

Summary
Nasu-Hakola disease (NHD) is a rare autosomal recessive disorder characterized by sclerosing leukoencephalopathy and multifocal bone cysts, caused by a loss-of-function mutation of either TYROBP (DAP12) or TREM2. TREM2 and DAP12 constitute a receptor/adaptor signaling complex expressed exclusively on osteoclasts, dendritic cells, macrophages, and microglia. Premortem molecular diagnosis of NHD requires genetic analysis of both TYROBP and TREM2, in which 20 distinct NHD-causing mutations have been reported. Due to genetic heterogeneity, it is often difficult to identify the exact mutation responsible for NHD. Recently, the revolution of the next-generation sequencing (NGS) technology has greatly advanced the field of genome research. A targeted sequencing approach allows us to investigate a selected set of disease-causing genes and mutations in a number of samples within several days. By targeted sequencing using the TruSight One Sequencing Panel, we resequenced genetic mutations of seven NHD cases with known molecular diagnosis and two control subjects. We identified homozygous variants of TYROBP or TREM2 in all NHD cases, composed of a frameshift mutation of c.141delG in exon 3 of TYROBP in four cases, a missense mutation of c.2T>C in exon 1 of TYROBP in two cases, or a splicing mutation of c.482+2T>C in intron 3 of TREM2 in one case. The results of targeted resequencing corresponded to those of Sanger sequencing. In contrast, causative variants were not detected in control subjects. These results indicate that targeted sequencing is a useful approach to precisely identify genetic mutations responsible for NHD in a comprehensive manner.
Keywords: DAP12, Nasu-Hakola disease, targeted sequencing, TREM2, TYROBP
1. Introduction
Nasu-Hakola disease (NHD), also designated polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; OMIM 221770), is a rare autosomal recessive disorder, characterized by progressive presenile dementia and formation of multifocal bone cysts (1,2). Although NHD patients are clustered in Japan and Finland, approximately 200 NHD cases are presently reported worldwide. Clinically, patients show pathological bone fractures during the third decade of life, and a frontal lobe syndrome during the fourth decade of life, followed by progressive dementia and death until the fifth decade of life (3). Pathologically, the brains of NHD patients exhibit extensive demyelination, accumulation of axonal spheroids, microglia activation and intense astrogliosis predominantly in the white matter of frontal and temporal lobes and the basal ganglia (4).
NHD is caused by various homozygous loss-of-function mutations located in one of two genes, TYRO protein tyrosine kinase-binding protein (TYROBP), alternatively named DNAX-activation protein 12 (DAP12) on chromosome 19q13.1 or triggering receptor expressed on myeloid cells 2 (TREM2) on chromosome 6p21.1 (5,6). TREM2 and DAP12 constitute a receptor/adaptor signaling complex expressed exclusively on osteoclasts, dendritic cells, macrophages, and microglia (7). Premortem diagnosis of NHD requires genetic analysis of both TYROBP and TREM2. Up to the present, a panel of 20 different NHD-causing mutations was reported (Figure 1) (8,9). Due to genetic heterogeneity, it is often difficult to identify the exact mutation responsible for NHD. Furthermore, homozygous or compound heterozygous mutations, comprised of c.40+3delAGG, p.Q33X, p.Y38C, p.T66M, p.D86V and p.W198X in TREM2, cause frontotemporal dementia (FTD)-like syndrome without bone involvement (10,11). Importantly, accumulating evidence indicates that heterogeneous variants of p.R47H and p.R62H in TREM2, confer a substantial risk for development of late-onset Alzheimer's disease (LOAD) (11,12).
Figure 1.
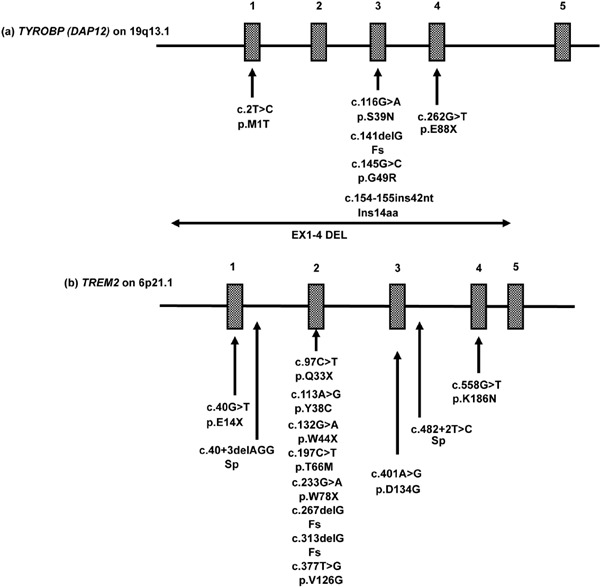
Twenty different mutations causative of NHD. NHD-causing mutations in (a) TYROBP (DAP12) or (b) TREM2 are shown with genetic and protein definitions and the position of exons.
Recently, the revolution of the next-generation sequencing (NGS) technology has had a great impact on the field of genome research. Targeted sequencing with focused gene panels, which are designed to include genomic regions causative of genetic diseases, allows us to investigate rapidly and efficiently a selected set of candidate genes in many samples (13). By using the TruSight One Sequencing Panel, we resequenced genetic mutations of seven NHD cases with known molecular diagnosis.
2. Materials and Methods
2.1. Ethics
The Human Research Ethics Committee (HREC) of the Meiji Pharmaceutical University (MPU) approved this study (No. 1904, 2015), which follows the Ethical Guidelines for Analytical Research on the Human Genome/Genes, Japan. Written informed consent was taken from all participants.
2.2. Targeted resequencing
All patients showed clinical characteristics of NHD, such as multiple bone cysts and diffuse leukoencephalopathy. Before targeted sequencing, the causative mutations were characterized by Sanger sequencing of the TYROBP or TREM2 gene. Fifty ng of genomic DNA extracted from peripheral blood mononuclear cells (PBMC) was processed for DNA library preparation by using MiSeq Reagent Kit V3 (Illumina, San Diego, CA, USA) and target enrichment by the TruSight One Sequencing Panel that contains coding regions of 4,813 genes associated with known clinical phenotypes (Illumina). The genes were selected from the Human Gene Mutation Database (HGMD; http://www.hgmd. cf.ac.uk), the Online Mendelian Inheritance in Man database (OMIM; http://omim.org/search/advanced/entry), the GeneTests database (http://www.genetests.org), the Illumina TruSight Exome content set, and other available sequencing panels. The full list of the genes is available online (http://www.illumina.com/content/dam/illumina-marketing/documents/products/gene_lists/gene_list_trusight_one.zip). The concentration of target DNA was quantified by Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). The prepared library was loaded on the Flowcell of MiSeq (Illumina) for sequencing.
2.3. Processing of sequencing data
The sequencing data, composed of 150 bp paired-end reads, were processed for MiSeq Reporter (Illumina), which is endowed with Burrows-Wheeler Aligner (BWA) for alignment on a reference sequence of hg19 and Genome Analysis Toolkit (GATK) for variant calling. Then, the Variant Call format (VCF) file was processed for analyzing on VariantStudio (Illumina), which contains annotation databases, such as the ClinVar database (ClinVar; http://www.ncbi.nlm.nih.gov/clinvar), OMIM, the dbSNP database (dbSNP; http://www.ncbi.nlm.nih.gov/SNP), the Polymorphism Phenotyping v2 database (Polyphen-2; http://genetics.bwh.harvard.edu/pph2), and the Sorting Intolerant from Tolerant database (SIFT; http://sift.jcvi.org). The genetic mutations were visualized by importing a BAM format file into Integrative Genomics Viewer (IGV, Broad Institute, Cambridge, MA, USA).
3. Results and Discussion
Variant calling data enclosed in a VCF file were filtered by VariantStudio under the condition of homozygous mutations, quality value > 1000, read depth > 30, and only variants with ClinVar annotation. Pathogenic and probably pathogenic mutations with ClinVar significance were selected. Benign mutations on PolyPhen-2 and tolerated mutations on SIFT were excluded. Finally, we were able to narrow down the set of disease causing-mutations in all NHD cases, composed of a frameshift mutation of c.141delG in exon 3 of TYROBP (14), causing premature termination at amino acid residue 52 in four cases (Figure 2), a missense mutation of c.2T>C at the initiation codon in exon 1 of TYROBP (14) in two cases (Figure 3), or a splicing mutation of c.482+2T>C at the splice-donor consensus site in intron 3 of TREM2 (15) in one case (Figure 4). The entire process was completed within a couple of days. The results of targeted resequencing corresponded completely to those of Sanger sequencing. Causative variants were not detected in control subjects.
Figure 2.
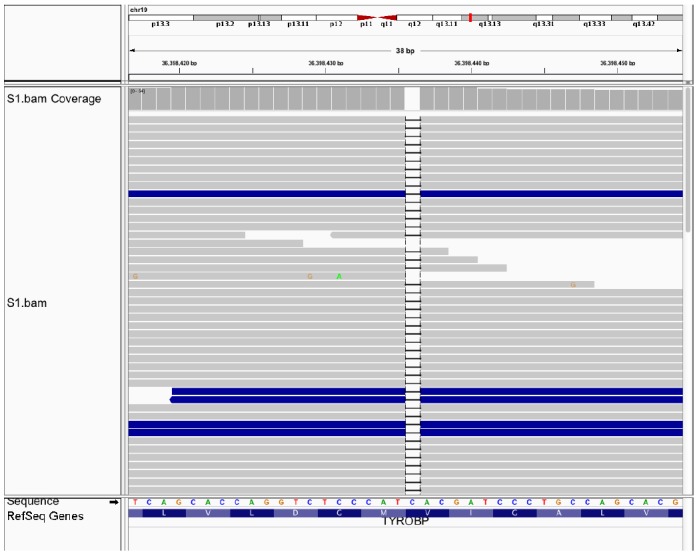
A frameshift mutation of c.141delG in exon 3 of TYROBP. By targeted resequencing, we identified a frameshift mutation of c.141delG in exon 3 of TYROBP, causing premature termination at amino acid residue 52 in four NHD cases. The genetic mutation was visualized by importing the sequence alignment data into IGV.
Figure 3.

A missense mutation of c.2T>C in exon 1 of TYROBP. By targeted resequencing, we identified a missense mutation of c.2T>C at the initiation codon in exon 1 of TYROBP in two NHD cases. The genetic mutation was visualized by importing the sequence alignment data into IGV.
Figure 4.

A splicing mutation of c.482+2T>C in intron 3 of TREM2. By targeted resequencing, we identified a splicing mutation of c.482+2T>C at the splice-donor consensus site in intron 3 of TREM2 in one NHD case. The genetic mutation was visualized by importing the sequence alignment data into IGV.
In the present study, by targeted sequencing on a commercially available focused gene panel, we resequenced TYROBP or TREM2 mutations in seven NHD cases with known molecular diagnosis. We identified genetic variants in all seven NHD cases, which were previously validated by Sanger sequencing, indicating that this approach is highly reliable and reproducible. Thus, targeted sequencing provides a rapid, convenient, high throughput, and comprehensive platform for molecular diagnosis of NHD in a clinical setting. Furthermore, targeted sequencing is capable of detecting novel unpredicted variants located in focused genes, including noncausative but disease-modifying ones. In contrast, Sanger sequencing of individual genes and exons is often laborious with low throughput, although it does not require expensive NGS machines. With respect to the disadvantage, the results of targeted sequencing are derived exclusively from a limited set of focused genes. Furthermore, this approach could not detect noncoding or deep intronic mutations and copy number variations. However, targeted sequencing produces a much smaller dataset size, compared with that of whole genome or whole exome sequencing, making interpretation of the data more concise (16).
A recent study sequenced genomic DNA of 79 patients with sporadic inclusion body myositis (sIBM) on a panel of 38 target genes, and identified 27 rare coding missense variants with those including disease-causing mutations in the valosin containing protein (VCP) gene, suggesting that targeted sequencing is a clinically meaningful approach for genetic evaluation of sIBM (17). More recently, targeted sequencing with the TruSight One sequencing panel, causative mutations were found in six of 17 families of pediatric patients with various genetic diseases, such as Sotos syndrome, Joubert syndrome, and neurofibromatosis type 1A (16). In different studies, targeted sequencing with the TruSight One Sequencing Panel showed somatic mosaicism of a disease-causing mutation in a patient with CDKL5-related encephalopathy (18), and served as a prenatal diagnostic tool for LAMA2-related muscular dystrophy (19). Furthermore, targeted sequencing with the TruSight One Sequencing Panel identified three distinct rare homozygous and compound heterozygous variants causative of nephronophthisis-related ciliopathy (NPHP-RC), an autosomal recessive cystic kidney disease, in three patients (20).
In conclusion, targeted sequencing is a useful approach for the exact molecular diagnosis of rare Mendelian diseases, including NHD, by investigating genetic mutations in a comprehensive manner.
Acknowledgements
This work was supported by grants from the Research on Intractable Diseases, entitled “Clinicopathological and genetic studies of Nasu-Hakola disease” (H21-Nanchi-Ippan-201; H22-Nanchi-Ippan-136), the Ministry of Health, Labour and Welfare of Japan, and the JSPS KAKENHI (C25430054 and 16K07043) and the Dementia Drug Development Research Center (DRC) project (S1511016), the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. We thank Ms. Shiho Saito for technical assistance.
References
- 1. Nasu T, Tsukahara Y, Terayama K. A lipid metabolic disease - “Membranous lipodystrophy” - an autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Acta Pathol Jpn. 1973; 23: 539-558. [DOI] [PubMed] [Google Scholar]
- 2. Hakola HP. Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr Scand Suppl. 1972; 232: 1-173. [PubMed] [Google Scholar]
- 3. Bianchin MM, Capella HM, Chaves DL, Steindel M, Grisard EC, Ganev GG, da Silva Júnior JP, Neto Evaldo S, Poffo MA, Walz R, Carlotti Júnior CG, Sakamoto AC. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy - PLOSL): A dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol. 2004; 24: 1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Satoh J, Tabunoki H, Ishida T, Yagishita S, Jinnai K, Futamura N, Kobayashi M, Toyoshima I, Yoshioka T, Enomoto K, Arai N, Arima K. Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology. 2011; 31: 363-375. [DOI] [PubMed] [Google Scholar]
- 5. Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002; 71: 656-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klünemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE, Peltonen L, Paloneva J. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. 2005; 64: 1502-1507. [DOI] [PubMed] [Google Scholar]
- 7. Colonna M, Wang Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci. 2016; 17: 201-207. [DOI] [PubMed] [Google Scholar]
- 8. Xing J, Titus AR, Humphery MB. The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: A molecular genetics perspective. Res Rep Biochem. 2015; 5: 89-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walter J. The triggering receptor expressed on myeloid cells 2: A molecular link of neuroinflammation and neurodegenerative diseases. J Biol Chem. 2016; 291: 4334-4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chouery E, Delague V, Bergougnoux A, Koussa S, Serre JL. Mégarbané A. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum Mutat. 2008; 29: E194-204. [DOI] [PubMed] [Google Scholar]
- 11. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013; 368: 117-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013; 368: 107-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zemojtel T, Köhler S, Mackenroth L, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Transl Med. 2014; 6: 252ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kondo T, Takahashi K, Kohara N, Takahashi Y, Hayashi S, Takahashi H, Matsuo H, Yamazaki M, Inoue K, Miyamoto K, Yamamura T. Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): Three genetic forms. Neurology. 2002; 59: 1105-1107. [DOI] [PubMed] [Google Scholar]
- 15. Numasawa Y, Yamaura C, Ishihara S, Shintani S, Yamazaki M, Tabunoki H, Satoh JI. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur J Neurol. 2011; 18: 1179-1183. [DOI] [PubMed] [Google Scholar]
- 16. Okazaki T, Murata M, Kai M, Adachi K, Nakagawa N, Kasagi N, Matsumura W, Maegaki Y, Nanba E. Clinical diagnosis of Mendelian disorders using a comprehensive gene-targeted panel test for next-generation sequencing. Yonago Acta Med. 2016; 59: 118-125. [PMC free article] [PubMed] [Google Scholar]
- 17. Weihl CC, Baloh RH, Lee Y, Chou TF, Pittman SK, Lopate G, Allred P, Jockel-Balsarotti J, Pestronk A, Harms MB. Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromuscul Disord. 2015; 25: 289-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kato T, Morisada N, Nagase H, Nishiyama M, Toyoshima D, Nakagawa T, Maruyama A, Fu XJ, Nozu K, Wada H, Takada S, Iijima K. Somatic mosaicism of a CDKL5 mutation identified by next-generation sequencing. Brain Dev. 2015; 37: 911-915. [DOI] [PubMed] [Google Scholar]
- 19. Russo CD, Di Giacomo G, Cignini P, Padula F, Mangiafico L, Mesoraca A, D'Emidio L, McCluskey MR, Paganelli A, Giorlandino C. Comparative study of aCGH and Next Generation Sequencing (NGS) for chromosomal microdeletion and microduplication screening. J Prenat Med. 2014; 8: 57-69. [PMC free article] [PubMed] [Google Scholar]
- 20. Yamamura T, Morisada N, Nozu K, Minamikawa S, Ishimori S, Toyoshima D, Ninchoji T, Yasui M, Taniguchi-Ikeda M, Morioka I, Nakanishi K, Nishio H, Iijima K. Rare renal ciliopathies in non-consanguineous families that were identified by targeted resequencing. Clin Exp Nephrol. 2016; DOI: 10.1007/s10157-016-1256-x. [DOI] [PubMed] [Google Scholar]