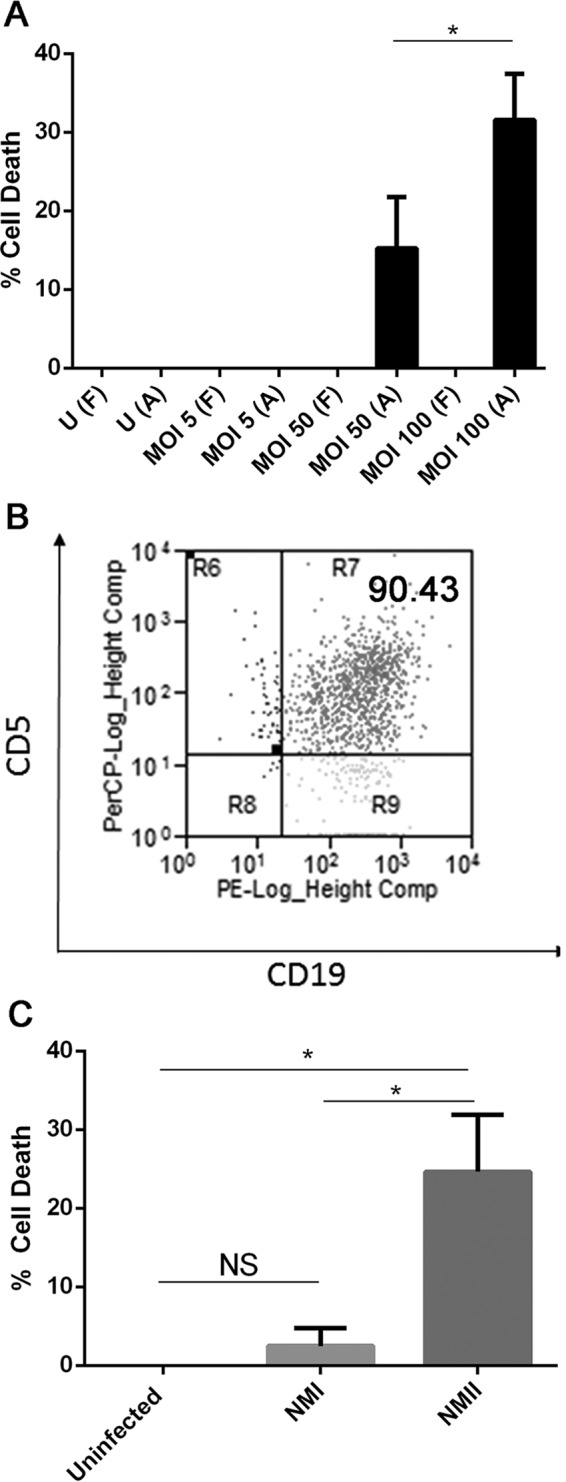
NMII induces cell death in peritoneal B1a B cells in a dose-dependent manner, but virulent NMI does not. Purified B cells were infected with either NMI or NMII for 1 and 3 days. At each time point, an MTS assay was utilized to assay for cell death. (A) Different doses of NMII infection for 3 days were evaluated for the rate of cell death that they caused using MTS assays. U, uninfected cells; F, floating cells; A, adhered cells. (B) Cells that were positive by the MTS assays and staining by TUNEL were adhered cells. Flow cytometry staining indicates that adhered cells were mostly CD19+ CD11b+ CD5+ B1a B cells. The value at top right indicates the percentage of stained cells. (C) MTS assays were performed on adhered cells infected with either NMI or NMII for 3 days. *, P < 0.05; NS, not significant. These data indicate that NMII induces cell death in peritoneal B1a B cells in a dose-dependent manner but NMI does not.
FIG 5.

NMII induces cell death in peritoneal B1a B cells. Peritoneal B cells were purified by MACS and infected with NMII. At day 3 postinfection, cells were stained by use of the TUNEL reaction and with antibodies against NMII and CD5. This figure shows CD5+ B1a cells that are TUNEL positive and that colocalize with NMII (white arrows). These data indicate that NMII induces cell death in peritoneal B1a B cells.
NMII-induced B1a cell death depends on activation of caspase-1.
To understand the signaling pathway of NMII-induced B1a cell death, we examined if activation of the apoptosis executioner caspase-3 and its downstream substrate, PARP, occurred in NMII-infected B1a cells by Western blotting. As shown in Fig. 6A, both cleaved caspase-3 and cleaved PARP were undetectable in NMII-infected B1a cells at 1 and 3 days postinfection. Since activation of caspase-3 is a major characteristic of apoptosis, this observation suggests that NMII-induced B1a cell death may not be through a caspase-3-mediated apoptotic signaling pathway. Recently, it has been shown that several Gram-negative bacterial pathogens, such as Salmonella, Francisella, and Legionella, were able to induce a caspase-1-dependent programmed cell death (17) in infected host cells. To determine if NMII-induced B1a cell death is dependent on caspase-1 activation, the caspase-1 activity in NMII-infected B1a cells was analyzed by Western blotting. As shown in Fig. 6B, cleaved caspase-1 (20-kDa protein) was detected in NMII-infected B1a cells but not uninfected cells at both 1 and 3 days postinfection. Since IL-1β and IL-18 are often released following caspase-1 cleavage, we also examined whether NMII-infected B1a cells secreted IL-1β and IL-18 at 1 or 3 days postinfection. As shown in Fig. 6C, NMII-infected B1a cells secreted a significantly higher level of IL-18 than uninfected control cells at both 1 day and 3 days postinfection. The level of IL-1β secretion was also measured by ELISA. At 1 day postinfection, no IL-1β was detected in the supernatants. However, at 3 days postinfection, NMII-infected cells had significantly higher levels of IL-1β in the cell supernatant than uninfected control cells (Fig. 6D). These results suggest that NMII infection may induce a caspase-1-dependent programmed cell death in B1a cells. To prove this hypothesis, we tested whether the caspase-1 inhibitor Ac-YVAD-cmk can inhibit NMII-induced caspase-1-dependent programmed cell death in B1a cells. As shown in Fig. 6E, the percentage of dead cells among the caspase-1 inhibitor Ac-YVAD-cmk-treated NMII-infected cells was significantly lower than that among NMII-infected cells not treated with Ac-YVAD-cmk at 3 days postinfection. This result demonstrates that NMII-induced B1a cell death depends on the activation of caspase-1. To further confirm this observation, we also examined if NMII bacteria can induce cell death in B1a cells from caspase-1-deficient mice. As shown in Fig. 7A, apoptotic DNA was observed in NMII-infected B1a cells from wild-type (WT) mice but was undetectable in NMII-infected B1a cells from caspase-1-deficient mice at 3 days postinfection. The results also indicate that the NMII infection rate in caspase-1-deficient B1a cells was significantly higher than the infection rate in WT B1a cells at 3 days postinfection (Fig. 7B), suggesting that caspase-1-deficient B1a cells are more permissive to infection with NMII bacteria than WT B1a cells. In addition, the percentage of dead cells among NMII-infected caspase-1-deficient B1a cells was no different from that among uninfected control cells, but it was significantly lower than the percentage of dead cells among NMII-infected WT B1a cells at 3 days postinfection (Fig. 7C). Since cell death mediated by caspase-1 activation is an important characteristic of the programmed cell death form known as pyroptosis, collectively, these data suggest that avirulent NMII bacteria can induce a caspase-1-dependent pyroptosis in murine peritoneal B1a cells.
FIG 6.

NMII-induced B1a B cell death depends on activation of caspase-1. Protein was extracted from purified B1a B cells at day 1 and day 3 postinfection. (A and B) Analysis of caspase-3 (Casp 3) and PARP-1 cleavage (A) and caspase-1 (Casp 1) cleavage (B) in NMII-infected B1a cells by Western blotting. Lanes U, uninfected cells; lanes I, NMII-infected cells; lanes +, staurosporine-treated uninfected cells as an apoptotic cell positive control. (C) IL-18 levels in infected and uninfected cell supernatants at both 1 day and 3 days postinfection were measured using ELISA. (D) IL-1β levels in infected and uninfected cell supernatants were measured using ELISA. (E) The MTS assay was used to measure cell death at day 3 postinfection with NMII following treatment with or without a caspase-1 inhibitor. *, P < 0.05; **, P < 0.01; ***, P < 0.001. These data indicate that NMII-induced cell death in B1a B cells is dependent on cleavage of caspase-1 and causes secretion of the cytokines IL-1β and IL-18.
FIG 7.

NMII does not induce death in caspase-1-deficient B1a B cells. B1a B cells were harvested and purified from either wild-type or caspase-1-deficient mice and infected with NMII. (A) Staining by TUNEL was used to visualize DNA fragmentation following NMII infection. (B) Infected cell numbers were counted at 3 days postinfection. (C) The MTS assay was used to measure the numbers of viable cells among caspase-1-knockout (KO) or wild-type B1a B cells infected with NMII at day 3 postinfection. **, P < 0.01. These data indicate that caspase-1 is required for B1a cell death induction and that caspase-1-deficient cells are more permissive to infection with NMII.
NMII-infected B1a cells secreted TNF-α, but neutralization of secreted TNF-α did not prevent NMII-induced pyroptosis.
TNF-α is a proinflammatory cytokine that has demonstrated the ability to induce cell death in many cell types via binding with the TNF-α receptor on the cell surface (40). One previous study demonstrated that secreted TNF-α contributes to NMII-induced apoptosis in undifferentiated THP-1 cells (22). Our recent work indicated that virulent NMI-infected B1a cells were able to produce high levels of TNF-α in vitro (30). However, it remains unknown whether avirulent NMII-infected B1a cells can secrete TNF-α and whether secreted TNF-α is involved in NMII-induced pyroptosis in B1a cells. In this study, ELISA was used to measure the concentration of TNF-α in the supernatant from NMII-infected B1a cells. A significantly higher level of TNF-α was detected in the supernatant from NMII-infected B1a cells (350 pg/ml) than in that from uninfected control cells (105 pg/ml) at 3 days postinfection. This result suggests that NMII bacteria can stimulate B1a cells to produce TNF-α (Fig. 8A). To determine whether secreted TNF-α contributed to NMII-induced B1a cell death, the MTS assay was used to examine if the neutralization of secreted TNF-α by anti-TNF-α antibodies can significantly inhibit NMII-induced B1a cell death. There was no significant difference in the percentage of dead cells between untreated and anti-TNF-α antibody-treated NMII-infected B1a cells at 3 days postinfection (Fig. 8B). This result suggests that secreted TNF-α may not contribute to NMII-induced B1a cell death.
FIG 8.
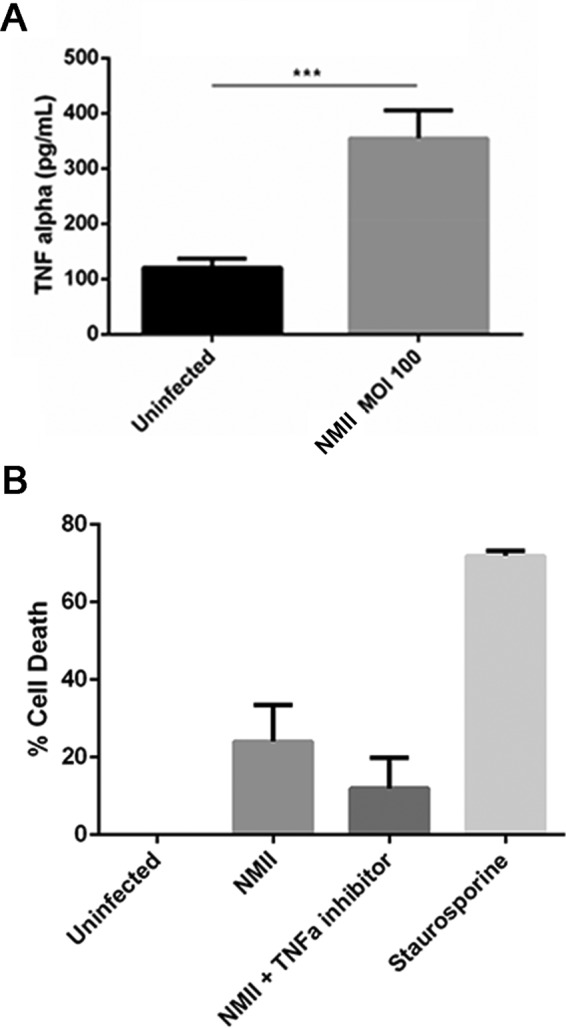
NMII-infected purified B1a B cells secrete the proinflammatory cytokine TNF-α. (A) B1a B cells were purified by MACS and evaluated for TNF-α secretion at day 1 postinfection using ELISA. ***, P < 0.001. (B) Purified B1a B cells were infected with NMII and treated with an anti-TNF-α antibody prior to the MTS assay. These data indicate that NMII induces the secretion of TNF-α by B1a B cells, but this does not contribute significantly to cell death.
NMII-induced B1a cell death depends on bacterial replication.
To determine if bacterial replication is responsible for NMII-induced B1a cell death, we examined if inhibition of C. burnetii replication could prevent NMII-induced cell death in B1a cells. Since rifampin has been demonstrated to be the antibiotic that most effectively inhibits C. burnetii replication in a cell culture system (41), we used 10 μg/ml of rifampin to inhibit C. burnetii replication in NMII-infected B1a cells. Staining by TUNEL was used to detect fragmented cell DNA in NMII-infected B1a cells at 1 and 3 days postinfection. Figure 9A shows the results of staining by TUNEL of NMII-infected B1a cells either untreated or treated with 10 μg/ml of rifampin at 3 days postinfection. Fewer TUNEL-positive cells were observed among rifampin-treated NMII-infected B1a cells than untreated NMII-infected B1a cells. Additionally, although the percentage of TUNEL-positive cells was similar between untreated and rifampin-treated NMII-infected B1a cells at 1 day postinfection, the percentage of TUNEL-positive cells among rifampin-treated NMII-infected B1a cells was significantly lower than the percentage of TUNEL-positive cells among untreated NMII-infected B1a cells at 3 days postinfection (Fig. 9B). These results suggest that NMII-induced B1a cell death may depend on bacterial replication. To further test this hypothesis, we also used the MTS assay to examine if heat-killed NMII bacteria were able to induce B1a cell death. As shown in Fig. 9C, the percentage of dead cells among heat-killed NMII-infected B1a cells was no different from that among uninfected control cells, but it was significantly lower than the percentage of dead cells among NMII-infected B1a cells. These results provide further support to demonstrate that NMII-induced B1a cell death depends on bacterial replication. C. burnetii possesses a type 4 Dot/Icm secretion system (T4SS), and it is considered that T4SS-secreted factors may have the ability to modulate apoptotic cell signaling within host cells (42). To determine whether T4SS-secreted factors are involved in NMII-induced B1a cell death, we also investigated if dotA mutant NMII bacteria, which lack a functional T4SS, could induce cell death in B1a cells. The result indicated that the dotA mutant NMII bacteria were unable to induce cell death in B1a cells (Fig. 9C), suggesting that NMII-induced B1a cell death may depend on its T4SS.
FIG 9.

NMII induction of B1a B cell death is dependent on bacterial replication and a functional T4SS. (A) Staining by TUNEL was measured in B1a B cells infected with NMII and treated with rifampin or mock infected by use of PBS. (B) The number of cells with positive DNA fragmentation by TUNEL was counted at day 1 and day 3 postinfection. (C) The MTS assay was used to measure cell viability following infection with NMII, heat-killed NMII, and dotA-deficient NMII. *, P < 0.05; ***, P < 0.001. These data indicate that NMII replication and a functional T4SS are required in order to induce B1a B cell death.
NMII bacteria did not induce cell death in peritoneal B cells from TLR-2-deficient mice.
It has been shown that bacterial lipoproteins can induce apoptosis through TLR-2 signaling (43). To determine whether the TLR-2 signaling pathway is involved in NMII-induced B1a cell pyroptosis, staining by TUNEL was used to examine if NMII bacteria can induce cell death in B1a cells from TLR-2 deficient mice. As shown in Fig. 10A, TUNEL-positive cells were observed among NMII-infected WT B1a cells, but there was no TUNEL-positive staining of NMII-infected TLR-2-deficient B1a cells at 3 days postinfection. This observation suggests that the induction of NMII-induced B1a cell pyroptosis may be through the TLR-2 signaling pathway. In addition, the MTS assay was used to measure the percentage of dead cells among NMII-infected TLR-2-deficient B1a cells. As shown in Fig. 10B, a significantly higher percentage of cell death was detected among NMII-infected WT B1a cells than uninfected control cells, but cell death was undetectable among NMII-infected TLR-2-deficient B1a cells. This result provides additional support to the hypothesis that NMII-induced B1a cell pyroptosis is through the TLR-2 signaling pathway. In addition, IFA and real-time quantitative PCR were used to examine if there were differences in NMII infection and replication between WT and TLR-2-deficient B1a cells. As shown in Fig. 10C, the NMII infection rate among TLR-2 deficient B1a cells was significantly higher than the infection rate among WT B1a cells at 3 days postinfection. Interestingly, a significantly higher number of C. burnetii genome copies was detected in NMII-infected TLR-2-deficient B1a cells than NMII-infected WT B1a cells at 1 and 3 days postinfection (Fig. 10D). These results indicate that TLR-2-deficient B1a cells are more permissive to NMII infection, suggesting that activation of TLR-2 signaling may be required for the control of NMII bacterial infection and replication in B1a cells in vitro. Collectively, these data indicate that there was no cell death among NMII-infected TLR-2-deficient B1a cells regardless of the higher infection and replication rate. This finding demonstrates that NMII-induced B1a cell pyroptosis is through the TLR-2 signaling pathway.
FIG 10.

TLR-2-deficient peritoneal B1a B cells do not undergo death following NMII infection and are more permissive to infection. (A) Peritoneal B cells were adhered to poly-d-lysine-coated coverslips and infected with NMII. At day 1 and day 3 postinfection, cells were stained by use of the TUNEL reaction and anti-NMII antibodies. (B) The percentage of dead cells among TLR-2-knockout B1a cells was measured by the MTS assay. (C) The number of infected cells among TLR-2-knockout and WT mouse B1a cells was counted microscopically at day 3 postinfection. (D) At day 1 and day 3 postinfection, DNA was extracted from cells and evaluated for bacterial replication by examining the com1 gene copy numbers by real-time PCR. *, P < 0.05; ***, P < 0.001. These data indicate that cell death is induced by TLR-2 signaling and that, in the absence of this signaling, TLR-2-knockout B cells are more permissive to NMII infection.
The NLRP3 inflammasome is involved in NMII-induced B1a cell pyroptosis.
Several types of inflammasomes that activate caspase-1, including NLRP3, NLRC4, and AIM2, have been described in the mouse (16). The NLRP3 inflammasome is well studied and can be activated by a variety of factors found in bacteria and viruses (44, 45). To determine whether activation of the inflammasome plays a role in NMII-induced B1a cell pyroptosis, we examined if NMII bacteria can induce cell death in NLRP3-deficient B1a cells. As shown in Fig. 11A, apoptotic DNA was detected in NMII-infected WT B1a cells but was undetectable in NMII-infected NLRP3-deficient B1a cells at 3 days postinfection. In addition, cell death in NMII-infected NLRP3-deficient B1a cells was undetectable at 3 days postinfection by the MTS assay (Fig. 11B). These results indicate that NLRP3 inflammasome activation is required in NMII-induced B1a cell pyroptosis, suggesting that NMII bacteria stimulate the NLRP3 inflammasome in murine B1a cells, resulting in caspase-1 activation and eventually leading to pyroptosis.
FIG 11.

NLRP3-knockout B1a B cells do not undergo cell death following NMII infection. B1a cells were harvested and purified from NLRP3-knockout mice. (A) The TUNEL assay was used to measure DNA fragmentation in infected NLRP3-knockout B1a B cells. (B) The MTS assay was used to measure the viability of wild-type cells infected with NMII and NLRP3-knockout cells infected with NMII. ***, P < 0.001. These data indicate that stimulation of the NLRP3 inflammasome is critical for NMII-induced cell death in purified B1a B cells.
DISCUSSION
In this study, we examined if virulent NMI and avirulent NMII can differentially activate host cell apoptotic signaling in B1a cells. The results demonstrated that NMII induced a dose-dependent cell death in murine peritoneal B1a cells but NMI did not, suggesting that NMI and NMII may differentially activate B1a cell death signaling. Western blotting indicated that NMII-induced B1a cell death was not dependent on either caspase-3 or PARP-1 cleavage, but cleavage of caspase-1 was detected in NMII-infected B1a cells. In addition, the observation that inhibition or a deficiency of caspase-1 activity blocked NMII-induced B1a cell death suggests that NMII induces caspase-1-dependent pyroptosis in B1a cells. Collectively, these results demonstrate that NMII induces a caspase-1-dependent pyroptosis in murine peritoneal B1a cells through activation of the TLR-2 and NLRP3 signaling pathways.
Although the ability of C. burnetii NMII to inhibit and induce cell death in different cells has been reported (21, 22), the observation that NMII induced dose-dependent cell death but NMI did not in murine peritoneal B1a cells suggests that virulent NMI and avirulent NMII may differentially activate host cell death signaling. Interestingly, we also found that NMII infection was unable to induce cell death in TLR-2-deficient B1a cells. This result suggests that NMII binding with the TLR-2 receptor and activation of the TLR-2 signaling pathway are required for NMII-induced B1a cell death. Thus, the difference between NMI and NMII in inducing B1a cell death can be explained by the possibility that NMI and NMII differentially activate the TLR-2 signaling pathway. Since NMI can cause infection in wild-type animals but NMII cannot, the difference in the establishment of a persistent infection between NMI and NMII organisms may be related to their ability to modulate host cell death signaling and host cell death signaling may play an important role in host defense against C. burnetii infection.
Pyroptosis is inherently an inflammatory process of caspase-1-dependent programmed cell death, which is an important form of cell death during infection, particularly during infection with intracellular pathogens. It can result in not only the production of inflammatory cytokines but also rapid cell death with plasma membrane rupture and the release of proinflammatory intracellular contents (17). Unlike apoptosis, which is anti-inflammatory, pyroptosis leads to the production of proinflammatory cytokines, such as IL-1β and TNF-α, to stimulate an immune response (17), which eliminates the pathogen's replicative niche and signals to alert nearby cells of the infectious threat. The observations that (i) the cleavage of caspase-1 was detected in NMII-infected B1a cells, (ii) NMII-infected B1a cells secreted a significantly higher level of IL-1β and IL-18 than uninfected control cells, (iii) the caspase-1 inhibitor Ac-YVAD-cmk was able to inhibit NMII-induced B1a cell death, and (iv) NMII infection did not induce cell death in caspase-1-deficient B1a cells demonstrate that NMII infection induces a caspase-1-dependent pyroptosis in murine peritoneal B1a cells, while NMI infection does not induce cell death in B1a cells. Although it remains unknown if NMII infection can also induce caspase-1-dependent pyroptosis in other cells, the difference in inducing B1a cell pyroptosis between NMI and NMII may be responsible for the difference in their ability to establish a persistent infection in vivo. This hypothesis was supported by a previous study demonstrating that NMI and NMII differentially activated primary human alveolar macrophages, which produced different levels of pro-IL-1β and mature IL-1β (46). It has been reported that Shigella flexneri induced caspase-1-dependent host cell death, leading to the release of IL-1β, which causes acute inflammation and ultimately helps clear the infection (47). However, Salmonella-induced apoptosis was correlated to the severity of disease in vivo (48). This observation suggests that Salmonella-induced apoptosis is important for bacterial escape from the host cell and facilitates it spread to other cells. Thus, the induction of pyroptosis in NMII-infected B1a cells may lead to the clearance of NMII more efficiently in vivo, while the ability of NMI to avoid activation of cell death signaling may be responsible for its ability to establish a persistent infection and cause disease in wild-type animals. Future studies to test if NMII can induce a severity of disease similar to that induced by NMI in caspase-1-deficient mice will help determine what role that caspase-1-dependent pyroptosis plays in vivo during C. burnetii infection.
It has been shown that TNF-α can induce cell death in many cell types via binding with the TNF-α receptor on the cell surface (40). In addition, the observation that the neutralization of TNF-α in NMII-infected undifferentiated THP-1 cells partially blocked PARP cleavage suggests that secreted TNF-α may be one of the upstream factors involved in NMII-induced caspase-independent apoptosis in THP-1 cells (22). We also examined if NMII-infected B1a cells can secrete TNF-α and whether secreted TNF-α is involved in NMII-induced pyroptosis in B1a cells. The results indicate that although NMII-infected B1a cells are able to secrete TNF-α at 3 days postinfection, the neutralization of secreted TNF-α by anti-TNF-α antibodies did not affect NMII-induced B1a cell pyroptosis, suggesting that secreted TNF-α may not be involved in NMII-induced pyroptosis in B1a cells. This hypothesis is also supported by the observation that although NMI-infected B1a cells secreted high levels of TNF-α in vitro (30), no cell death was detected in NMI-infected B1a cells. Thus, in contrast to the involvement of secreted TNF-α in NMII-induced apoptosis in THP-1 cells, secreted TNF-α may not be the upstream factor that is involved in NMII-induced pyroptosis in B1a cells.
It has been shown that NMII-induced apoptosis in THP-1 cells depends on intracellular C. burnetii replication (22). To determine whether bacterial replication is also involved in NMII-induced B1a cell death, we examined if inhibition of C. burnetii replication by antibiotics could prevent NMII-induced cell death in B1a cells. The result that antibiotic treatment was able to significantly reduce the percentage of cell death in NMII-infected B1a cells at 3 days postinfection suggests that bacterial replication may be responsible for NMII-induced B1a cell death. This hypothesis was further supported by the result that although B1a cells were able to phagocytose heat-killed NMII bacteria (data not shown), cell death was undetectable in B1a cells infected with heat-killed NMII. In addition, it has been demonstrated that L. pneumophila induces pyroptosis via its T4SS by expression of flagellin in murine macrophages (49). Since C. burnetii possesses a T4SS similar to that in L. pneumophila (50) and it is considered that C. burnetii T4SS-secreted factors may have the ability to modulate apoptotic cell signaling within host cells (42), we also investigated if dotA mutant NMII bacteria, which lack a functional T4SS, could induce cell death in B1a cells. Although the dotA mutant NMII bacteria were observed inside B1a cells (data not shown), the observation that cell death was undetectable in dotA mutant NMII-infected B1a cells suggests that the T4SS of NMII bacteria may play a critical role in NMII-induced B1a cell death. Collectively, these results suggest that NMII-induced B1a cell death may depend on bacterial replication and its T4SS-secreted factors.
The Toll-like receptor (TLR) family has been demonstrated to have the ability to induce both apoptosis and pyroptosis (51). It was shown that Mycobacterium tuberculosis was able to induce a caspase-1-dependent cell death through the activation of TLR-2 signaling in macrophages (52). The observation that cell death was undetectable in NMII-infected TLR-2-deficient B1a cells suggests that activation of the TLR-2 signaling pathway by NMII stimuli is responsible for NMII-induced B1a cell pyroptosis. This is the first evidence to demonstrate that bacterial stimulus activation of the TLR-2 signaling pathway can induce a caspase-1-dependent pyroptosis in murine peritoneal B1a cells. On the other hand, interestingly, the observations that a significantly higher infection rate and significantly higher bacterial genome copy numbers were detected in NMII-infected TLR-2-deficient B1a cells than NMII-infected WT B1a cells indicate that TLR-2-deficient B1a cells are more permissive to NMII infection, suggesting that TLR-2 signaling is important for the control of NMII bacterial infection and replication in B1a cells in vitro. This hypothesis was supported by a previous study that demonstrated that TLR-2-knockout mice developed fever responses during infection with NMII (53). Since NMII does not induce infection in wild-type mice, these data suggest that TLR-2 may play an important role in host defense against C. burnetii infection.
The inflammasome consists of a variety of molecules which, when activated, induce the cleavage of pro-caspase-1 into the active form of caspase-1 (54–56). Several types of inflammasomes that induce the activation of caspase-1, including NLRP3, NLRC4, and AIM2, have been described in mice (54, 56). NLRP3 was demonstrated to play an important role against multiple Gram-negative bacterial pathogens in response to TLR activation (57). A study by Deng et al. demonstrated that NLRP3 is activated in response to Pseudomonas aeruginosa infection and leads to killing of the internalized bacteria (58). To determine whether the inflammasome is involved in NMII-induced caspase-1-dependent pyroptosis in B1a cells, we examined if NMII could induce cell death in B1a cells from NLRP3-deficient mice. The result that no cell death was detected in NMII-infected NLRP3-deficient B1a cells indicates that NLRP3 inflammasome activation may also be required for NMII-induced B1a cell pyroptosis. In contrast to this observation, Cunha et al. reported that NMII can inhibit caspase-1 activation in murine bone marrow-derived macrophages and the caspase-11-mediated noncanonical activation of the NLRP3 inflammasome induced by L. pneumophila (59). In addition, Graham et al. found that although NMII induces IL-1β production in human alveolar macrophages and IL-1β production correlated with caspase-dependent inflammasome activation, no lytic cell death was observed (60). Thus, these studies suggest that avirulent NMII may process the ability to activate or inhibit the inflammasome on the basis of the infected cell types and the stage of infection.
Although TLRs and inflammasomes can be activated during microbial infections, it is unclear how the TLR and inflammasome signaling pathways cooperate during a microbial infection. The expression of NLRP3 following activation of macrophages with TLR ligands and other TLR agonists has been documented previously (61–63). Recently, it has been shown that activation of TLR-2 contributes to rapid induction of the inflammasome independently of NLRP3 along with caspase-1 activation and host cell death in macrophages infected with Francisella novicida. In addition, macrophages from TLR-2−/− mice showed a significant delay in the inflammasome-dependent response following infection (64). Our results that NMII did not induce cell death in TLR-2- or NLRP3-deficient peritoneal B1a cells suggest that both the TLR-2 and NLRP3 signaling pathways are involved in NMII-induced caspase-1-dependent pyroptosis in murine peritoneal B1a cells. Thus, these data suggest that TLR and inflammasome signaling pathways may be connected during the response to microbial infections. However, Bradley et al. (65) demonstrated that TLR-2 activation neither leads to activation of inflammasome formation nor plays a role in NMII replication restriction in murine macrophages, suggesting that TLR-2 activation may not be connected to inflammasome activation. Thus, future study is required to confirm if the TLR-2 and NLRP3 signaling pathways are connected during NMII-induced caspase-1-dependent pyroptosis in B1a cells.
An important role for B cell death has been described in a murine model of sepsis. Sarkar et al. demonstrated that caspase-1-knockout mice are resistant to i.p. infection with Escherichia coli but WT mice succumb to infection (66). However, IL-1β/IL-18-dual-knockout mice succumbed to infection, as did WT mice. This indicates a role for pyroptosis in E. coli-induced sepsis. In addition, B cell apoptosis was suggested to play a role after histological staining of the spleen found apoptotic bodies in the white pulp, which stained positive for B cell-specific CD79 in wild-type and IL-1β/IL-18-dual-knockout mice. This observation was not found in caspase-1-deficient mice, indicating that B cell pyroptosis may be an important factor in disease progression in this model of sepsis. Although the role of NMII-induced B cell pyroptosis in vivo remains unclear, it is possible that pyroptosis may play a role during C. burnetii infection in vivo.
It is noteworthy that IFA analysis of NMII-infected B cells from WT mice indicates that several cells that were not stained with anti-C. burnetii antibodies were TUNEL positive, while several cells that were stained with anti-C. burnetii antibodies were TUNEL negative. Since pyroptosis is characterized by the formation of pores, it is possible that at the later stages of pyroptosis, cellular contents are leaked into the supernatant. Although the reason for the observation that several cells were not stained with anti-C. burnetii antibodies but were TUNEL positive is unclear, this may because, at the later stages of NMII-induced pyroptosis, cellular contents (including C. burnetii) leak into the supernatant and only nuclei remain on the cover glass; these nuclei are then stained by TUNEL but are not stained with C. burnetii antibody. On the other hand, the observation that several NMII-infected cells were TUNEL negative might be because these cells were in the early stage of C. burnetii infection and the DNA damage was undetectable by the TUNEL assay at this stage.
In summary, our results demonstrated that (i) avirulent NMII bacteria but not virulent NMI bacteria induce dose-dependent cell death in murine peritoneal B1a cells, (ii) NMII-induced B1a cell death is dependent on the activation of caspase-1, (iii) bacterial replication and a functional T4SS are required for NMII-induced B1a cell death, and (iv) both the TLR-2 and NLRP3 signaling pathways are involved in NMII-induced B1a cell death. This is the first study to demonstrate that NMII induces a caspase-1-dependent pyroptosis in murine peritoneal B1a cells and requires its T4SS and activation of the TLR-2 and NLRP3 signaling pathways. The predicted mechanism underlying C. burnetii NMII-induced caspase-1-dependent pyroptosis is summarized in Fig. 12. Future studies are necessary to understand how the TLR-2 and NLRP3 signaling pathways cooperate to induce caspase-1-dependent pyroptosis in murine peritoneal B1a cells and to identify the bacterial factors that are responsible for NMII-induced pyroptosis.
FIG 12.

A model for NMII-induced pyroptosis in peritoneal B1a B cells. NMII infection and replication induce activation of TLR-2 signaling, which leads to the expression of inflammatory cytokines and the activation of the NLRP3 inflammasome. Inflammasome activation causes cleavage of pro-caspase-1 into active caspase-1. Caspase-1 cleaves pro-IL-1β and pro-IL-18 into active IL-1β and IL-18, respectively, which induce pyroptotic cell death. Pyroptotic cell death leads to lysis of infected cells, which releases their inflammatory contents. ?, possible linkage.
ACKNOWLEDGMENTS
This study was funded by Public Health Service grant RO1AI083364 from the National Institute of Allergy and Infectious Diseases, NIH, and a contract from the Defense Threat Reduction Agency (DTRA).
We thank the staff at the MU Laboratory for Infectious Disease Research for their assistance with these experiments. We also thank Alexander Jurkevich and the MU Molecular Cytology Core for their assistance with confocal microscopy.
REFERENCES
- 1.Maurin M, Raoult D. 1999. Q fever. Clin Microbiol Rev 12:518–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raoult D. 1993. Treatment of Q fever. Antimicrob Agents Chemother 37:1733–1736. doi: 10.1128/AAC.37.9.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raoult D. 1999. Afebrile blood culture-negative endocarditis. Ann Intern Med 131:144–146. doi: 10.7326/0003-4819-131-2-199907200-00012. [DOI] [PubMed] [Google Scholar]
- 4.Raoult D, Fenollar F, Stein A. 2002. Q fever during pregnancy. Arch Intern Med 162:701–704. doi: 10.1001/archinte.162.6.701. [DOI] [PubMed] [Google Scholar]
- 5.Dijkstra F, van der Hoek W, Wijers N, Schimmer B, Rietveld A, Wijkmans CJ, Vellema P, Schneeberger PM. 2012. The 2007-2010 Q fever epidemic in The Netherlands: characteristics of notified acute Q fever patients and the association with dairy goat farming. FEMS Immunol Med Microbiol 64:3–12. doi: 10.1111/j.1574-695X.2011.00876.x. [DOI] [PubMed] [Google Scholar]
- 6.Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. 1999. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med 159:167–173. doi: 10.1001/archinte.159.2.167. [DOI] [PubMed] [Google Scholar]
- 7.Hackstadt T, Peacock MG, Hitchcock PJ, Cole RL. 1985. Lipopolysaccharide variation in Coxiella burnetii: intrastrain heterogeneity in structure and antigenicity. Infect Immun 48:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moos A, Hackstadt T. 1987. Comparative virulence of intra- and interstrain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infect Immun 55:1144–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baca OG, Akporiaye ET, Aragon AS, Martinez IL, Robles MV, Warner NL. 1981. Fate of phase I and phase II Coxiella burnetii in several macrophage-like tumor cell lines. Infect Immun 33:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howe D, Barrows LF, Lindstrom NM, Heinzen RA. 2002. Nitric oxide inhibits Coxiella burnetii replication and parasitophorous vacuole maturation. Infect Immun 70:5140–5147. doi: 10.1128/IAI.70.9.5140-5147.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howe D, Melnicakova J, Barak I, Heinzen RA. 2003. Maturation of the Coxiella burnetii parasitophorous vacuole requires bacterial protein synthesis but not replication. Cell Microbiol 5:469–480. doi: 10.1046/j.1462-5822.2003.00293.x. [DOI] [PubMed] [Google Scholar]
- 12.Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. 2010. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun 78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fink SL, Cookson BT. 2005. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parmely MJ, Fischer JL, Pinson DM. 2009. Programmed cell death and the pathogenesis of tissue injury induced by type A Francisella tularensis. FEMS Microbiol Lett 301:1–11. doi: 10.1111/j.1574-6968.2009.01791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fink SL, Bergsbaken T, Cookson BT. 2008. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci U S A 105:4312–4317. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. 2010. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergsbaken T, Fink SL, Cookson BT. 2009. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jorgensen I, Miao EA. 2015. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev 265:130–142. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voth DE, Heinzen RA. 2007. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell Microbiol 9:829–840. doi: 10.1111/j.1462-5822.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- 20.Voth DE, Heinzen RA. 2009. Sustained activation of Akt and Erk1/2 is required for Coxiella burnetii antiapoptotic activity. Infect Immun 77:205–213. doi: 10.1128/IAI.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lührmann A, Roy CR. 2007. Coxiella burnetii inhibits activation of host cell apoptosis through a mechanism that involves preventing cytochrome c release from mitochondria. Infect Immun 75:5282–5289. doi: 10.1128/IAI.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Zhang G, Hendrix LR, Tesh VL, Samuel JE. 2012. Coxiella burnetii induces apoptosis during early stage infection via a caspase-independent pathway in human monocytic THP-1 cells. PLoS One 7:e30841. doi: 10.1371/journal.pone.0030841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DiLillo DJ, Mastsushita T, Tedder TF. 2010. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann N Y Acad Sci 1183:38–57. doi: 10.1111/j.1749-6632.2009.05137.x. [DOI] [PubMed] [Google Scholar]
- 24.Parra D, Rieger AM, Li J, Zhang YA, Randall LM, Hunter CA, Barreda DR, Sunyer JO. 2012. Pivotal advance: peritoneal cavity B-1 B cells have phagocytic and microbicidal capacities and present phagocytosed antigen to CD4+ T cells. J Leukoc Biol 91:525–536. doi: 10.1189/jlb.0711372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouaziz JD, Calbo S, Maho-Vaillant M, Saussine A, Bagot M, Bensussan A, Musette P. 2010. IL-10 produced by activated human B cells regulates CD4(+) T-cell activation in vitro. Eur J Immunol 40:2686–2691. doi: 10.1002/eji.201040673. [DOI] [PubMed] [Google Scholar]
- 26.Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, Mauri C. 2007. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol 178:7868–7878. doi: 10.4049/jimmunol.178.12.7868. [DOI] [PubMed] [Google Scholar]
- 27.Bouaziz J-D, Yanaba K, Tedder TF. 2008. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev 224:201–214. doi: 10.1111/j.1600-065X.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- 28.Hahne M, Renno T, Schroeter M, Irmler M, French L, Bornand T, MacDonald HR, Tschopp J. 1996. Activated B cells express functional Fas ligand. Eur J Immunol 26:721–724. doi: 10.1002/eji.1830260332. [DOI] [PubMed] [Google Scholar]
- 29.Lundy SK. 2009. Killer B lymphocytes: the evidence and the potential. Inflamm Res 58:345–357. doi: 10.1007/s00011-009-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoenlaub L, Elliott A, Freches D, Mitchell WJ, Zhang G. 2015. The role of B cells in host defense against primary Coxiella burnetii infection. Infect Immun 83:4826–4836. doi: 10.1128/IAI.01073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin F, Oliver AM, Kearney JF. 2001. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity 14:617–629. doi: 10.1016/S1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 32.Weber GF, Chousterman BG, Hilgendorf I, Robbins CS, Theurl I, Gerhardt LM, Iwamoto Y, Quach TD, Ali M, Chen JW, Rothstein TL, Nahrendorf M, Weissleder R, Swirski FK. 2014. Pleural innate response activator B cells protect against pneumonia via a GM-CSF-IgM axis. J Exp Med 211:1243–1256. doi: 10.1084/jem.20131471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maglione PJ, Chan J. 2009. How B cells shape the immune response against Mycobacterium tuberculosis. Eur J Immunol 39:676–686. doi: 10.1002/eji.200839148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mangan NE, Fallon RE, Smith P, van Rooijen N, McKenzie AN, Fallon PG. 2004. Helminth infection protects mice from anaphylaxis via IL-10-producing B cells. J Immunol 173:6346–6356. doi: 10.4049/jimmunol.173.10.6346. [DOI] [PubMed] [Google Scholar]
- 35.Mannick J, Asano K, Izumi K, Kieff E, Stamler J. 1994. Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein-Barr virus reactivation. Cell 79:1137–1146. doi: 10.1016/0092-8674(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 36.Popi AF, Zamboni DS, Mortara RA, Mariano M. 2009. Microbicidal property of B1 cell derived mononuclear phagocyte. Immunobiology 214:664–673. doi: 10.1016/j.imbio.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Ho T, Htwe KK, Yamasaki N, Zhang GQ, Ogawa M, Yamaguchi T, Fukushi H, Hirai K. 1995. Isolation of Coxiella burnetii from dairy cattle and ticks, and some characteristics of the isolates in Japan. Microbiol Immunol 39:663–671. doi: 10.1111/j.1348-0421.1995.tb03254.x. [DOI] [PubMed] [Google Scholar]
- 38.Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Hansen B, Samuel JE, Heinzen RA. 2011. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol 77:3720–3725. doi: 10.1128/AEM.02826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray A, Dittel BN. 2010. Isolation of mouse peritoneal cavity cells. J Vis Exp 2010:1488. doi: 10.3791/1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wajant H, Pfizenmaier K, Scheurich P. 2003. Tumor necrosis factor signaling. Cell Death Differ 10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 41.Brennan RE, Samuel JE. 2003. Evaluation of Coxiella burnetii antibiotic susceptibilities by real-time PCR assay. J Clin Microbiol 41:1869–1874. doi: 10.1128/JCM.41.5.1869-1874.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE. 2013. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat Rev Microbiol 11:561–573. doi: 10.1038/nrmicro3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. 1999. Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science 285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 44.Kim J-J, Jo E-K. 2013. NLRP3 inflammasome and host protection against bacterial infection. J Korean Med Sci 28:1415–1423. doi: 10.3346/jkms.2013.28.10.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Storek KM, Monack DM. 2015. Bacterial recognition pathways that lead to inflammasome activation. Immunol Rev 265:112–129. doi: 10.1111/imr.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graham JG, MacDonald LJ, Hussain SK, Sharma UM, Kurten RC, Voth DE. 2013. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell Microbiol 15:1012–1025. doi: 10.1111/cmi.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schroeder GN, Jann NJ, Hilbi H. 2007. Intracellular type III secretion by cytoplasmic Shigella flexneri promotes caspase-1-dependent macrophage cell death. Microbiology 153:2862–2876. doi: 10.1099/mic.0.2007/007427-0. [DOI] [PubMed] [Google Scholar]
- 48.Yrlid U, Wick MJ. 2000. Salmonella-induced apoptosis of infected macrophages results in presentation of a bacteria-encoded antigen after uptake by bystander dendritic cells. J Exp Med 191:613–624. doi: 10.1084/jem.191.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casson CN, Shin S. 2013. Inflammasome-mediated cell death in response to bacterial pathogens that access the host cell cytosol: lessons from Legionella pneumophila. Front Cell Infect Microbiol 3:111. doi: 10.3389/fcimb.2013.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voth DE, Broederdorf LJ, Graham JG. 2012. Bacterial type IV secretion systems: versatile virulence machines. Future Microbiol 7:241–257. doi: 10.2217/fmb.11.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salaun B, Romero P, Lebecque S. 2007. Toll-like receptors' two-edged sword: when immunity meets apoptosis. Eur J Immunol 37:3311–3318. doi: 10.1002/eji.200737744. [DOI] [PubMed] [Google Scholar]
- 52.Underhill DM, Ozinsky A, Smith KD, Aderem A. 1999. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A 96:14459–14463. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ochoa-Reparaz J, Sentissi J, Trunkle T, Riccardi C, Pascual DW. 2007. Attenuated Coxiella burnetii phase II causes a febrile response in gamma interferon knockout and Toll-like receptor 2 knockout mice and protects against reinfection. Infect Immun 75:5845–5858. doi: 10.1128/IAI.00901-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. 2006. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 56.Martinon F, Burns K, Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 57.Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. 2012. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deng Q, Wang Y, Zhang Y, Li M, Li D, Huang X, Wu Y, Pu J, Wu M. 2016. Pseudomonas aeruginosa triggers macrophage autophagy to escape intracellular killing by activation of the NLRP3 inflammasome. Infect Immun 84:56–66. doi: 10.1128/IAI.00945-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cunha LD, Ribeiro JM, Fernandes TD, Massis LM, Khoo CA, Moffatt JH, Newton HJ, Roy CR, Zamboni DS. 2015. Inhibition of inflammasome activation by Coxiella burnetii type IV secretion system effector IcaA. Nat Commun 6:10205. doi: 10.1038/ncomms10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Graham JG, Winchell CG, Kurten RC, Voth DE. 2016. Development of an ex vivo tissue platform to study the human lung response to Coxiella burnetii. Infect Immun 84:1438–1445. doi: 10.1128/IAI.00012-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bauernfeind F, Ablasser A, Bartok E, Kim S, Schmid-Burgk J, Cavlar T, Hornung V. 2011. Inflammasomes: current understanding and open questions. Cell Mol Life Sci 68:765–783. doi: 10.1007/s00018-010-0567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 63.Arend WP, Palmer G, Gabay C. 2008. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x. [DOI] [PubMed] [Google Scholar]
- 64.Jones CL, Weiss DS. 2011. TLR2 signaling contributes to rapid inflammasome activation during F. novicida infection. PLoS One 6:e20609. doi: 10.1371/journal.pone.0020609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bradley WP, Boyer MA, Nguyen HT, Birdwell LD, Yu J, Ribeiro JM, Weiss SR, Zamboni DS, Roy CR, Shin S. 2016. Primary role for Toll-like receptor-driven tumor necrosis factor rather than cytosolic immune detection in restricting Coxiella burnetii phase II replication within mouse macrophages. Infect Immun 84:998–1015. doi: 10.1128/IAI.01536-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sarkar A, Hall MW, Exline M, Hart J, Knatz N, Gatson NT, Wewers MD. 2006. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1beta and interleukin-18. Am J Respir Crit Care Med 174:1003–1010. doi: 10.1164/rccm.200604-546OC. [DOI] [PMC free article] [PubMed] [Google Scholar]