

Summary
Metachromatic leukodystrophy (MLD) is a lysosomal storage disease caused by Arylsulfatase A (ASA) deficiency. The hallmark of the disease is central and peripheral neurodegeneration. More than 200 mutations have been identified in ARSA gene so far. Some of these mutations were characterized. The aim of this study is to reinforce genotype-phenotype correlation and to understand the effect of mutations on the enzyme by biochemical characterization. Two missense mutations (c.919G→A, p.307Glu→Lys and c.954G→T, p.318Trp→Cys in exon 5) were constructed on WT-ASA cDNA and were confirmed by DNA sequence analysis. Plasmid DNA carrying mutant or normal ASA cDNA was transferred to Chinese Hamster Ovary (CHO) cells through transient transfection. ASA protein was produced by CHO cells. Hexosaminidase beta-subunit gene was cotransfected into the CHO cells as a control gene of transfection efficiency. 48 hours after transfection, cells were collected and homogenized. ASA and hexosaminidase activities were measured in supernatant. ASA enzyme activity is decreased 100% according to the control by the effect of both mutations. The mutations are located in the higly conserved region of the protein. In this study, we showed that both mutations result in null ASA activity in CHO cells making the protein nonfunctional. We confirmed that p.307Glu→Lys and p.318Trp→Cys mutations cause late infantile form of MLD disease.
Keywords: Missense mutations, in vitro mutagenesis, transfection, CHO cells, genotype-phenotype correlation
1. Introduction
Metachromatic leukodystrophy (MLD) is an autosomal recessive sphingolipid storage disease that occurs as a result of deficiency of lysosomal Arylsulfatase A (ASA) or its activator protein. Its frequency is estimated to be 1 in 69,890 newborns in Turkey (1) and 1 in 40,000 newborns in Northern Sweden (2).
ASA catalyses the desulfation of sphingolipid sulfatide. Sulfatide is found in high concentrations in myelin sheat of the nervous system. As a result of ASA deficiency, sulfatide is accumulated in the nervous system and affects the oligodendrocytes and results in neurodegeneration. Sulfatide is also stored in various organs such as liver and kidney. According to the starting age MLD shows three clinical phenotypes: late infantile, juvenile and adult. Clinically, the most severe type is late infantile type. It starts around 2-year-old and ends in early childhood with death. Juvenile and adult types start between 4–16-year-old and any age after puberty. ASA can be 5–15% of reference value in some patients with pseudodeficiency which can lead to misdiagnosis. DNA-based methods for pseudodeficiency allele and mutation detection and activator protein detection can be useful for further diagnosis of MLD types (3).
To date 200 mutations have been described in the ARSA gene (4). Most of them are missense mutations. A few of these mutations occur only with high frequency. There is a genotype-phenotype correlation in MLD. Patients with late infantile phenotype usually have homozygous mutations. Mutations result in very low level of enzyme activity. However, patients with juvenile phenotype usually have one severe allele and one mild allele combinations and have some residual enzyme activity. Adult patients are frequently homozygous for alleles expressing residual enzyme activities. Severity of the disease correlates inversely with the residual enzyme activity (3).
In this study we constructed two missense (in exon 5 c.919G→A, p.307Glu→Lys and c.954G→T, p.318Trp→Cys) mutations by site-directed mutagenesis on wild-type ARSA gene, transiently transfected to the CHO cells, and characterized biochemically.
2. Materials and Methods
2.1. Materials
Cell culture media were obtained from Gibco (Germany). Taq DNA polymerase, oligonucleotides and restriction enzymes were purchased from Sigma Chemical Co. (Germany). DH5-alpha cells were purchased from Invitrogene (Germany), Quickchange site-directed mutagenesis kit was purchased from Qiagen (Germany). Wild-type ASA plasmid was kindly supplied by Prof. Dr. Volkmar Gieselmann (Bonn University). Other reagents were from Sigma and Merck.
2.2. In vitro mutagenesis, amplification of ARSA genes and DNA sequencing
In vitro mutagenesis was performed according to the protocol of QuickChange site-directed mutagenesis kit on the wild-type ARSA gene. The sequences of the oligonucleotides used for the introduction of the mutations were: c.919G→A, p.307Glu→Lys, 5′ GA AAGGGAACGACCTACAAGGGCGGTGTCCGAG AG 3′ and c.954G→T, p.318Trp→Cys, 5′ CTGCCTTG GCCTTCTGTCCAGGTCATATCGCTC 3′. Mutations were confirmed by DNA sequencing.
2.3. Cell culture and transfection
Chinese hamster ovary (CHO) cells were grown in Dulbecco's Modified Eagles Medium (DMEM) with 1% glutamine and 10% fetal calf serum (FCS) at 37°C in 5% CO2. Four µg of vector was transfected to the CHO cells (40% confluent) using Superfect Transfection Reagent (Qiagen). After 48 h of transfection the medium was discarded, the cells were washed 3×, scraped and centrifuged. Cell pellet dissolved in 100 µL Tris-HCl pH 7,8 and were mixed with protease inhibitor mix and immediately lysed by freezing and thawing 3X in liquid nitrogen. Then supernatants were used for protein measurement by bicinconinic acid method.
2.4. Enzyme analysis
Hexosaminidase and Arylsulfatase A activities were measured according to the protocol described before (5,6) by using fluorometric substrate 4MUG and spectrophotometric substrate p-nitrocathecol sulfate. As a control, β-hexosaminidase activity was assayed and used to correct variations in transfection efficiencies.
3. Results and Discussion
Metachromatic leukodystrophy is common in Turkey among patient with sphingolipid storage disease. Minimum calculated incidence is 1.43/100,000 live births in Turkey (1). Worldwide incidence of MLD is 1/40,000 to 1/160,000 (7). Previously we reported two ASA mutations causing late infantile MLD in two different Turkish patients (8): (a) a c.919G→A transition in exon 5 causing a p.307Glu→Lys and (b) a c.954G→T transition in exon 5 causing a p.318Trp→Cys. The patients were homozygotes for the mutations. Confirmation of the mutations' effect by in vitro mutagenesis and reinforcement genotype-phenotype correlation are important for using those mutations in prenatal diagnosis. It is also important for understanding the functional domains of ASA protein and underlying mechanism of the disease. Here we analyzed the effect of these mutations on the function of the protein in CHO cells. Two missense mutations (8) were created on ASA cDNA constructs by site-directed mutagenesis, and their DNA sequence confirmed using the automatic sequence analyzer from Applied Biosystem. CHO cells were transiently transfected with each of the cDNAs, collected after 48 h, mixed with protease inhibitors, lysed and then subjected to enzyme and protein analysis. Enzyme activities were measured in supernatants of two independent transfection experiments and the results were expressed as nmol/hr/mg pr. ASA activity was 2 folds of mock transfected cells in WT-ASA cDNA transfected CHO cells. In mutant ASA cDNA transfected CHO cells ASA activity was almost equal to the activity measured in mock transfected cells. On the other hand, lysates of both mutant transfected CHO cells contained only background ASA activity (the same activity with the mock transfected). The human Hex activity was found to be 1.8–2 times higher in transfected cells than in mock transfected cells and activities were found to be in the same range in all transfected cells. Activity measurements obtained from two independent transfection experiments were calculated as mean values, subtracted from mock transfected cells' value, and expressed as % activity of the normal control. ASA activity was found deficient in both mutant protein containing samples according to the control (Table 1). We found that 307Glu→Lys and 318Trp→Cys missense mutations impairs enzyme activity. On the other hand, in wild-type transfected CHO cells two-fold ASA activity was observed.
Table 1. Verification of ASA activities in transiently transfected CHO cells expressing two mutant ARSA gene as observed in Turkish late-infantile Metachromatic Leukodystrophy patients.
| ARSA gene expressed | Clinical phenotype | ASA activity* (nmol/hr/mg protein) | ASA activity** (% of wild type) |
|---|---|---|---|
| Wild-type | Positive control | 215 (n = 2) | 100 |
| c.919G→A, p.307Glu→Lys | Late infantile | 0 (n = 2) | 0 |
| c.954G→T, p.318Trp→Cys | Late infantile | 0 (n = 2) | 0 |
Mean of two independent transfection experiments.
100% of ASA activity equals the activity found in wild type. Each enzyme activity value subtracted from mock transfected activity. Enzyme activities are calculated as % activity of wild-type activity.
Secondary structure of ASA is very well-defined (9). It has a mixture of alpha-helix and beta-plated structure (26% α-helix and 310 helix, 16% β-sheet, 46% β and 310 turn and 12% primary structure). Center of the enzyme contains two significant β-sheets: minor and major. The minor includes 4 anti-parallel β-strands and the major includes 10 different beta strands. The major β-pleated sheet is located between A, D and E helices on one side and B, C, G and H helices on the other side. The short helix F connects anti parallel β11 and β12 (Figure 1). Major and minor β sheets linked by hydrogen bonding and a disulfide bond between Cys300-Cys414 (9,10). 919 G→A mutation in exon 5 of ASA gene leads to a substitution of a basic amino acid lysine into an acidic amino acid glutamate (p.307Glu→Lys) in the protein. Glu307 is a member of F helix (Figure 1) and it is located in a highly conserved region of human sulfatases. F helix connects the anti parallel beta sheets. The substitution of a basic amino acid (Lys) into an acidic amino acid (Glu) may result in changes in interaction of beta-sheets in the active center, cause deficient enzyme activity both in patients (8) and in vitro by probably disrupting the alpha-helix structure of F helix (Figure 1). ASA activity was found 0% of wild transfected in mutant-protein expressing CHO cells. Different mutations have been identified at the adjacent amino acids involved in alpha-helical structure in the literature. All of those mutations are caused late infantile type of MLD. Enzyme activity was found completely deficient in transiently transfected COS-1 cells carrying p.308Gly→Val substitution which is found in European and Japanese patients (11). On the other hand, enzyme activity was found 13% of control in in vitro mutagenesis study of p.309Gly→Ser mutation. Protein found to have entered to the lysosome but unstabile (12).
Figure 1.
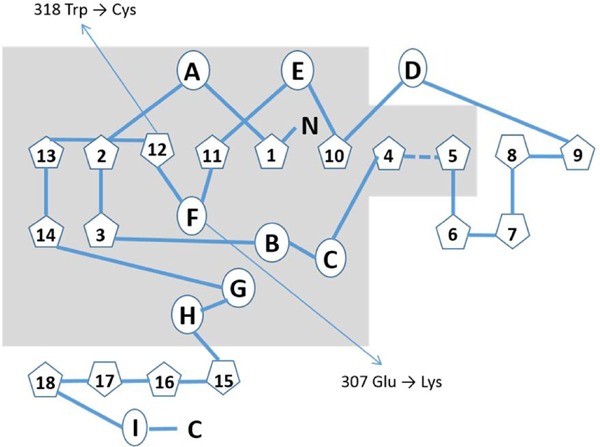
Simplified schematic represantation of the secondary structure of Arylsulfatase A modified from Lukatela G. et al., 1998 (9).  indicates alpha helices (A to I) and
indicates alpha helices (A to I) and  indicates beta-structures (1 to 18). Grey area shows the higly conserved structural elements among human sulfatases. The locations of two Turkish mutations are indicated with arrows.
indicates beta-structures (1 to 18). Grey area shows the higly conserved structural elements among human sulfatases. The locations of two Turkish mutations are indicated with arrows.
c.954 G→T mutation in ASA gene causes p.318Trp→Cys substitution. Instead of a hydrophobic amino acid tryptophan, SH group containing uncharged polar amino acid cysteine is located in the protein. 318Trp is the member of primary structure between beta-sheet 12 and anti-parallel beta-sheet 13 (Figure 1). ASA protein has 15 cysteine amino acids. 12 of them make disulfide bridges. One disulfide bond combines 2 beta-sheets, 2 stabilize the hairpin structure of beta6, and beta 7, 3 makes the node structure of 20 amino acids at the C-terminal. This 6 cysteine clusters are special and specific for ASA, the rest 3 are single. Cys38 and Cys294 are located at the intermediate surface of homodimer, Cys69 is converted to formylglycine after synthesis of the protein and Fgly69 is the active site amino acid of the protein. p.318Trp→Cys substitution creates a new Cys residue in the secondary structure of the protein. Deficient enzyme activity both in patient's samples and transfected CHO cells, show that p.318Trp→Cys substitution makes important structural changes. This effect can be a new disulfide bond with one of the free Cys residues or active site amino acid Cys prior to posttranslational modification to formyl glycine. The clinical phenotype of the patient is late infantile (8). Defect in the posttranslational modification of the Cys69 leads to deficiency of all sulfatases. Sulfate binding amino acid is FGly69, but Lys123, Ser150, His229 and Cys69 are the contributing amino acid residues for sulfate binding at the active center of the enzyme (9,10).
As a conclusion, p.307Glu→Lys and p.318Trp→Cys substitutions in the ASA protein affect enzyme activity and cause late-infantile type MLD. Biochemical characterization of the disease causing mutations is important for correlating genotype and phenotype, for understanding molecular basis of the disease, for prenatal diagnostic testing and for developing new therapeutic strategies like chaperone therapy.
Acknowledgements
Authors thank Prof. Dr. Volkmar Gieselmann for his gift of WT ASA plasmid. This study is supported by Hacettepe University Scientific Research Project Coordination Unit Grant number 727.
References
- 1. Ozkara HA, Topcu M. Sphingolipidoses in Turkey. Brain Dev 2004; 26:363-366. [DOI] [PubMed] [Google Scholar]
- 2. Gustavson KH, Hagberg B. The incidence and genetics of metachromatic leukodystrophy in Northern Sweden. Acta Pediatr Scand. 1971; 60:585-590. [DOI] [PubMed] [Google Scholar]
- 3. von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrophy. In: The metabolic and molecular bases of inherited disease (Scriver CR, Beaudet AL, Sly WS, Valle D, eds). McGraw-Hill, New York, USA, 2001; pp. 3695-3724. [Google Scholar]
- 4. Cesani M, Lorioli L, Grossi S, Amico G, Fumagalli F, Spiga I, Filocamo M, Biffi A. Mutation update of ARSA and PSAP genes causing metachromatic leukodystrophy. Hum Mutat. 2016; 37:16-27. [DOI] [PubMed] [Google Scholar]
- 5. Suzuki K. Enzymatic diagnosis of sphingolipidoses. Methods Enzymol. 1987; 138:727-762. [DOI] [PubMed] [Google Scholar]
- 6. Ozkara HA, Sandhoff K. Characterization of two Turkish beta-hexosaminidase mutations causing Tay-Sachs disease. Brain Dev. 2003; 25:191-194. [DOI] [PubMed] [Google Scholar]
- 7. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999; 281:249-254. [DOI] [PubMed] [Google Scholar]
- 8. Onder E, Sinici I, Müjgan Sönmez F, Topçu M, Ozkara HA. Identification of two novel arylsulfatase A mutations with a polymorphism as a cause of metachromatic leukodystrophy. Neurol Res. 2009; 31:60-66. [DOI] [PubMed] [Google Scholar]
- 9. Lukatela G, Krauss N, Theis K, Selmer T, Gieselmann V, von Figura K, Saenger W. Crystal structure of human arylsulfatase A: The aldehyde function and the metal ion at the active site suggest a novel mechanism for sulfate ester hydrolysis. Biochemistry. 1998; 37:3654-3664. [DOI] [PubMed] [Google Scholar]
- 10. von Bulow R, Schmidt B, Dierks T, von Figura K, Uson I. Crystal structure of an enzyme-substrate complex provides insight into the interaction between human arylsulfatase A and its substrates during catalysis. J Mol Biol. 2001; 305:269-277. [DOI] [PubMed] [Google Scholar]
- 11. Tsuda T, Hasegawa Y, Eto Y. Two novel mutations in a Japanese patient with the late-infantile form of metachromatic leukodystrophy. Brain & Dev. 1996; 18:400-403. [DOI] [PubMed] [Google Scholar]
- 12. Kreysing J, Bohne W, Bosenberg C, Marchesini S, Turpin JC, Baumann N, von Figura K, Gieselmann V. High residual arylsulfatase A (ARSA) activity in a patient with late-infantile metachromatic leukodystrophy. Am J Hum Genet. 1993; 53:339-346. [PMC free article] [PubMed] [Google Scholar]