

Summary
The superoxide-producing nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex of phagocytes (phox) plays a key role in production of reactive oxygen species (ROS) by microglia. The catalytic subunits of the NADPH oxidase are composed of p22phox and gp91phox. Nasu-Hakola disease (NHD) is a rare autosomal recessive disorder caused by a loss-of-function mutation of either TYROBP (DAP12) or TREM2. Pathologically, the brains of NHD patients exhibit extensive demyelination designated leukoencephalopathy, astrogliosis, accumulation of axonal spheroids, and remarkable activation of microglia predominantly in the white matter of frontal and temporal lobes. However, a pathological role of the gp91phox-p22phox complex in generation of leukoencephalopathy in NHD remains unknown. We clarified the expression of gp91phox and p22phox in the white matter of the frontal cortex derived from five NHD and eight control subjects. We identified the expression of p22phox and gp91phox immunoreactivity almost exclusively on microglia. Microglia overexpressed gp91phox in NHD brains and p22phox in myotonic dystrophy (MD) brains, when compared with non-neurological control (NC) brains. These results suggest that the enhanced expression of gp91phox by microglia might contribute to overproduction of ROS highly toxic to myelinating oligodendrocytes, resulting in oligodendrocyte cell death that induces leukoencephalopathy in NHD brains.
Keywords: gp91phox, leukoencephalopathy, microglia, Nasu-Hakola disease, p22phox
1. Introduction
The superoxide-producing nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) complex of phagocytes (phox) plays a key role in the elimination of invading pathogens by phagocytic cells (1). The core catalytic enzymes called flavocytochrome b558 are composed of a membrane-associated heterodimeric complex, consisting of p22phox (α subunit) and gp91phox (Nox2, β subunit) (2). Upon stimulation of phagocytes, regulatory subunits, including p40phox, p47phox, p67phox, and Rac1/2, are activated by posttranslational modification, conformational change, and protein-protein interaction, and translocates from the cytosol to the membrane, resulting in activation of the gp91phox-p22phox complex (2). Following the assembly of the oxidase complex, the flavocytochrome b558 transports electrons from intracellular NADPH to extracellular or phagosomal oxygen, leading to production of superoxide anion. Superoxide anion is short lived and reacts with various molecules to form reactive oxygen species (ROS), such as hydrogen peroxide, hydroxyl radicals, and hypochlorous acid, and peroxynitrite (1). An excessive amount of ROS causes oxidative stress and cell death by inducing oxidative modification of lipids, proteins, and nucleic acids, whereas a low level of ROS play a key role in redox-dependent intracellular signaling that amplifies the proinflammatory response (3,4).
Microglia, the resident myeloid cells in the central nervous system (CNS), have a capacity to constantly scavenge invading pathogens, apoptotic debris, unwanted synapses, and aggregated proteins by sensing them with a panel of pattern recognition receptors (PRRs) (5). Microglia serve as a major source of ROS through the expression of the phagocytic Nox in the lesions of cerebral ischemia, traumatic brain injury, multiple sclerosis (MS), and Alzheimer's disease (6–9). In these diseases, the microglial gp91phox-p22phox complex is promptly activated in response to neurotoxic stimuli, followed by ROS-mediated neuronal damage (3).
Nasu-Hakola disease (NHD), also designated polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; OMIM 221770), is a rare autosomal recessive disorder, characterized by progressive presenile dementia and formation of multifocal bone cysts, caused by genetic mutations of either TYROBP(DAP12) or TREM2 (10). TREM2 and DAP12 constitute a receptor/adaptor signaling complex expressed exclusively on osteoclasts, dendritic cells, macrophages, and microglia. Although NHD patients are clustered in Japan and Finland, approximately 200 NHD cases are presently reported worldwide. Clinically, the patients with NHD show recurrent bone fractures during the third decade of life, and a frontal lobe syndrome during the fourth decade of life, and progressive dementia and death until the fifth decade of life (11). Pathologically, the brains of NHD patients exhibit extensive demyelination designated leukoencephalopathy, astrogliosis, accumulation of axonal spheroids, and remarkable activation of microglia predominantly in the white matter of frontal and temporal lobes and the basal ganglia (12). However, a pathological role of the gp91phox-p22phox complex with relevance to leukoencephalopathy in NHD remains largely unknown. In the present study, we have attempted to clarify the expression of gp91phox and p22phox in the white matter lesions of NHD brains by immunohistochemistry.
2. Materials and Methods
2.1. Human brain tissues
The brain autopsies were performed at the National Center Hospital, National Center of Neurology and Psychiatry (NCNP), Japan, Kohnodai Hospital, National Center for Global Health and Medicine (NCGM), Japan, and affiliated hospitals of Research Resource Network (RRN), Japan. The comprehensive examination by established neuropathologists (YS and TI) validated the pathological diagnosis. In all cases, written informed consent was obtained. The Ethics Committee of the NCNP for the Human Brain Research, the Ethics Committee of the NCGM on the Research Use of Human Samples, and the Human Research Ethics Committee (HREC) of the Meiji Pharmaceutical University (MPU) approved the present study.
For immunohistochemical studies, serial sections containing the white matter of the frontal cortex were prepared from four subjects who died of non-neurological causes (NC), composed of a 63-year-old man who died of prostate cancer and acute myocardial infarction (NC1), a 67-year-old man who died of dissecting aortic aneurysm (NC2), a 57-year-old man who died of alcoholic liver cirrhosis (NC3), and a 61-year-old man who died of rheumatoid arthritis with interstitial pneumonia (NC4), four neuropsychiatric disease controls affected with myotonic dystrophy (MD), composed of a 68-year-old man (MD1), a 61-year-old man (MD2), a 60-year-old man (MD3), and a 53-year-old woman (MD4), and five NHD patients, composed of a 42-year-old man (NHD1), a 48-year-old woman (NHD2), a 44-year-old man (NHD3), a 32-year-old woman (NHD4), and a 38-year-old man (NHD5). The homozygous mutation of a single base deletion of 141G (141delG) in exon 3 of DAP12 was identified in NHD1, NHD2, and NHD5, while the genetic analysis was not performed in NHD3 or NHD4, as described previously (13).
2.2. Immunohistochemistry
After deparaffination, tissue sections were heated in 10 mM citrate sodium buffer, pH 6.0 by autoclave at 110°C for 15 min in a temperature-controlled pressure chamber (Biocare Medical, Concord, CA, USA). They were treated at room temperature (RT) for 15 min with 3% hydrogen peroxide-containing methanol to block the endogenous peroxidase activity. They were then incubated with phosphate-buffered saline (PBS) containing 10% normal goat serum at RT for 15 min to block non-specific staining, followed by incubation in a moist chamber at 4°C overnight with rabbit polyclonal anti-p22phox antibody (sc-20781, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or with mouse monoclonal anti-gp91phox antibody (ab139371, Abcam, Cambridge, UK). After washing with PBS, tissue sections were incubated at RT for 30 min with horseradish peroxidase (HRP)-conjugated secondary antibodies (Nichirei, Tokyo, Japan), followed by incubation with diaminobenzidine tetrahydrochloride (DAB) substrate (Vector, Burlingame, CA, USA). They were processed for a counterstain with hematoxylin. Negative controls underwent all the steps except for exposure to primary antibody. In limited experiments, double immunolabeling of mouse anti-gp91phox antibody and rabbit antibodies against p22phox, Iba1 (Wako Pure Chemical, Tokyo, Japan; a marker of microglia/macrophages), GFAP (Dako, Tokyo, Japan; a marker of astrocytes), or NeuN (Abcam; a marker of neurons), was performed, followed by incubation with HRP-conjugated or alkaline phosphatase-conjugated anti-mouse or anti-rabbit secondary antibody and exposure to DAB substrate and Warp Red chromogen (Biocare Medical).
2.3. Quantification of gp91phox and p22phox immunoreactivity
To quantify immunolabeled areas, the images derived from three fields of the white matter were captured at a 200 X magnification on the Olympus BX51 universal microscope. They were then processed for quantification by using ImageJ software (National Institute of Health, Bethesda, MD, USA). The gp91phox- or p22phox-immunolabeled area was calibrated by the Iba1-immunolabeled area of the identical field. The difference in the average of immunopositive areas between NHD and the controls was evaluated statistically by one-way analysis of variance (ANOVA) followed by post-hoc Tukey's test.
3. Results and Discussion
By immunohistochemistry, we identified the intense expression of gp91phox and p22phox immunoreactivity chiefly on microglia/macrophages in the white matter of the frontal cortex derived from NC, MD and NHD subjects (Figure 1, panels a–d and Figure 2, panels a–d). By double immunolabeling, the expression pattern of gp91phox overlapped with that of p22phox (Figure 3, panel a) and Iba1-positive microglia expressed gp91phox (Figure 3, panel b), whereas myelinating oligodendrocytes, GFAP-positive astrocytes and NeuN-positive neurons did not express gp91phox (Figure 3, panels c, d), suggesting that oligodendrocytes, astrocytes and neurons do not express discernible levels of the gp91phox-p22phox complex in the human brain. By quantitative analysis, the immunolabeled area of gp91phox-positive microglia was increased significantly in NHD brains, when compared with NC brains but not with MD brains (Figure 4, panel a; p = 0.0338). In contrast, the immunolabeled area of p22phox-positive microglia was not significantly different between NC and NHD brains, whereas it was increased significantly in MD brains, when compared with NC brains (Figure 4, panel b; p = 0.0096).
Figure 1.
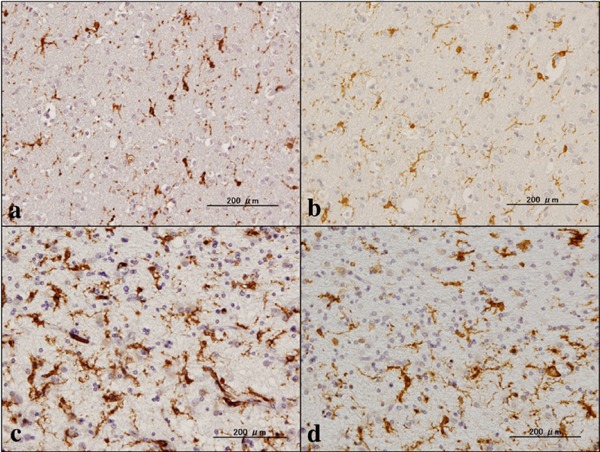
Expression of gp91phox and Iba1. (a) NC, gp91phox, (b) NC, the same area of (a), Iba1, (c) NHD, gp91phox, and (d) NHD, the same area of (c), Iba1.
Figure 2.
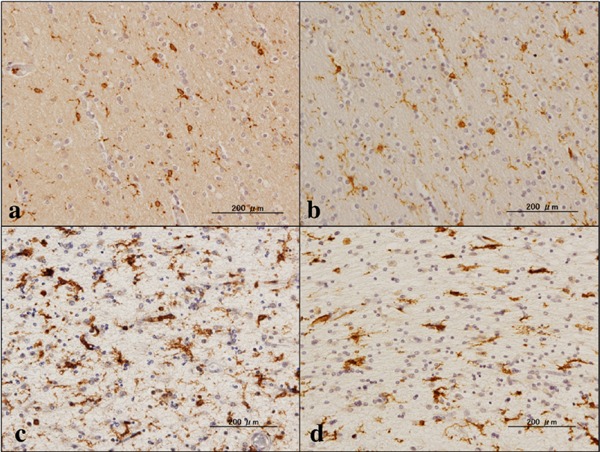
Expression of p22phox and Iba1. (a) NC, p22phox, (b) NC, the same area of (a), Iba1, (c) NHD, p22phox, and (d) NHD, the same area of (c), Iba1.
Figure 3.
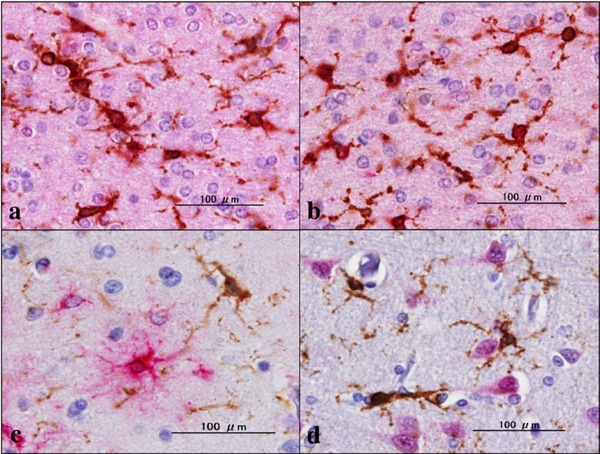
Double labeling of gp91Phox and cell type-specific markers. (a) MD, gp91phox (brown), p22phox (red), (b) MD, gp91phox (brown), Iba1 (red), (c) MD, g91phox (brown), GFAP (red), and (d) gp91phox (brown), NeuN (red).
Figure 4.
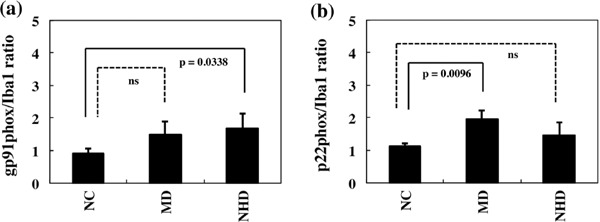
Quantitative analysis of gp91phox and p22phox expression on microglia in NC, MD, and NHD brains. (a) The ratio of immunolabeled area of gp91phox/Iba1, and (b) The ratio of immunolabeled area of p22phox/Iba1.
The enhanced expression of the gp91phox-p22phox complex plays a key role in microglial production of superoxide anion and ROS (3,7). We previously found accumulation of numerous Iba1-positive microglia in NHD brains and showed that mRNA levels of microglia/macrophage marker genes, such as CD68, CD163, and MSR1, were elevated in NHS brains (12,14). The present study indicated that microglia overexpressed gp91phox in NHD brains and p22phox in MD brains, when compared with NC brains. The elevation of p22phox expression levels in MD brains is attributable to a high prevalence of white matter damage in this disease (15). More importantly, our observations raise a possible scenario that the enhanced expression of gp91phox in NHD brains contributes to overproduction of ROS highly toxic to myelinating oligodendrocytes (16,17), resulting in oligodendrocyte cell death, followed by development of leukoencephalopathy. Supporting this, previous studies showed that the expression of gp91phox and p22phox was upregulated in activated microglia accumulating in initial lesions of MS, an inflammatory demyelinating disease in the CNS, suggesting a role of oxidative tissue damage in progression of demyelination (8,18).
Notably, a population of activated microglia expressed gp91 in the penumbra of transient cerebral ischemia (6). The expression of gp91 was upregulated in ameboid microglia, which generate superoxide anion and peroxynitrite, distributed in the pericontusional area following traumatic brain injury (7). Microglia play a key role in lipopolysaccharide (LPS)-induced dopaminergic cell death in a manner dependent on the expression of gp91phox (19). All of these observations suggest that microglia contain a functionally active NADPH oxidase capable of generating ROS. Microglia, promptly activated by proinflammatory cytokines, disease-associated proteins, and environmental toxins, express high levels of gp91phox and produce a large amount of ROS that cause oxidative stress leading to neuronal and myelin damage (3,16,17).
In conclusion, the enhanced expression of gp91phox on microglia, a central component of the NADPH oxidase, might be involved in development of leukoencephalopathy in NHD brains via myelin damage caused by superoxide anion-inducible ROS.
Acknowledgements
The authors thank Drs. Kenji Jinnai, Nobutaka Arai, Kiyotaka Nakamagoe, Nobutaka Motohashi, and Saburo Yagishita for providing us brain samples. This work was supported by grants from the Research on Intractable Diseases, entitled “Clinicopathological and genetic studies of Nasu-Hakola disease” (H21-Nanchi-Ippan-201; H22-Nanchi-Ippan-136), the Ministry of Health, Labour and Welfare of Japan, and grants from the JSPS KAKENHI (C25430054 and 16K07043) and the Dementia Drug Development Research Center (DRC) project (S1511016), the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
References
- 1. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. 2007; 87:245-313. [DOI] [PubMed] [Google Scholar]
- 2. Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med. 2014; 76:208-226. [DOI] [PubMed] [Google Scholar]
- 3. Surace MJ, Block ML. Targeting microglia-mediated neurotoxicity: The potential of NOX2 inhibitors. Cell Mol Life Sci. 2012; 69:2409-2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr Opin Cell Biol. 2015; 33:8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol. 2014; 88:594-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Green SP, Cairns B, Rae J, Errett-Baroncini C, Hongo JA, Erickson RW, Curnutte JT. Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J Cereb Blood Flow Metab. 2001; 21:374-384. [DOI] [PubMed] [Google Scholar]
- 7. Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation. 2010; 7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, Lassmann H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. 2012; 135:886-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilkinson BL, Landreth GE. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer's disease. J Neuroinflammation. 2006; 3:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klünemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE, Peltonen L, Paloneva J. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. 2005; 64:1502-1507. [DOI] [PubMed] [Google Scholar]
- 11. Bianchin MM, Capella HM, Chaves DL, Steindel M, Grisard EC, Ganev GG, da Silva JP, Júnior, Neto Evaldo S, Poffo MA, Walz R, Carlotti CG, Júnior, Sakamoto AC. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy - PLOSL): A dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol. 2004; 24:1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Satoh J, Tabunoki H, Ishida T, Yagishita S, Jinnai K, Futamura N, Kobayashi M, Toyoshima I, Yoshioka T, Enomoto K, Arai N, Arima K. Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology. 2011; 31:363-375. [DOI] [PubMed] [Google Scholar]
- 13. Satoh J, Motohashi N, Kino Y, Ishida T, Yagishita S, Jinnai K, Arai N, Nakamagoe K, Tamaoka A, Saito Y, Arima K. LC3, an autophagosome marker, is expressed on oligodendrocytes in Nasu-Hakola disease brains. Orphanet J Rare Dis. 2014; 9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Numasawa Y, Yamaura C, Ishihara S, Shintani S, Yamazaki M, Tabunoki H, Satoh JI. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur J Neurol. 2011; 18:1179-1183. [DOI] [PubMed] [Google Scholar]
- 15. Itoh K, Mitani M, Kawamoto K, Futamura N, Funakawa I, Jinnai K, Fushiki S. Neuropathology does not correlate with regional differences in the extent of expansion of CTG repeats in the brain with myotonic dystrophy type 1. Acta Histochem Cytochem. 2010; 43:149-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu J, Tian D, Murugan M, Eyo UB, Dreyfus CF, Wang W, Wu LJ. Microglial Hv1 proton channel promotes cuprizone-induced demyelination through oxidative damage. J Neurochem. 2015; 135:347-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roth AD, Núñez MT. Oligodendrocytes: Functioning in a delicate balance between high metabolic requirements and oxidative damage. Adv Exp Med Biol. 2016; 949:167-181. [DOI] [PubMed] [Google Scholar]
- 18. van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L, Gerritsen W, Kooi EJ, Witte ME, Geurts JJ, de Vries HE, Peferoen-Baert R, van den Elsen PJ, van der Valk P, Amor S. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflammation. 2012; 9:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004; 279:1415-1421. [DOI] [PubMed] [Google Scholar]