

Abstract
There is great interest in therapeutically harnessing endogenous regenerative mechanisms to increase the number of β cells in people with diabetes. By performing whole‐genome expression profiling of zebrafish islets, we identified 11 secreted proteins that are upregulated during β‐cell regeneration. We then tested the proteins' ability to potentiate β‐cell regeneration in zebrafish at supraphysiological levels. One protein, insulin‐like growth factor (Igf) binding‐protein 1 (Igfbp1), potently promoted β‐cell regeneration by potentiating α‐ to β‐cell transdifferentiation. Using various inhibitors and activators of the Igf pathway, we show that Igfbp1 exerts its regenerative effect, at least partly, by inhibiting Igf signaling. Igfbp1's effect on transdifferentiation appears conserved across species: Treating mouse and human islets with recombinant IGFBP1 in vitro increased the number of cells co‐expressing insulin and glucagon threefold. Moreover, a prospective human study showed that having high IGFBP1 levels reduces the risk of developing type‐2 diabetes by more than 85%. Thus, we identify IGFBP1 as an endogenous promoter of β‐cell regeneration and highlight its clinical importance in diabetes.
Keywords: β cell, diabetes, insulin, regeneration, zebrafish
Subject Categories: Molecular Biology of Disease, Signal Transduction, Stem Cells
Introduction
Diabetes is characterized by hyperglycemia that results from insulin deficiency, insulin resistance, or a combination of both. Insulin deficiency can be caused by a reduction in the number of insulin‐producing β cells—not only through autoimmune destruction of β cells in type‐1 diabetes (T1D) but also through stress‐induced apoptosis of β cells in type‐2 diabetes (T2D) (Weir & Bonner‐Weir, 2013)—at which point there is a need for insulin therapy or transplantation of pancreas or islets. However, it is difficult to adequately control glycemia with insulin injections, and there is a shortage of transplant donors, a shortage that is being progressively accentuated by a rise in the number of people with diabetes. Alternative strategies, preferably curative ones, are therefore needed. One potentially curative strategy would be to stimulate the endogenous mechanisms by which β cells regenerate. Experimental depletion of β cells in zebrafish and rodents is followed by significant recovery of the β‐cell mass (Curado et al, 2007; Nir et al, 2007; Pisharath et al, 2007). Likewise, the human β‐cell mass is known to adapt to the high demand for insulin that occurs in states such as pregnancy and obesity (Butler et al, 2003; Ackermann & Gannon, 2007; Rahier et al, 2008; Hanley et al, 2010). Identifying the signals that drive this pancreatic adaptability could enable the development of a regenerative treatment for diabetes, as a complement to immunomodulatory treatment for T1D or as a preventive intervention for T2D.
The types of signals that could be used to increase the β‐cell mass include small molecules or secreted proteins that potentiate β‐cell proliferation, β‐cell neogenesis from ductal progenitors, or transdifferentiation of mature cell types to β cells. Human β cells are not as proliferative as rodent β cells, although proliferation of human β cells can be seen as obesity develops (Butler et al, 2003; Hanley et al, 2010; Wang et al, 2015). As for β‐cell neogenesis, it is currently unclear whether there are adult human pancreatic progenitors from which β‐cell neogenesis could occur. By contrast, there is an abundance of mature cells—for example, α cells, δ cells, and acinar cells—that have been shown to transdifferentiate into β cells in mouse models of diabetes (Thorel et al, 2010; Al‐Hasani et al, 2013; Baeyens et al, 2014; Chera et al, 2014). Because no signal for β‐cell regeneration has yet been shown to be useful in humans, there is a need to identify new signals that can promote β‐cell regeneration in model organisms and to then translate such findings to humans.
In vivo screens for signals that can promote β‐cell regeneration are warranted because in vitro screens cannot reproduce the endogenous environment of a living organism—including signaling between different cell types and tissues, the existence of various progenitors and other sources of β cells, and physiological responses to β‐cell depletion. Ideally, such in vivo screens would identify endogenous factors that mediate regeneration, because regenerative drugs based on endogenous factors are likely to have fewer side effects than those based on exogenous factors. An excellent model organism in which to perform such in vivo screens is the zebrafish (Seth et al, 2013). Not only do zebrafish have only one islet in their first week of life (Kinkel & Prince, 2009), making them extremely efficient for pancreatic screening, but they also have prominent regenerative capacity (Goldman, 2014). We previously used a zebrafish model of diabetes to screen for small molecules that can potentiate β‐cell regeneration and identified the adenosine signaling pathway as a potent promoter of β‐cell proliferation following β‐cell ablation (Andersson et al, 2012). This regenerative pathway is conserved in mouse, rat, and pig (Andersson et al, 2012; Annes et al, 2012; Zhao et al, 2014) and is now a therapeutic candidate. Zebrafish screens have likewise generated therapeutic candidates in other areas, such as hematopoiesis (North et al, 2007).
In this study, we first performed an unbiased genetic screen in zebrafish to identify genes that are upregulated during β‐cell regeneration. We then over‐expressed in zebrafish the 11 candidate genes we identified that encode secreted proteins, and assessed their effects on β‐cell regeneration in vivo. Using this setup, we identified Igfbp1a as a potent stimulator of β‐cell regeneration and showed that Igfbp1a stimulates β‐cell regeneration by promoting transdifferentiation of α to β cells. Complementary studies in human islets and analysis of data from a prospective human study indicate that this effect is conserved in humans and that high serum levels of IGFBP1 are associated with a reduced risk of developing T2D. Together, our data suggest that IGFBP1 can stimulate regeneration and expansion of β cells and may protect against the development of diabetes.
Results
Genetic screening in zebrafish identifies igfbp1a as a promoter of β‐cell regeneration
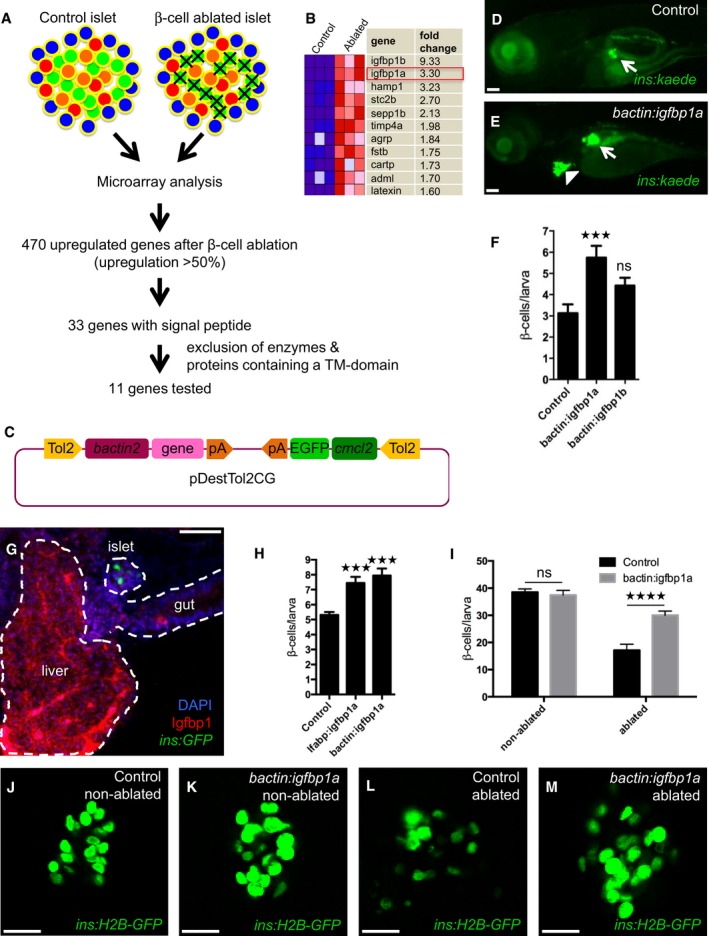
We used a zebrafish model of regeneration to identify endogenous pathways that could potentially be harnessed to promote the regeneration of β cells. The model is based on transgenic zebrafish larvae expressing nitroreductase (NTR)—an enzyme that converts metronidazole (MTZ) to a cytotoxic product—under the control of the insulin promoter; incubating these Tg(ins:CFP‐NTR) larvae in MTZ results in the specific ablation of their β cells (Curado et al, 2007). We ablated the β cells of larvae in this way from 3 to 4 days postfertilization (dpf) and directly thereafter isolated the islets. RNA was extracted from both control and β‐cell‐ablated islets and subjected to microarray analysis. We found that 470 genes were upregulated in response to β‐cell ablation, of which 33 contained a signal‐peptide sequence mediating secretion. To enrich for ligands, we excluded from this 33‐member regenerative secretome those proteins that were either enzymes or contained at least one transmembrane domain (schema outlined in Fig 1A), narrowing our candidate list to 11 genes (Fig 1B).
Figure 1. Genetic screening in zebrafish identifies igfbp1a as a promoter of β‐cell regeneration.
-
ASchema of the analysis of gene expression in islets isolated from control larvae and larvae subjected to β‐cell ablation. β cells were ablated by exposing nitroreductase (NTR)‐expressing transgenic larvae to metronidazole (MTZ) from 3 to 4 dpf. Islets were then isolated, and their RNA extracted and analyzed by microarray. Out of the 470 genes that were upregulated more than 50%, 33 genes encoded proteins that harbored a signal peptide for secretion (according to the algorithm of SignalP). Excluding genes that encode enzymes or proteins with a transmembrane (TM) domain, we selected 11 genes for overexpression studies in zebrafish larvae (C–E).
-
BMicroarray heat map showing the upregulation in expression of the 11 candidate genes in β‐cell‐ablated versus control islets. Igfbp1a and b were the genes whose expression increased the most after β‐cell ablation.
-
CSchema of the construct used for overexpression of the candidate genes (under the control of the beta‐actin promoter), and expression of GFP in the heart (as an internal control for genomic integration).
-
D, ERepresentative images at 6 dpf of Tg(ins:kaede);Tg(ins:CFP‐NTR) transgenic larvae that had been injected at the 1–2 cell stage with transposase mRNA (control) or transposase mRNA + bactin:igfbp1a (bactin:igfbp1a), subjected to β‐cell ablation by metronidazole (MTZ) during 3–4 dpf, and subsequently allowed to regenerate for 2 days. The GFP+ heart (arrowhead) visualizes successful integration of the construct. Islets are indicated by white arrows. Scale bars: 100 μm.
-
FQuantification of β‐cell regeneration at 6 dpf in control (n = 23), bactin:igfbp1a‐overexpressing (n = 13), and bactin:igfbp1b‐overexpressing (n = 8) Tg(ins:kaede);Tg(ins:CFP‐NTR) larvae; ***P = 0.0002, ns = non‐significant (P = 0.3106).
-
GImmunohistochemistry showing Igfbp1 protein expression in 6 dpf Tg(ins:GFP) following β‐cell ablation between 3 and 4 dpf. Scale bar: 50 μm.
-
HBoth liver‐specific (lfabp promoter; n = 46) and widespread (bactin promoter; n = 18) overexpression of igfbp1a increase β‐cell regeneration when compared to control (n = 77); ***P < 0.001.
-
I–MQuantification of β cells with or without β‐cell ablation and igfbp1a overexpression by confocal microscopy, which detects even weakly insulin‐expressing β cells; ****P < 0.0001. Scale bars: 15 μm.
We next generated constructs for the mosaic overexpression of the 11 candidate genes in zebrafish larvae. We reasoned that mosaic overexpression of secreted factors can affect the whole organism and thereby the vast majority of the different cellular mechanisms of β‐cell regeneration. Each of the 11 genes was cloned into a vector backbone containing a ubiquitous promoter driving overexpression, as well as an internal control (the cmlc2 promoter driving GFP expression in the heart) for visualizing the transposon‐mediated integration of the construct into the genome (Fig 1C). Each construct was injected, together with mRNA encoding transposase, into 1–2 cell‐stage Tg(ins:CFP‐NTR);Tg(ins:Kaede) embryos, giving rise to larvae in which the β cells are visualized by the green‐fluorescent protein Kaede. Larvae that successfully integrated the constructs in a mosaic fashion were identified through their expression of GFP in the heart at 3 dpf. From 3 to 4 dpf, we used MTZ to ablate the β cells of mosaically overexpressing larvae and control larvae (i.e., larvae injected with only transposase mRNA), and at 6 dpf, we examined whether overexpression of any of the 11 secreted proteins had increased β‐cell regeneration. One protein, insulin‐like growth factor binding‐protein 1a (Igfbp1a), strikingly increased the regeneration of β cells (Fig 1D and E). The Igfbp1 gene is duplicated in zebrafish, and both of its paralogs, igfbp1a and igfbp1b, were upregulated after β‐cell ablation (Fig 1B), although only Igfbp1a robustly increased β‐cell regeneration when overexpressed (Fig 1F). In addition to being transcriptionally upregulated in islets, igfbp1a was also transcriptionally upregulated 1.6‐fold in purified α cells, 0.9‐fold in hepatocytes, and 1.7‐fold in whole larvae, which together with its strong protein expression in liver after β‐cell ablation (Fig 1G), indicates that igfbp1a is produced in several cell types and organs following β‐cell ablation.
Given the strong expression of igfbp1a in the liver, we asked whether liver‐specific overexpression of igfbp1a is sufficient to increase β‐cell regeneration. Overexpressing igfbp1a under the control of the liver‐specific lfabp promoter, we found that liver‐specific overexpression was approximately as efficient at inducing β‐cell regeneration as widespread overexpression under the control of the bactin promoter (Fig 1H). This finding indicates that igfbp1a can be secreted by the liver, circulate, and potentiate β‐cell regeneration in the pancreas.
To determine whether overexpression of igfbp1a also increases the number of β cells during normal β‐cell development and compare its effect to that during β‐cell regeneration, we quantified the β cells in both ablated and non‐ablated igfbp1a‐overexpressing larvae by using confocal microscopy, which detects even weakly insulin‐expressing β cells (Fig 1I–M). We found that whereas igfbp1a overexpression increased β‐cell regeneration, it had no effect on the total number of β cells during development.
Igfbp1a's effect on β‐cell regeneration is specific and functionally relevant
To determine whether Igfbp1a increases the regeneration of β cells by promoting β‐cell survival rather than the generation of new β cells, we followed the fate of β cells during ablation and regeneration via cell labeling. Using Tg(ins:CFP‐NTR);Tg(ins:Kaede) larvae, we converted the fluorescence of the Kaede protein expressed in the larval β cells from green to red by exposing the larvae to UV light (Ando et al, 2002) at 3 dpf; this conversion marked all β cells that were present before the ablation step. We then treated the larvae from 3 to 4 dpf with MTZ to ablate the β cells and examined whether the number of red‐fluorescent β cells at 6 dpf was greater in the bactin:igfbp1a‐overexpressing larvae than in the control larvae. On average, fewer than two β cells per larva survived the ablation in control and bactin:igfbp1a‐overexpressing larvae (Fig 2A–C). By contrast, many β cells were labeled green only, indicating that they were newly formed. Thus, Igfbp1a increases the regeneration of β cells by promoting the generation of new β cells rather than the survival of existing ones.
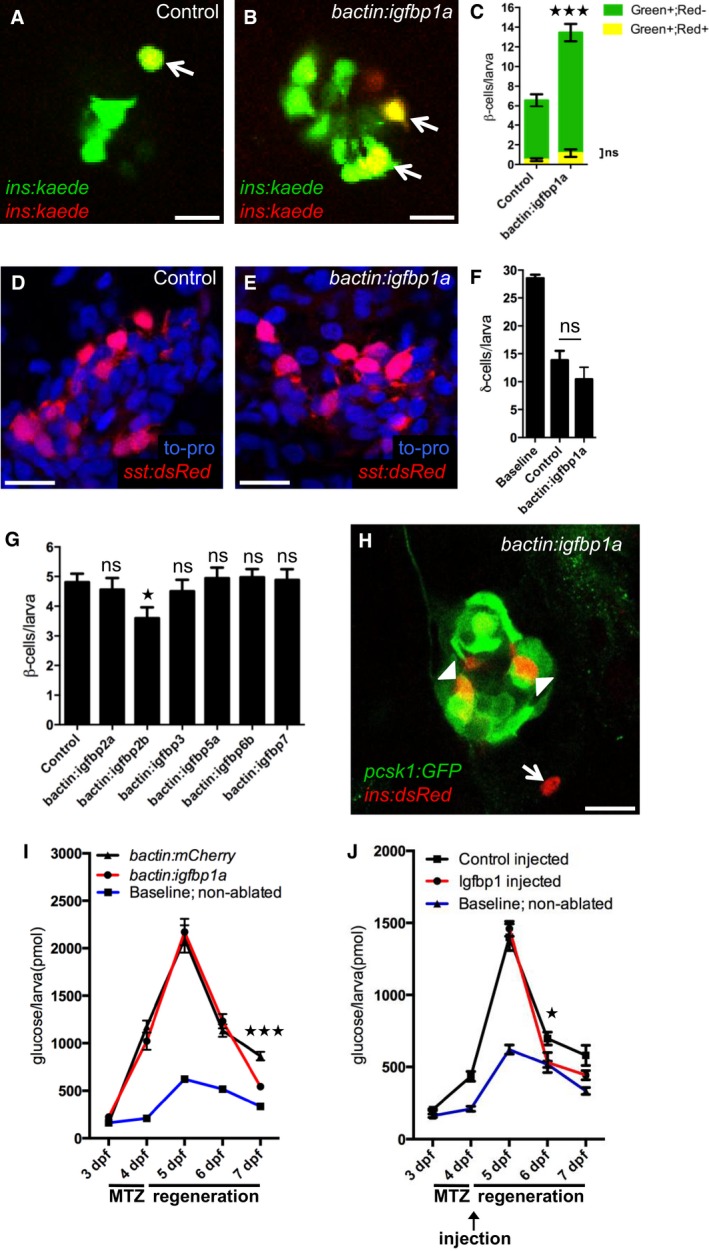
Figure 2. Igfbp1a's effect on β‐cell regeneration is specific and functionally relevant.
-
A–CIgfbp1a promotes β‐cell regeneration, rather than β‐cell survival. To trace β cells, we exposed Tg(ins:kaede) larvae to UV light (which causes the existing Kaede protein to switch from emitting green fluorescence to emitting red fluorescence) just before ablating the β cells by MTZ treatment from 3 to 4 dpf. After regeneration, the newly formed β cells are green, whereas the β cells that survived ablation are yellow (overlap of green and red). Representative confocal images (A, B) at 6 dpf of control and bactin:igfbp1a‐overexpressing Tg(ins:kaede);Tg(ins:CFP‐NTR) transgenic larvae; arrows indicate surviving (yellow) β cells. Scale bars: 10 μm. (C) Quantification of β‐cell regeneration (green bars) and β‐cell survival (yellow bars) per larva at 6 dpf. ***P < 0.001; n = 20 larvae in the control group, n = 27 larvae in the bactin:igfbp1a‐overexpressing group.
-
D–FIgfbp1a does not promote δ‐cell regeneration. We treated control and bactin:igfbp1a‐overexpressing Tg(sst:flag‐NTR);Tg(sst:dsRed) larvae with MTZ from 3 to 4 dpf to ablate δ cells, and then allowed them to regenerate until 6 dpf. Representative confocal images (D, E) at 6 dpf of control and bactin:igfbp1a‐overexpressing larvae showing comparable number of δ cells after 2 days of regeneration. Scale bars: 15 μm. (F) Quantification of the total number of δ cells per δ‐cell‐ablated larva at 6 dpf compared to the baseline number of δ cells in non‐ablated control larvae. ns, P = 0.2325; n = 13 in the control group, n = 7 in the bactin:igfbp1a group.
-
GOther Igfbps do not promote β‐cell regeneration. We injected Tg(ins:kaede);Tg(ins:CFP‐NTR) transgenics with either transposase mRNA (control; n = 31) or transposase mRNA + one of six different igfbps, igfbp2a (n = 25), igfbp2b (n = 25), igfbp3 (n = 26), igfbp5a (n = 17), igfbp6b (n = 34), and igfbp7 (n = 17), treated them with MTZ from 3 to 4 dpf to ablate their β cells, and then quantified their β cells after 2 days of regeneration, at 6 dpf; ns=non‐significant (igfbp2a P = 0.986, igfbp3 P = 0.979, igfbp5a P = 0.999, igfbp6b P = 0.997, and igfbp7 P = 0.999); igfbp2b *P = 0.037.
-
HTg(pcsk1:GFP) is expressed in regenerating β cells within the islet (arrowheads), but not outside the islet (arrow), at 6 dpf in larvae overexpressing igfbp1a. Scale bar: 15 μm.
-
I, JFree‐glucose levels during β‐cell regeneration in control (bactin:mCherry) and bactin:igfbp1a‐overexpressing Tg(ins:kaede);Tg(ins:CFP‐NTR) larvae (I), as well as in Tg(ins:kaede);Tg(ins:CFP‐NTR) larvae injected in the pericardial sac at 4 dpf with 1 ng of recombinant mouse Igfbp1 (J). We treated larvae with MTZ from 3 to 4 dpf to ablate their β cells and monitored their free‐glucose levels at 3–7 dpf. Free‐glucose levels were significantly lower after genetic igfbp1a overexpression or Igfbp1‐protein injection (red lines) than in controls (black lines) at 7 and 6 dpf, respectively. Baseline reference levels of free glucose throughout development are shown for a different set of larvae without β‐cell ablation. n = 24 larvae (four pools of six larvae) per data point; ***P < 0.001, *P < 0.05.
To determine whether Igfbp1a elicits a generalized increase in regeneration of the endocrine pancreas, we examined its effect on δ‐cell regeneration. We created a transgenic line called Tg(sst:NTR), in which the expression of NTR is under the control of the somatostatin promoter. We ablated the δ cells in these larvae from 3 to 4 dpf and then analyzed δ‐cell regeneration at 6 dpf. There was no difference in δ‐cell regeneration between control and bactin:igfbp1a‐overexpressing larvae, as determined in Tg(sst:NTR) larvae that also carry the δ‐cell marker Tg(sst:dsRed) (Fig 2D–F), indicating that Igfbp1a does not have a generalized effect on regeneration of the endocrine pancreas.
To determine whether Igfbp1a's regenerative properties are shared by other Igfbps, we overexpressed several different Igfbps in Tg(ins:CFP‐NTR);Tg(ins:Kaede) larvae (Fig 2G). None of the 6 Igfbps tested could potentiate β‐cell regeneration (although Igfbp2b significantly decreased β‐cell regeneration), indicating that the ability to stimulate β‐cell regeneration is specific to Igfbp1.
We next examined the functionality of the regenerated β cells. First, we examined whether the regenerated β cells express pcsk1, the enzyme that processes proinsulin to its active form and is therefore considered a requirement for a functional β cell, by generating a zebrafish line expressing GFP under the control of the pcsk1 promoter, Tg(pcsk1:GFP). We found that β cells regenerated within the islet (which presumably include β cells formed both through transdifferentiation of other endocrine cell types and through β‐cell proliferation) expressed Tg(pcsk1:GFP), whereas the few β cells that were formed outside the islet did not express Tg(pcsk1:GFP), at least not after 2 days of regeneration from 4 to 6 dpf (Fig 2H). Second, we tested the ability of Igfbp1a to restore normoglycemia in our β‐cell regeneration model. By measuring levels of free glucose, that is, glucose that has not been phosphorylated intracellularly by hexokinases, we get an estimate of glycemia. Control and bactin:igfbp1a‐over‐expressing Tg(ins:CFP‐NTR) larvae were exposed to MTZ from 3 to 4 dpf and then allowed to recover from 4 to 7 dpf. Free‐glucose levels were measured from pooled clutches of larvae every 24 h throughout the experiment (Fig 2I). After 3 days of β‐cell regeneration, the free‐glucose level was significantly lower in the bactin:Igfbp1a‐overexpressing larvae than in the controls. As with genetic overexpression of igfbp1a, injection of recombinant Igfbp1 protein also decreased the levels of free glucose (Fig 2J). Thus, Igfbp1a increases the number of functional β cells and is associated with an accelerated restoration of normal free‐glucose levels.
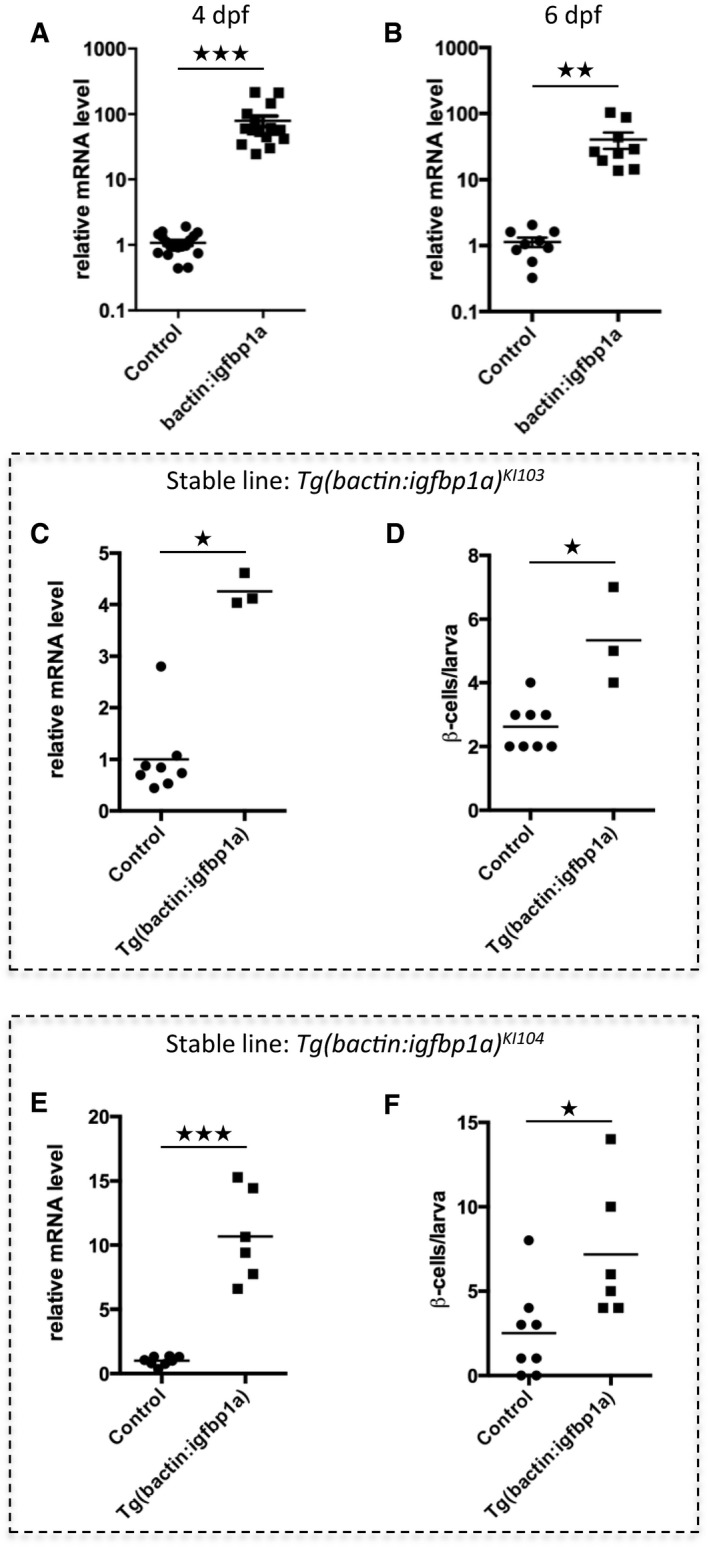
To validate our findings from the mosaic‐overexpression experiments, we established stable transgenic lines overexpressing igfbp1a, that is, Tg(bactin:igfbp1a) lines and found that these larvae also had an increase in β‐cell regeneration (Fig EV1). However, whereas mosaically overexpressing larvae expressed around 80 times more igfbp1a than control larvae, stable transgenic lines overexpressed lower levels of igfbp1a (Fig EV1C and E) but nonetheless had significantly greater levels of β‐cell regeneration than their corresponding controls (Fig EV1D and F). The stable transgenic lines did not grow and breed as well as wild‐type zebrafish, perhaps because of the importance of IGF signaling in the gonad (Li et al, 2015), and were therefore not amenable to certain types of experiments. Hereinafter, to distinguish between the two different modes of Igfbp1a overexpression, we use the term Tg(bactin:igfbp1a) when referring to a stable transgenic line expressing 16 times the endogenous level of igfbp1a (with the exception of Fig EV1 where the other stable lines are characterized), and the term bactin:Igfbp1a when referring to mosaically overexpressing larvae.
Figure EV1. Expression level of igfbp1a in mosaic over‐expressing larvae and stable transgenic lines.
-
A, BRelative igfbp1a mRNA levels in mosaic bactin:igfbp1a transgenic larvae. Control or bactin:igfbp1a‐injected embryos were collected for RNA purification and qPCR analysis at 4 dpf (A) or 6 dpf (B). Igfbp1a expression was approximately 80 times higher in bactin:igfbp1a overexpressing larvae than in control larvae. ***P < 0.0001 and **P = 0.0024. Each data point represents one larva; thus, it can be seen that substantial Igfbp1a overexpression was attained in all bactin:igfbp1a‐injected embryos that generated mosaically overexpressing larvae.
-
C–FRelative igfbp1a mRNA levels (C, E) in two independent stable Tg(bactin:igfbp1a) lines, which have the line designations KI103 and KI104. Control and Tg(bactin:igfbp1a) larvae were processed for qPCR analysis at 6 dpf. Both transgenic lines (data for each line are shown separately, in panels C and E) have moderately, but significantly, higher expression of igfbp1a than sibling controls; *P = 0.0121 and ***P = 0.0007, respectively. (D, F) Like the larvae mosaically overexpressing igfbp1a, the stable Tg(bactin:igfbp1a) lines also have an increase in β‐cell regeneration. We crossed the Tg(bactin:igfbp1a) lines into the double transgenic Tg(ins:H2B‐GFP);Tg(ins:Flag‐NTR) to allow visualization of β‐cell regeneration. We treated the resulting offspring with metronidazole (MTZ) from 3 to 4 dpf to ablate the β cells and then allowed them to regenerate until 6 dpf. Both Tg(bactin:igfbp1a)KI103 and Tg(bactin:igfbp1a)KI104 stable lines had a significant increase in β‐cell regeneration (*P = 0.0121 and *P = 0.0107, respectively).
Cellular mechanisms of Igfbp1a's effect on β‐cell regeneration
To determine Igfbp1a's cellular effect on β‐cell regeneration, we examined the three main regenerative mechanisms described to date, that is, β‐cell neogenesis from ductal cells, β‐cell proliferation, and α‐ to β‐cell transdifferentiation.
To determine whether Igfbp1a‐induced β‐cell regeneration occurs through β‐cell neogenesis from ductal cells, we used the notch‐responsive tp1 promoter to drive the expression of the stable fluorescent protein H2BmCherry and thereby label the ductal cells. We then ablated the β cells in Tg(ins:flag‐NTR);Tg(tp1:H2BmCherry) transgenics and compared β‐cell regeneration in those that additionally carried the Tg(bactin:igfbp1a) to those that did not. There was no significant increase in the number of ductal‐derived β cells after overexpression of igfbp1a (Fig 3A–C).
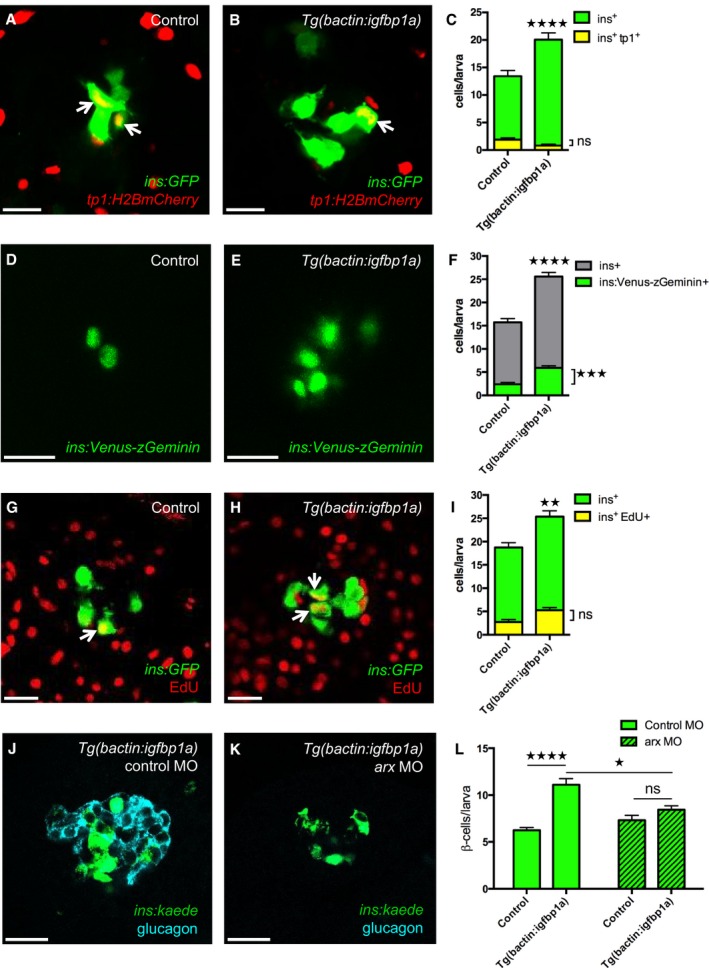
Figure 3. Cellular mechanisms of Igfbp1a's effect on β‐cell regeneration.
-
A–CTg(ins:GFP);Tg(tp1:H2B‐mCherry);Tg(ins:Flag‐NTR) transgenics, with or without Tg(bactin:igfbp1a), were treated with MTZ from 3 to 4 dpf to ablate the β cells and were then allowed to regenerate from 4 to 6 dpf. Representative confocal images at 6 dpf of control (A) and Tg(bactin:igfbp1a) (B) larvae showing a modest number of ins + tp1 + co‐expressing cells, indicated by arrows, after 2 days of regeneration. Scale bars: 15 μm. (C) Quantification of the total number of β cells (green bars) at 6 dpf, and of β cells expressing Tg(tp1:H2B‐mCherry), that is, of ductal origin (yellow bars). ****P < 0.0001, ns= non‐significant. n = 23 larvae in the control group, n = 17 larvae in the Tg(bactin:igfbp1a) group.
-
D–FTg(ins:Venus‐zGeminin) was examined in control and Tg(bactin:igfbp1a) larvae at 6 dpf, after β‐cell ablation 3–4 dpf by using Tg(ins:Flag‐NTR). Representative confocal images (D, E) at 6 dpf of control and Tg(bactin:igfbp1a) larvae showing cell cycle activation of β cells in green. Scale bars: 15 μm. (F) Quantification of the total number of β cells with activated cell cycle at 6 dpf, ***P < 0.001, n = 32 larvae in the control group, n = 41 larvae in the Tg(bactin:igfbp1a) group. The number of β cells with activated cell cycle is displayed together with the total number of β cells in experiments with the same setup, ****P < 0.0001. n = 39 larvae in the control group, n = 33 larvae in the Tg(bactin:igfbp1a) group.
-
G–IEdU was used as a marker for cell cycle progression. Tg(ins:H2B‐GFP);Tg(ins:Flag‐NTR) transgenics, with or without Tg(bactin:igfbp1a), were treated with MTZ from 3 to 4 dpf to ablate their β cells, and subsequently incubated with EdU during regeneration from 4 to 6 dpf. Representative confocal images (G, H) at 6 dpf of control and Tg(bactin:igfbp1a) larvae showing β cells in green and the β cells that had incorporated EdU in yellow (green and red overlap; arrowheads). Scale bars: 20 μm. (I) Quantification of the total number of β cells (green bars) at 6 dpf and of β cells that incorporated EdU (yellow bars) during β‐cell regeneration from 4 to 6 dpf. **P < 0.01, ns = non‐significant. n = 16 in both the control and the Tg(bactin:igfbp1a) group.
-
J–LTg(ins:kaede);Tg(ins:CFP‐NTR) transgenics, with or without Tg(bactin:igfbp1a), were injected with a control morpholino or a morpholino that knocked down arx, which is necessary for α‐cell differentiation and thus glucagon expression (J, K). Scale bars: 20 μm. (L) Quantification of the total number of regenerating β cells at 4 dpf (after β‐cell ablation at 2–3 dpf). ****P < 0.0001, *P < 0.05, ns=non‐significant. n = 42, 20, 48 and 20, respectively.
To determine whether β‐cell proliferation contributes to the Igfbp1a‐induced increase in β‐cell regeneration, we used a dual approach. First, we used a reporter line that visualizes cell cycle activation specifically in β cells, that is, Tg(ins:Venus‐zGeminin), a system based on FUCCI (fluorescence ubiquitination cell cycle indicator) (Sakaue‐Sawano et al, 2008). Using this reporter line, we found that Igfbp1a overexpression significantly increases cell cycle activation 2 days after β‐cell ablation, that is, at 6 dpf (Fig 3D–F), though the increase is small compared to the increase in β‐cell regeneration. However, because a cell can activate the cell cycle without actually going on to replicate its DNA and divide into 2 cells, we next assessed Igfbp1a's effect on EdU incorporation, as a measure of DNA replication. We ablated the β cells in Tg(ins:flag‐NTR);Tg(ins:H2B‐GFP) transgenics and compared DNA replication in those that additionally carried the Tg(bactin:igfbp1a) to those that did not, by exposing the larvae to EdU during the regenerative period, that is, from 4 to 6 dpf (Fig 3G–I). This approach afforded a cumulative assessment of β‐cell proliferation, which did not significantly differ between control and Tg(bactin:igfbp1a) larvae between 4 and 6 dpf. Thus, the small increase in cell cycle activation does not lead to an increase in cell division. These data suggest that β‐cell proliferation is not the main contributor to Igfbp1a‐induced β‐cell regeneration.
To get a first indication of whether α‐ to β‐cell transdifferentiation might underlie the Igfbp1a‐induced increase in β‐cell regeneration, we determined whether α cells are necessary for Igfbp1a‐induced β‐cell regeneration. We depleted the α cells (Fig 3J and K) by injecting Tg(ins:kaede);Tg(ins:CFP‐NTR) transgenics with an antisense morpholino to knock down arx, a transcription factor necessary for the development of α cells (Collombat et al, 2003; Djiotsa et al, 2012). Because the effect of morpholinos is short‐lived, we shortened the setup of the experiment by ablating the β cells during 2–3 dpf and allowing them to regenerate from 3 to 4 dpf. Whereas Igfbp1a overexpression induced significant β‐cell regeneration in larvae injected with control morpholino, it failed to do so in larvae depleted of α cells (Fig 3L). Thus, Igfbp1a‐induced β‐cell regeneration depends mainly on α cells, at least during the first day of regeneration when there are few β cells present.
Igfbp1a increases β‐cell regeneration by potentiating α‐ to β‐cell transdifferentiation
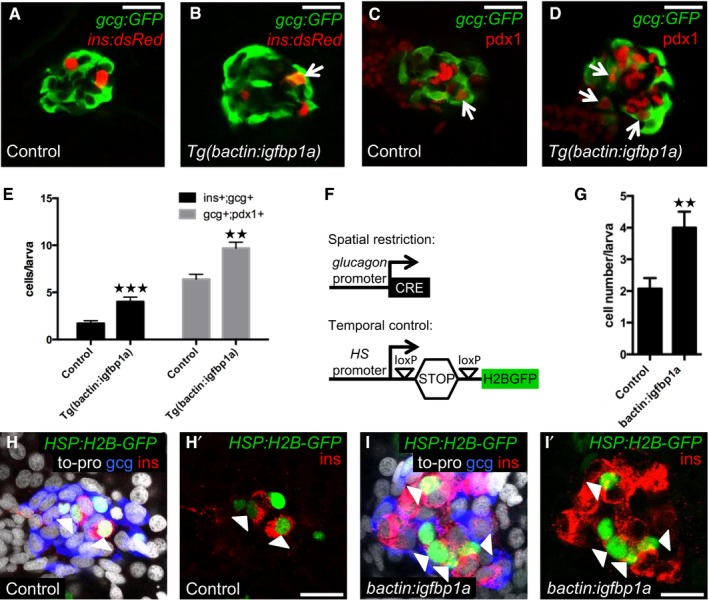
To further explore whether Igfbp1a potentiates β‐cell regeneration in zebrafish by increasing α‐ to β‐cell transdifferentiation, we examined the cellular co‐expression of glucagon and insulin, and that of glucagon and the β‐cell transcription factor pdx1. We found that Tg(bactin:igfbp1a) larvae had a greater number of bihormonal (glucagon‐ and insulin‐co‐expressing) cells during β‐cell regeneration than did control larvae, as evidenced by the co‐expression of Tg(ins:dsRed) and Tg(gcg:GFP) (Fig 4A and B). Moreover, they also had a greater number of cells co‐expressing Tg(gcg:GFP) and pdx1 (Fig 4C and D). Compared to control larvae, larvae overexpressing Igfbp1a had more than twice as many cells co‐expressing Tg(ins:dsRed) and Tg(gcg:GFP), and a 50% increase in cells co‐expressing Tg(gcg:GFP) and pdx1 (Fig 4E). These data indicate, but do not formally prove, that Igfbp1a potentiates β‐cell regeneration by inducing α‐ to β‐cell transdifferentiation.
Figure 4. Igfbp1a increases β‐cell regeneration by potentiating α‐ to β‐cell transdifferentiation.
-
A–EControl and Tg(bactin:igfbp1a)‐overexpressing larvae were treated with MTZ from 3 to 4 dpf to ablate β cells and analyzed at 6 dpf, after 2 days of regeneration. Representative confocal images (A, B) of control and Tg(bactin:igfbp1a)‐overexpressing Tg(ins:dsRed);Tg(gcg:GFP);Tg(ins:Flag‐NTR) larvae. A bihormonal glucagon‐ and insulin‐expressing cell (gcg+;ins+) is indicated by an arrow. Representative confocal images (C, D) of control and Tg(bactin:igfbp1a)‐overexpressing Tg(gcg:GFP);Tg(ins:Flag‐NTR) larvae stained for Pdx1. Pdx1‐ and glucagon‐expressing cells (pdx1+;gcg+) are indicated by arrows. Scale bars: 20 μm. (E) Quantification of gcg+;ins+ and pdx1+;gcg+ cells in the control and Tg(bactin:igfbp1a) group; ***P < 0.001, **P = 0.0012; n = 18 and 18 in the control groups, n = 15 and 10 in the Tg(bactin:igfbp1a) groups, for the gcg+;ins+ and pdx1+;gcg+ quantification, respectively.
-
F–ILineage‐tracing evidence supports α‐ to β‐cell transdifferentiation. (F) Schema for temporal and spatial lineage tracing. loxP‐mediated excision of the STOP cassette permits heat‐inducible expression of the stable fusion‐protein H2B‐GFP. Control and bactin:igfbp1a‐overexpressing Tg(hsp:loxP‐mCherry‐STOP‐loxP‐H2B‐GFP);Tg(gcg:CRE);Tg(ins:Flag‐NTR) larvae were first heat‐shocked at 3 dpf to label the glucagon‐expressing cells with the stable fluorescent protein H2B‐GFP, thereafter treated with MTZ from 3 to 4 dpf to ablate the β cells, and analyzed at 6 dpf after 2 days of regeneration. Note that the temporal control of the lineage tracing is mediated by a heat‐shock. (G) Quantification of insulin‐expressing cells that originate from α cells. Control larvae, n = 13; bactin:igfbp1a larvae, n = 16. **P = 0.0051. Representative confocal images (H, I) at 6 dpf (after 2 days of regeneration) of control and bactin:igfbp1a‐overexpressing larvae stained for insulin, glucagon, to‐pro, and H2B‐GFP. ins+;H2B‐GFP+ cells are indicated by the arrowheads, and H' and I' show staining against only insulin and H2B‐GFP. Moreover, most ins+;H2B‐GFP+ cells no longer express glucagon. Scale bars: 15 μm.
To formally test whether Igfbp1a can induce α‐ to β‐cell transdifferentiation, we lineage traced a proportion of the α cells during β‐cell regeneration, by using a previously developed Cre/loxP strategy that enables both temporal control and spatial restriction for the labeling of cells (Ye et al, 2015). The Tg(gcg:CRE) line restricts CRE expression to glucagon‐expressing cells (i.e., α cells and bihormonal cells expressing both insulin and glucagon), and the Tg(hsp:loxP‐mCherry‐STOP‐loxP‐H2B‐GFP) reporter line enables heat‐inducible expression of the stable fusion‐protein H2B‐GFP once the STOP cassette has been excised by CRE (Fig 4F). Using triple transgenic Tg(gcg:CRE);Tg(hsp:loxP‐mCherry‐STOP‐loxP‐H2B‐GFP);Tg(ins:flag‐NTR) larvae, we labeled glucagon‐expressing cells with H2B‐GFP by subjecting the larvae to a heat‐shock. Thereafter, we exposed the larvae to MTZ from 3 to 4 dpf to ablate their β cells. Because we ablate essentially all insulin‐expressing cells, including preexisting bi‐hormonal cells that express insulin and glucagon, we can in this manner faithfully follow the destiny of the lineage‐traced α cells.
Using this setup, we found that bactin:igfbp1a‐overexpressing larvae had a greater number of β cells originating from α cells than did control larvae (Fig 4G–I). When normalized to the efficiency of Tg(gcg:CRE)‐lineage tracing of α cells, 17% (Ye et al, 2015), this result suggests that overexpression of igfbp1a causes approximately 10 additional α cells to transdifferentiate to β cells. Comparing this increase in number of β cells with the results of experiments analyzed in similar ways (e.g., results shown in Fig 1I, which were also analyzed by confocal microscopy and obtained using the same fluorescent protein) suggests that Igfbp1a increases β‐cell regeneration mainly by potentiating α‐ to β‐cell transdifferentiation.
Inhibition of the Igf pathway mimics Igfbp1a's stimulatory effect on β‐cell regeneration
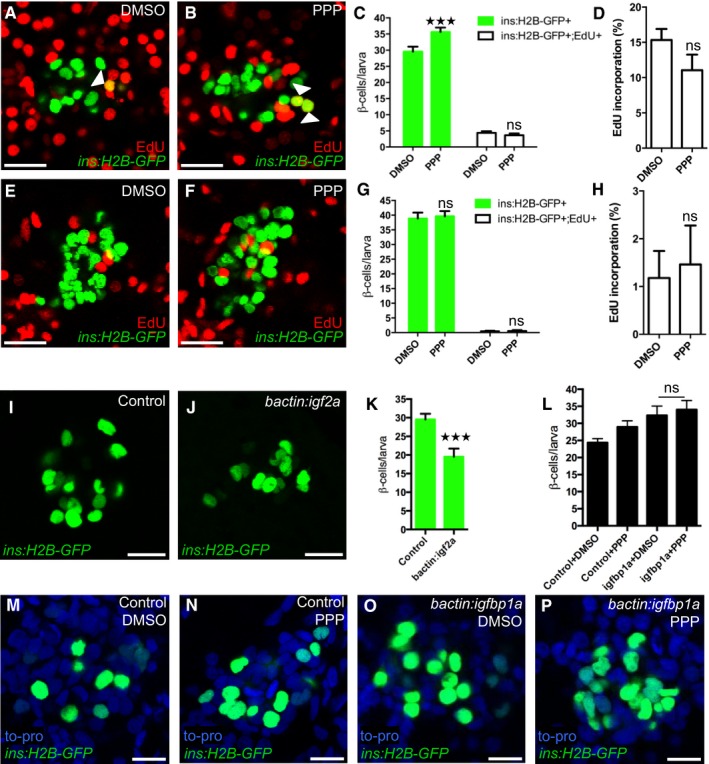
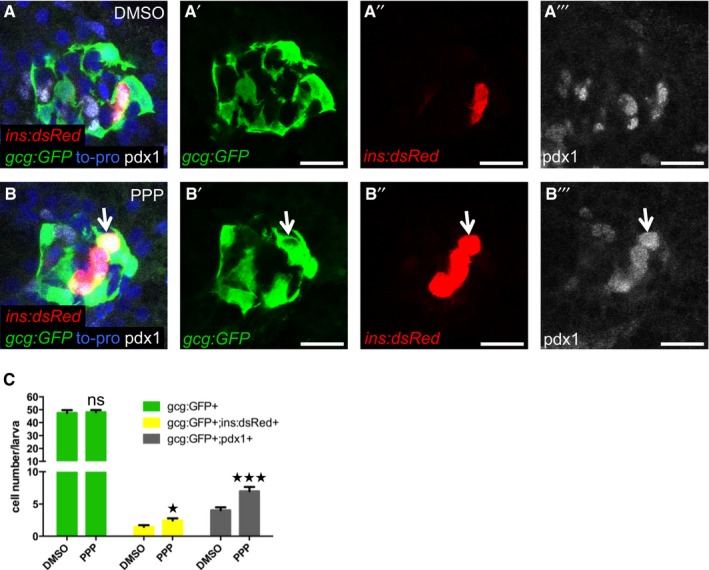
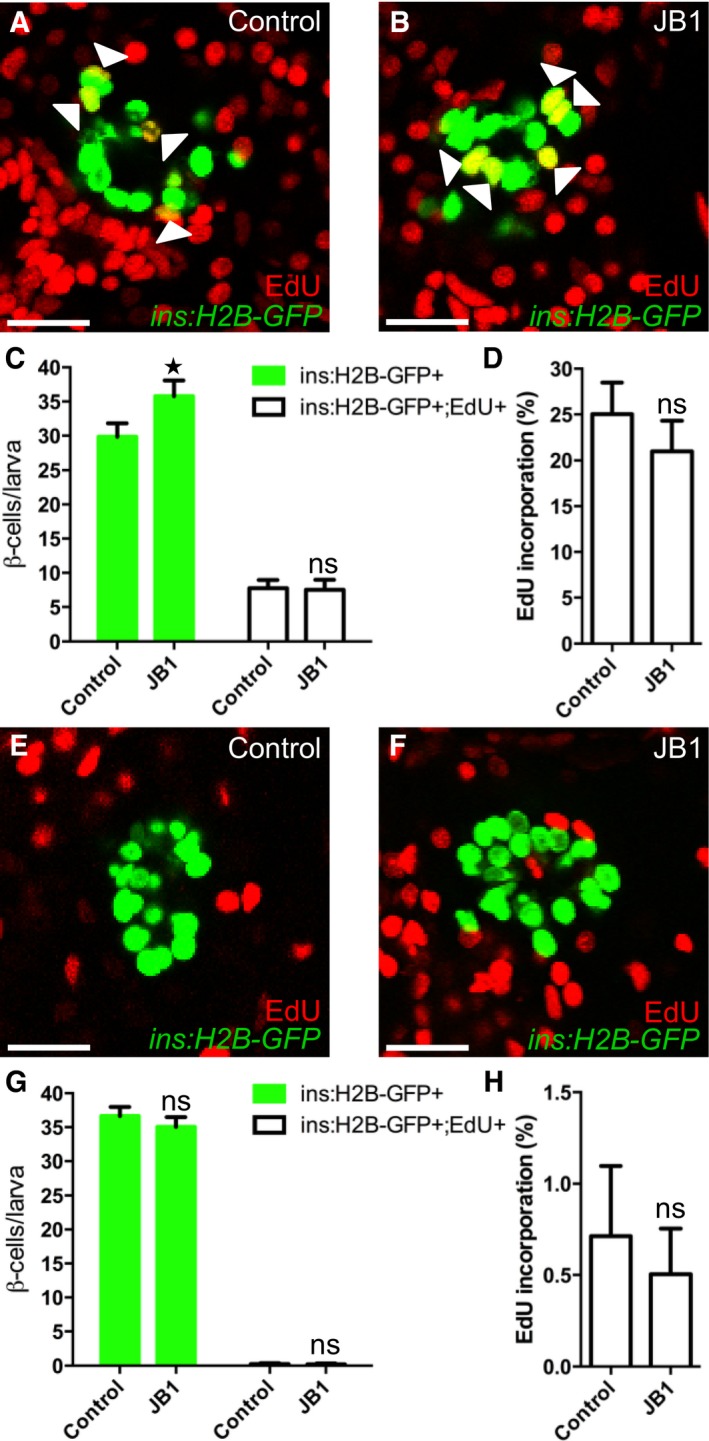
Igfbp1 can either inhibit or activate Igf signaling, depending on the context (Baxter, 2014). Therefore, it was important to determine whether inhibition or activation of Igf signaling increases β‐cell regeneration in zebrafish larvae. Inhibition of the Igf receptor Igf1r by picropodophyllin (PPP) increased β‐cell regeneration without markedly affecting β‐cell proliferation, mimicking the effect of Igfbp1a, though PPP was not as effective as Igfbp1a (Fig 5A–D). Like Igfbp1a, PPP also had no effect on the number of β cells during normal development (Fig 5E–H). Conversely, activation of the Igf pathway by overexpression of the ligand Igf2a, using the same setup as was used for Igfbp1a (i.e., mosaic overexpression of bactin:igf2a in zebrafish larvae), significantly decreased β‐cell regeneration (Fig 5I–K). Moreover, there was no synergistic effect between Igfbp1a and PPP, indicating that these factors stimulate β‐cell regeneration via the same mechanism (Fig 5L–P). We also found that PPP can increase the number of cells co‐expressing Tg(ins:dsRed) and Tg(gcg:GFP), and of cells co‐expressing Tg(gcg:GFP) and pdx1, findings suggestive of transdifferentiation (Fig EV2A–C). Furthermore, we found that yet another inhibitor of the Igf1r, the specific peptidergic inhibitor JB1, had a similar effect on β‐cell regeneration and proliferation as PPP and Igfbp1a (Fig EV3A–H). Thus, inhibiting the Igf pathway stimulates β‐cell regeneration in zebrafish.
Figure 5. Inhibition of the Igf pathway mimics Igfbp1a's stimulatory effect on β‐cell regeneration.
-
A–HPPP, an IGF1R inhibitor, promotes β‐cell regeneration. Tg(ins:H2B‐GFP);Tg(ins:Flag‐NTR) larvae were treated with MTZ from 3 to 4 dpf to ablate the β cells and then treated with EdU and DMSO or with EdU and PPP during regeneration from 4 to 6 dpf. Representative confocal images (A, B) at 6 dpf of DMSO‐ and PPP‐treated larvae displaying β cells in green and the β cells that had incorporated EdU as yellow overlap (arrowheads). (C) Quantification of the total number of β cells (green bars) at 6 dpf, and β cells that had incorporated EdU (white bars) from 4 to 6 dpf during β‐cell regeneration. ***P = 0.0003, P = 0.8607, respectively. (D) Rate of β‐cell proliferation displayed as the percentage of β cells that incorporated EdU. P = 0.1194. n = 18 larvae for the DMSO‐treated group, n = 17 larvae for the PPP‐treated group. (E–H) To examine whether PPP affected β‐cell proliferation during regular development, Tg(ins:H2B‐GFP) larvae were treated with EdU and DMSO or PPP from 4 to 6 dpf. Representative confocal images (E, F) of 6 dpf DMSO‐ and PPP‐treated larvae displaying β cells in green and the β cells that had incorporated EdU as yellow overlap. Scale bars: 20 μm. (G) Quantification of the total number of β cells (green bars) and β cells that had incorporated EdU (white bars) per larva from 4 to 6 dpf. P = 0.9098 and 0.9976, respectively. (H) Rate of β‐cell proliferation displayed as the percentage of β cells that incorporated EdU. P = 0.7822. n = 16 larvae for DMSO‐treated group, 18 larvae for PPP‐treated group.
-
I–KActivation of the Igf pathway reduces β‐cell regeneration. Control and bactin:igf2a‐overexpressing Tg(ins:H2B‐GFP);Tg(ins:Flag‐NTR) larvae were treated with MTZ from 3 to 4 dpf to ablate β cells and subsequently let to regenerate from 4 to 6 dpf. Representative confocal images (I, J) of 6 dpf control and bactin:igf2a‐overexpressing larva displaying β cells in green. Scale bars: 15 μm. (K) Quantification of the total number of β cells per larva at 6 dpf following β‐cell regeneration from 4 to 6 dpf. ***P < 0.0001. n = 28 larvae for control, n = 15 larvae for bactin:igf2a.
-
L–PNo synergistic effect was observed for igfbp1a and PPP. Control and bactin:igfbp1a‐overexpressing Tg(ins:H2B‐GFP);Tg(ins:Flag‐NTR) larvae were treated with MTZ from 3 to 4 dpf to ablate β cells and subsequently treated with DMSO or PPP during regeneration from 4 to 6 dpf. (L) Quantification of the total number of β cells per larva. n > 10 (n = 23, 20, 14, 13). P = 0.9546. Representative confocal images (M–P) of 6 dpf control or bactin:igfbp1a overexpressing larvae treated with either DMSO or PPP, displaying β cells after 2 days regeneration. Scale bars: 10 μm.
Figure EV2. The IGF1R inhibitor PPP increases β‐cell regeneration and co‐expression of insulin and pdx1 with glucagon.
-
A–CPdx1 expression in 6‐dpf larvae during regeneration. Tg(ins:dsRed);Tg(gcg:GFP);Tg(ins:Flag‐NTR) larvae were treated with MTZ from 3 to 4 dpf to ablate the β cells and subsequently treated with DMSO or the IGF1R inhibitor PPP from 4 to 6 dpf. Representative confocal images (A, B) at 6 dpf of DMSO‐ or PPP‐treated larvae. A gcg+ins+pdx1+ cell is indicated by an arrow. Scale bars: 15 μm. (C) Quantification of gcg+, gcg+ins+, and pdx1+gcg+ cells (P = 0.8409, *P = 0.0232, and ***P = 0.0007, respectively) in DMSO‐ and PPP‐treated groups. n = 20 for each group. Results are presented as mean values ± SEM and analyzed with ANOVA. Related to Fig 4.
Figure EV3. The IGF1R inhibitor JB1 promotes β‐cell regeneration.
-
A–DTg(ins:H2B‐GFP);Tg(ins:Flag‐NTR) larvae were treated with MTZ from 3 to 4 dpf to ablate the β cells. DMSO or JB1 was then injected into the larval pericardial cavity at 4 dpf, and the larvae were treated with EdU from 4 to 6 dpf. Representative confocal images (A, B) at 6 dpf of DMSO‐ and JB1‐injected larvae, showing β cells in green and the β cells that incorporated EdU as yellow (green and red overlap; see arrowheads). Scale bars: 20 μm. (C) Quantification of the number of all β cells (green bars) and of β cells that incorporated EdU (white bars) per larva at 6 dpf. *P = 0.0422; ns=non‐significant (P = 0.9944). (D) Rate of β‐cell proliferation, shown as the percentage of β cells that incorporated EdU. ns=non‐significant (P = 0.5885). n = 12 larvae in the DMSO‐injected group; n = 19 larvae in the JB1‐injected group.
-
E–HTo determine whether inhibition of Igf signaling affects β‐cell proliferation during regular development, we treated DMSO‐ or JB1‐injected Tg(ins:H2B‐GFP) larvae with EdU in the absence of β‐cell ablation. DMSO or JB1 was injected into pericardial cavity of Tg(ins:H2B‐GFP) larvae at 4 dpf, and the larvae were then treated with EdU from 4 to 6 dpf. Representative confocal images (E, F) at 6 dpf of DMSO‐ and JB1‐injected larvae, showing β cells in green and the cells that incorporated EdU in red. Scale bars: 20 μm. (G) Quantification of the number of all β cells (green bars) and of β cells that incorporated EdU (white bars) from 4 to 6 dpf. ns=non‐significant (P = 0.4088 and 0.9997, respectively). (H) Rate of β‐cell proliferation, shown as the percentage of β cells that incorporated EdU. P = 0.8504. n = 20 larvae in the DMSO‐injected group, n = 27 larvae in the JB1‐injected group.
Igfbp1a also increases β‐cell regeneration in 1‐month‐old zebrafish
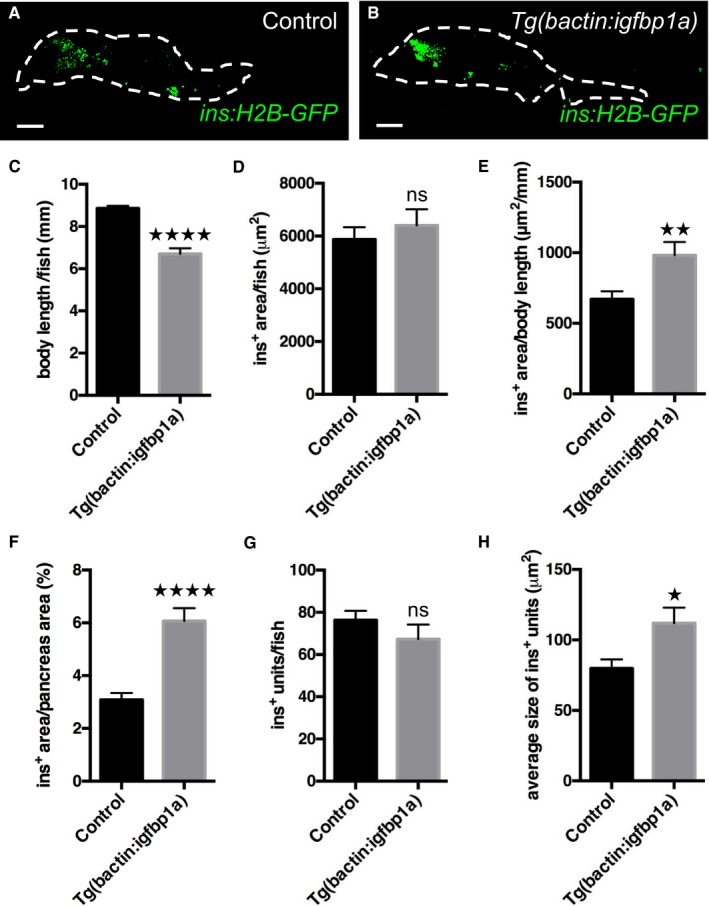
To determine whether the effect of igfbp1a in larval zebrafish holds true in older zebrafish, we ablated the β cells in 1‐month‐old Tg(ins:flag‐NTR);Tg(ins:H2B‐GFP) zebrafish and compared β‐cell regeneration in those that additionally carried Tg(bactin:igfbp1a) to those that did not. We allowed the zebrafish to regenerate for 4 days before we analyzed their pancreata. We scanned through the whole pancreas with confocal microscopy and displayed the insulin‐positive area in a flattened projection of the pancreas (Fig 6A and B). Because the Tg(bactin:igfbp1a) zebrafish do not grow as big as their wild‐type siblings (Fig 6C), we show the analysis of β‐cell regeneration in both absolute and relative terms. As such, the absolute β‐cell area did not differ between Tg(bactin:igfbp1a) and wild‐type siblings (Fig 6D). However, we found that β‐cell area was markedly increased in Tg(bactin:igfbp1a) zebrafish compared to wild‐type siblings when related to body length (Fig 6E), as well as when related to the size of the whole pancreas (Fig 6F); that is, in the Tg(bactin:igfbp1a) zebrafish, the β cells make up a larger proportion of the pancreas and organism as a whole. Moreover, we found that Igfbp1a overexpression did not change the number of units of adjacent insulin‐positive pixels (ranging from single β cells to β‐cell clusters) (Fig 6G), but it did significantly increase the average size of the units (Fig 6H), indicating that β‐cell regeneration takes place in existing islets rather than through neogenesis outside the islets. Thus, igfbp1a can potentiate β‐cell regeneration also in 1‐month‐old zebrafish, at least when related to body length or whole‐pancreas size.
Figure 6. Igfbp1a also increases β‐cell regeneration in 1‐month‐old zebrafish.
-
A, BRepresentative confocal projections of the whole pancreas (dashed lines) of 35‐day‐old Tg(ins:H2B‐GFP);Tg(ins:Flag‐NTR) transgenics, with or without Tg(bactin:igfbp1a), that were subjected to β‐cell ablation between day 30 and 31 and then allowed to regenerate for 4 days. Scale bars represent 100 μm.
-
CBody length of control and Tg(bactin:igfbp1a) zebrafish; ****P < 0.0001.
-
D–HQuantification of β‐cell regeneration was automated with an ImageJ script. (D) Insulin‐positive area per zebrafish. (E) Relative insulin‐positive area per body length; **P < 0.01. (F) The percentage of the pancreas area that was insulin‐positive was significantly larger in Tg(bactin:igfbp1a) than in controls; ****P < 0.0001. (G) The total number of recorded units of adjacent ins:H2B‐GFP+ pixels (units ranging from single β cells to β‐cell clusters) did not differ between control and Tg(bactin:igfbp1a) zebrafish. (H) The size of recorded ins:H2B‐GFP+ units was on average larger in Tg(bactin:igfbp1a) than in controls; *P = 0.039. n = 26 in the control group, n = 27 in the Tg(bactin:igfbp1a) group.
IGFBP1 increases the prevalence of insulin‐ and glucagon‐expressing bihormonal cells in both mouse and human islets
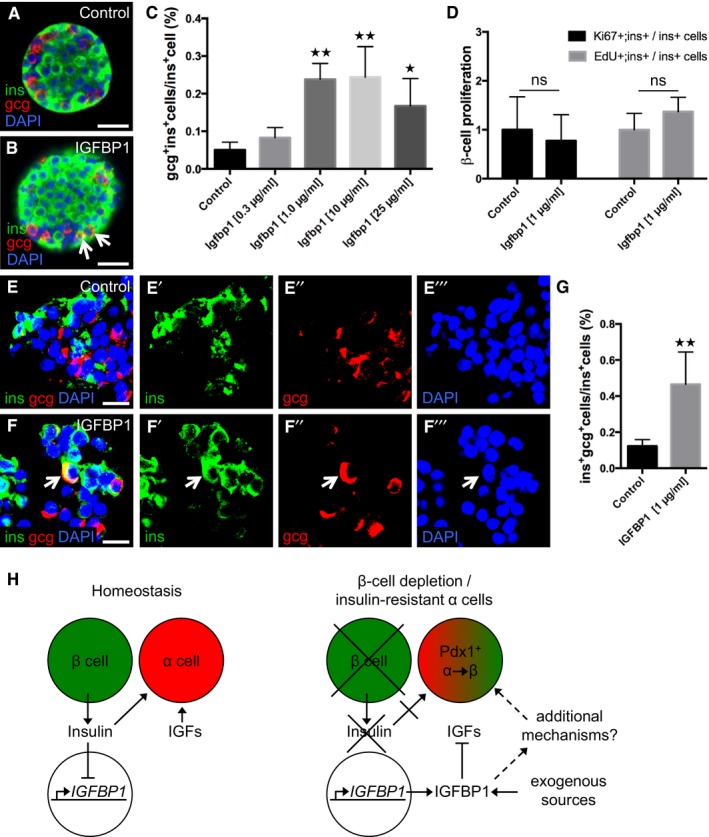
To determine whether IGFBP1's ability to induce transdifferentiation is conserved across species, we first assessed IGFBP1's effect on the prevalence of insulin‐ and glucagon‐expressing bihormonal cells in isolated mouse islets. The presence of such bihormonal cells indicates that transdifferentiation is taking place, though it cannot distinguish between α‐ and β‐cell transdifferentiation and the converse, β‐ to α‐cell transdifferentiation. We found that islets treated in vitro with recombinant mouse IGFBP1 for 3 days had up to four times as many glucagon‐ and insulin‐expressing cells as did vehicle‐treated islets (Fig 7A–C), without having any differences in β‐cell proliferation (Fig 7D).
Figure 7. IGFBP1 increases the prevalence of insulin‐ and glucagon‐expressing bihormonal cells in both mouse and human islets.
-
A, BRepresentative confocal images of control and Igfbp1‐treated mouse islets. gcg+ins+ cells are indicated by arrows. Scale bars: 25 μm.
-
CQuantification of the percentage of gcg+ins+ bihormonal cells per ins+ cells in mouse islets after treatment with increasing concentrations of Igfbp1. P = 0.7304 (Igfbp1 0.3 μg/ml), **P = 0.0023 (1 μg/ml), **P = 0.0013 (10 μg/ml), *P = 0.0410 (25 μg/ml), respectively. Multiple optical sections from 10–50 islets per group, from 4–8 different mice, were used for the quantifications.
-
DQuantification of β‐cell proliferation in mouse islets after treatment with Igfbp1; data displayed as relative values so that the proliferation markers EdU and Ki67 can be compared. Multiple optical sections from 14–22 islets, from 3 different mice, were used for the quantifications.
-
E, FRepresentative confocal images of control or IGFBP1‐treated human islets. A gcg+ins+ cell is indicated by an arrow. For clarity, individual colors for denoting insulin (green), glucagon (red), and DAPI (blue) are also shown separately. Scale bars: 15 μm.
-
GQuantification of the percentage of gcg+ins+ bihormonal cells per ins+ cells in human islets from five donors. **P = 0.0036. Co‐expression of insulin and glucagon is indicative of transdifferentiation, which could reflect either α cells transdifferentiating to β cells or vice versa. **P < 0.0028; n = 115 sections in the control group, n = 147 sections in the IGFBP1 group.
-
HOur proposed mechanism by which the extracellular signals insulin, IGFs, and IGFBP1 can regulate α‐ to β‐cell transdifferentiation.
To determine whether this effect is conserved from zebrafish to humans, we tested whether IGFBP1 can induce the formation of bihormonal cells in human islets. Human islets from five independent donors (without diabetes) were treated with 1 μg/ml of recombinant human IGFBP1 for 3 days and then analyzed by immunohistochemistry (Fig 7E and F). As in mouse islets, the prevalence of insulin‐ and glucagon‐expressing bihormonal cells was significantly greater in IGFBP1‐treated human islets than in vehicle‐treated islets (Fig 7G). Interestingly, although not directly comparable owing to differences in experimental conditions, the baseline occurrence of bihormonal cells was three times as high in human islets as in mouse islets. Thus, IGFBP1 can increase the prevalence of insulin‐ and glucagon‐expressing bihormonal cells in both mouse and human islets cultured in vitro, a finding indicative of transdifferentiation.
High levels of IGFBP1 are associated with a lower risk of developing type‐2 diabetes
To examine a possible role for IGFBP1 in the development of T2D in humans, we reinvestigated the association between serum levels of IGFBP1 and the development of diabetes in a 10‐year prospective study (Lewitt et al, 2008, 2010). Serum levels of IGFBP1 were measured in 1,190 individuals (from a cohort of 7,949 individuals) who were without diabetes at baseline, approximately half of whom had a family history of diabetes (FHD). In both men and women who later developed T2D, fasting levels of IGFBP1 at baseline were significantly lower than in their respective age‐ and FHD‐matched controls, who had normal glucose tolerance (NGT) at follow‐up. Among men, the mean level of IGFBP1 was 14 μg/l (95% CI: 13–17 μg/l) in cases and 21 μg/l (95% CI: 18–24 μg/l) in controls; among women, it was 25 μg/l (95% CI: 21–29 μg/l) in cases and 44 μg/l (95% CI 39–50 μg/l) in controls (P < 0.001 for both sexes). Fasting levels of IGFBP1 were higher among women than among men, both in cases and in controls (P < 0.001). Moreover, there was an inverse correlation between fasting levels of IGFBP1 at baseline and the risk of developing T2D: in both men and women, as the IGFBP1 levels increased the risk of T2D decreased (P for trend < 0.001; odds ratios shown in Table 1). Among subjects in the highest quartile of IGFBP1 levels, there was an estimated risk reduction of 86% in men and 98% in women, compared to subjects in the lowest quartile. Furthermore, there was a progressive risk reduction with increasing levels of IGFBP1 between the quartiles.
Table 1.
High levels of IGFBP1 are associated with a lower risk of developing type‐2 diabetes
| NGT | T2D | P for trend | |||
|---|---|---|---|---|---|
| n | n | OR | 95% CI | ||
| Men (IGFBP1, μg/l) | |||||
| < 11 | 13 | 36 | 1.00 | ||
| 11–18 | 29 | 29 | 0.30 | 0.12–0.76 | |
| 19–28 | 28 | 25 | 0.33 | 0.14–0.77 | |
| > 28 | 37 | 17 | 0.14 | 0.05–0.38 | < 0.001 |
| Women (IGFBP1, μg/l) | |||||
| < 22 | 3 | 26 | 1.00 | ||
| 22–32 | 14 | 15 | 0.17 | 0.04–0.85 | |
| 33–48 | 18 | 12 | 0.07 | 0.01–0.39 | |
| > 48 | 25 | 7 | 0.02 | 0.01–0.13 | < 0.001 |
Odds ratios (ORs) and 95% confidence intervals (CIs) for the association of IGFBP1 serum levels measured at baseline and the development of T2D at 10‐year follow‐up. Men and women were grouped into quartiles according to their baseline level of IGFBP1.
Conditional logistic regression analysis was performed. Cases (T2D; type‐2 diabetes) were pair matched to their respective controls (NGT; normal glucose tolerance) by exact age and by family history of diabetes (negative or positive).
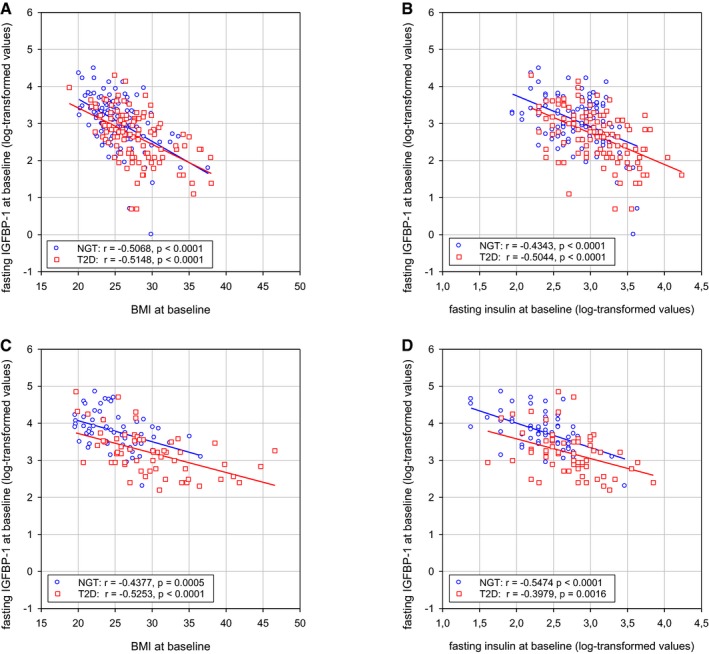
We also found that BMI and fasting levels of insulin correlate inversely with IGFBP1 levels at baseline, both in subjects who later developed T2D and in controls with a NGT at follow‐up (Fig EV4). Because increasing BMI and insulin levels are associated with decreasing insulin sensitivity, and α cells become insulin resistant during the development of T2D (Lee et al, 2014), we propose a model in which neither insulin nor IGF signals to the α cells when IGFBP1 levels are high, thereby enabling the α cells to transdifferentiate to β cells (Fig 7H).
Figure EV4. Correlations of IGFBP1 levels with BMI and insulin levels.
-
A–DCorrelations between baseline levels of fasting IGFBP1 levels and BMI (A, C) or fasting insulin levels (B, D) in men (A, B) and women (C, D). The correlations between these baseline values are significant both in subjects who developed T2D (red squares) and in controls with a normal glucose tolerance (NGT) (blue circles) at follow‐up after 8–10 years. Results are presented as individual values with linear regression. Related to Table 1.
Discussion
In this study, we show that Igfbp1 promotes β‐cell regeneration and that high levels of IGFBP1 are associated with a reduction in the risk of developing diabetes. Using an unbiased, whole‐organism screen, we identified Igfbp1 as a stimulator of β‐cell regeneration in zebrafish. Through overexpression and lineage‐tracing experiments in zebrafish, we then showed that Igfbp1 promotes β‐cell regeneration by increasing α‐ to β‐cell transdifferentiation. Translating these findings, we acquired evidence that IGFBP1 increases α‐ to β‐cell transdifferentiation in mice and humans also: IGFBP1 increased the number of cells co‐expressing insulin and glucagon in cultured mouse and human islets in vitro. Finally, we highlight the relevance of IGFBP1 in diabetes by analyzing data from a large prospective study of the development of T2D, showing that high levels of IGFBP1 are associated with a reduced risk of developing T2D. Our results indicate that Igfbp1 plays a previously unappreciated role in increasing the β‐cell number, a role that may possibly be important in protecting against the development of T2D and in regenerating the β cells in T1D.
Igfbp1 belongs to a family of seven Igfbp members, which affect IGF and insulin signaling in various ways, depending on the context and on their expression patterns (Baxter, 2014). We found that the ability to stimulate β‐cell regeneration was specific to Igfbp1: We overexpressed an additional six Igfbp members and none of them potentiated β‐cell regeneration. Igfbp1‐deficient mice have no phenotype during development or adult homeostasis, but after hepatectomy, they cannot regenerate their livers to the same extent as their wild‐type littermates (Leu et al, 2003). This indicates that Igfbp1 may be a regenerative signal with importance for both hepatocytes and β cells. Given our findings, it would be interesting to examine whether Igfbp1‐deficient mice are more prone to develop diabetes after various challenges (α‐ to β‐cell transdifferentiation might not be relevant for development or at homeostasis in the adult mouse, but only following challenges). Moreover, we did not see an effect of Igfbp1 on δ‐cell regeneration, suggesting that Igfbp1 can promote regeneration of some but not all cell types, even though the IGF1R and the insulin receptor are widely expressed in the pancreas (http://sandberg.cmb.ki.se/pancreas/; Segerstolpe et al, 2016).
In recent years, several important studies of endocrine cell plasticity have emerged, highlighting transdifferentiation as a possible mechanism for expanding the β‐cell mass. Most notably, α‐ to β‐cell transdifferentiation was shown to occur in mice in response to β‐cell ablation (Thorel et al, 2010), a finding that was recently replicated in zebrafish (Ye et al, 2015). In another pioneering study, δ‐ to β‐cell transdifferentiation was shown to be crucial for β‐cell regeneration in newborn mice (Chera et al, 2014). A few studies have characterized factors that can induce transdifferentiation, mainly α‐ to β‐cell transdifferentiation, and emphasized the importance of activation of the transcription factors pdx1 and pax4 (Collombat et al, 2009; Yang et al, 2011; Al‐Hasani et al, 2013), downregulation of the transcription factors arx and foxo1 (Courtney et al, 2013; Chera et al, 2014), and epigenetic modulation (Bramswig et al, 2013). These intracellular factors are indeed likely to be part of a transcriptional program that increases endocrine cell plasticity and β‐cell differentiation. But which are the extracellular signals that regulate the transdifferentiation of α to β cells? From a therapeutic perspective, extracellular signals might be more druggable than transcription factors. Our current findings make a conceptual advance by identifying a targetable link between a physiological response (i.e., to β‐cell depletion) and cellular transformation (i.e., transdifferentiation and ultimately β‐cell regeneration), rather than reflecting the cell‐autonomous networks of transdifferentiation (as is the case with transcription factors). Thus, as an extracellular signal that stimulates α‐ to β‐cell transdifferentiation, Igfbp1 is itself a potential candidate for future development.
IGFBP1 can either stimulate or suppress IGF signaling, depending on the context (Baxter, 2014). We found that inhibiting the IGF signaling pathway mimicked the effect of IGFBP1 (whereas activating it had the opposite effect), indicating that IGFBP1 acts, at least in part, by inhibiting IGF signaling. Because insulin and IGF ligands signal through similar receptors and have some intracellular second messengers in common, it is likely that insulin itself also suppresses α‐ to β‐cell transdifferentiation. However, inhibiting the insulin receptor (or activating the glucagon receptor, which in many cases counteracts insulin signaling) is not a viable approach to increasing the number of β cells in diabetes because decreases in insulin signaling cause glucose levels to rise. By contrast, we found that igfbp1 overexpression accelerated the restoration of a normal glucose level, suggesting that inhibiting IGF signaling can simultaneously increase β‐cell regeneration and restore normoglycemia, though the effect on glucose levels could also depend on Igfbp1's ability to increase insulin sensitivity (Rajwani et al, 2012). Intriguingly, pancreas‐specific inactivation of Igf1 in mice results in islet enlargement and in resistance to high‐fat diet‐induced and streptozotocin‐induced diabetes (Lu et al, 2004); it would be interesting to examine the cellular origin of β cells in such mouse models. Overexpression of Igfbp1 in the mouse has actually been shown to increase the ratio of insulin:glucagon in islets, although genetic lineage tracing was not developed at that time, precluding any conclusion about α‐ to β‐cell transdifferentiation (Dheen et al, 1996, 1997). Whether IGFBP1 itself, direct IGF1R inhibitors, or IGF‐blocking antibodies will prove the most useful promoters of α‐ to β‐cell transdifferentiation will depend on their specificity, in vivo kinetics and tolerability, as well as on whether IGFBP1 can additionally stimulate α‐ to β‐cell transdifferentiation via mechanisms other than inhibition of IGF signaling.
Together with findings from other studies (Thorel et al, 2010; Ye et al, 2015, 2016), our data suggest that it is the balance between insulin/IGFs and glucagon that regulates α‐ to β‐cell transdifferentiation. Insulin signaling can repress IGFBP1 expression (Brismar et al, 1994; Kelley et al, 1996), and as such, it is likely that IGFBP1 is upregulated in a variety of diabetic models and tissues, that is, in response to β‐cell ablation or a decrease in insulin supply. In such a scenario, upregulation of IGFBP1 acts as an effector of insulin deficiency, leading to inhibition of IGFs and further attenuation of the common downstream mediators of insulin and IGF signaling—and thereby potentiating α‐ to β‐cell transdifferentiation (as schematically outlined in Fig 7H). In line with such a role, when IGFBP1 is overexpressed in the mouse under the control of its endogenous promoter, it improves glucose homeostasis (Crossey et al, 2000). Thus, it seems plausible that IGFBP1 serves as a link between insulin deficiency and inhibition of IGF signaling, a synergistic circuit that may come into play during β‐cell regeneration.
To examine a possible role for IGFBP1 in human diabetes, we turned to a 10‐year prospective study, in which we previously showed that having low serum levels of IGFBP1 predicts the development of T2D (Lewitt et al, 2008, 2010), casting IGFBP1 as a possible biomarker for the onset of T2D. Now we show that high serum levels of IGFBP1 are associated with a reduced risk of developing T2D in both men and women with and without a family history of diabetes and provide a mechanistic explanation for this risk reduction (i.e., IGFBP1 increases α‐ to β‐cell transdifferentiation and thus increases the number of β cells). It is possible that sex‐dependent differences in estrogen levels account for our finding that fasting levels of IGFBP1 are higher among women than among men, particularly since oral administration of estrogen to postmenopausal women is associated with an increase in IGFBP1 levels (Heald et al, 2005; Isotton et al, 2012). Conversely, however, circulating estrogen levels have been shown to correlate inversely with IGFBP1 levels in premenopausal women (Unden et al, 2005). Regardless of a possible influence of estrogen on IGFBP1 levels, we do not consider it a confounder for this study because the association between high IGFBP1 levels and reduced risk for developing T2D is seen in both men and women.
Despite individuals with impaired glucose tolerance or prediabetes having hyperinsulinemia, recent findings indicate that development of T2D is associated with the appearance of insulin‐resistant α cells (Lee et al, 2014). Thus, we propose that in individuals who have high levels of IGFBP1, a combined lack of insulin and IGF signaling in α cells in the prediabetes state allows transdifferentiation of α to β cells and thereby counteracts the development of T2D. An alternative explanation could be that the association between IGFBP1 and T2D is due to differences in IGFBP1 expression as a result of altered insulin response/sensitivity or BMI (Brismar et al, 1991; Kotronen et al, 2008). Indeed, we found that both fasting insulin levels and BMI correlate inversely with IGFBP1 levels. Moreover, because these factors are indicators of insulin sensitivity (and we favor a model in which α‐ to β‐cell transdifferentiation occurs when the α cells are insulin resistant), the relevance of these correlations is hard to interpret. Moreover, our new data—from in vivo zebrafish experiments to in vitro mouse and human experiments—all point to an evolutionarily conserved role for IGFBP1 in α‐ to β‐cell transdifferentiation.
Whether α‐ to β‐cell transdifferentiation indeed occurs in T2D is currently unknown. So far it has been shown that the incidence of bihormonal cells expressing glucagon and insulin (or somatostatin and insulin) is abnormally high in non‐diabetic obese individuals and in individuals with T2D (Butler et al, 2013; Yoneda et al, 2013; Mezza et al, 2014). In mice, it has been shown that δ cells transdifferentiate to β cells during the first weeks after birth (Chera et al, 2014), whereas α cells are the main cell type converting to β cells in adult mice (Thorel et al, 2010). In zebrafish, it has previously been shown that there is no δ‐ to β‐cell conversion at early stages (Ye et al, 2015). Thus, the cell types that transdifferentiate to β cells differ somewhat between zebrafish and mice, but it is unclear how this relates to humans. Furthermore, the relative contributions of transdifferentiation and β‐cell proliferation to expansion of the β‐cell mass also show species‐dependent differences. Contrary to rodent β cells, which rely largely on proliferation to expand their β‐cell mass in response to increasing demands for insulin (Porat et al, 2011), human β cells are less prone to proliferation and may rely more heavily on β‐cell neogenesis and transdifferentiation for compensatory increases in β‐cell mass (Butler et al, 2003; Rahier et al, 2008; Hanley et al, 2010). Indeed, whereas IGFs robustly promote proliferation (and thus regeneration) of rodent β cells (Agudo et al, 2008), they do not efficiently promote proliferation of human β cells (Wang et al, 2015). Perhaps reflecting these species‐dependent differences, the basal incidence of glucagon‐ and insulin‐coexpressing cells in our in vitro experiments was higher in human islets than in mouse islets. Together, these findings suggest that formation of new β cells could be critical to preventing the development of T2D and that transdifferentiation could play a greater role in such β‐cell formation than previously appreciated.
In sum, we show that IGFBP1 is a secreted factor that can increase the number of β cells by potentiating α‐ to β‐cell transdifferentiation. We suggest that IGFBP1 may in this way protect against the development of diabetes and is worth studying further in order to define its potential as an intervention for diabetes.
Materials and Methods
Zebrafish experiments
All studies involving animals were performed in accordance with local guidelines and regulations and approved by Stockholms djurförsöksetiska nämnd. The following previously published transgenic zebrafish lines were used: Tg(ins:CFP‐NTR)s892, Tg(ins:Kaede)s949, Tg(ins:H2B‐GFP; ins:dsRed)s960, Tg(ins:Flag‐NTR)s950, Tg(gcga:GFP)ia1, Tg(ins:GFP)zf5, Tg(gcga:Cre; cryaa:YFP)s962, Tg(hsp:loxp‐mCherry‐STOP‐loxP‐H2BGFP; cryaa:CFP)s925 , Tg(ins:dsRed)m1018 , Tg(tp1:H2BmCherry)s939 and Tg(sst2:RFP)gz19. The Tg(bactin:igfbp1a,cmlc2:eGFP)KI103, Tg(bactin:igfbp1a,cmlc2:eGFP)KI104 and Tg(bactin:igfbp1a,cmlc2:eGFP)KI105 lines (expressing 4, 10 and 16 times the endogenous level of igfbp1a, respectively) were generated by multisite gateway cloning (as described below) and stable integration of the resulting construct into the genome by injection into 1–2 cell‐stage embryos. The Tg(sst2:NTR,cryaa:Cerulean)KI102 line was generated by cloning the previously described somatostatin 2 promoter (Li et al, 2009) upstream of nfsB (the gene encoding NTR) in a vector also containing an eye marker (cryaa:Cerulean) transcribed in the reverse direction. The Tg(pcsk1:eGFP)KI106 line was generated by cloning 6,800 bp of genomic sequence upstream of the start codon into the E1b‐GFP‐Tol2‐Gateway vector. The Tg(ins:Venus‐zGeminin; cryaa:Venus)KI107 line was generated by replacing Cre with Venus‐zGeminin in the Tg(ins:Cre;cryaa:Venus)s924 vector (Hesselson et al, 2009).
Mosaic transgenic zebrafish were generated by using the Tol2 transposon system (Kwan et al, 2007). Specifically, 15–20 pg of the expression vector and 20 pg of transposase mRNA were injected into 1–2 cell‐stage embryos. GFP expression under the control of the cmlc2 promoter in the larval hearts was taken as evidence of successful integration of the expression construct (Fig 1D).
We ablated β cells in Tg(ins:CFP‐NTR) or Tg(ins:Flag‐NTR) zebrafish larvae by incubating the larvae in eggwater supplemented with 10 mM metronidazole (MTZ) (Sigma), 1% DMSO (VWR), and 0.2 mM 1‐phenyl‐2‐thiourea (PTU, Acros Organics) from 3 to 4 dpf. Free‐glucose levels were determined by grinding larvae in groups of six and analyzing the lysates in a glucose assay kit (BioVision), as previously described (Jurczyk et al, 2011).
Knockdown of arxa was performed by injecting zebrafish larvae at the one‐cell stage with 2 ng of a previously described morpholino targeting the second intron–exon splice junction, (5′–3′) GCGTCATATTTACCTGGTGAACACA (Gene Tools) (Djiotsa et al, 2012). The standard control morpholino, (5′–3′) CCTCTTACCTCAGTTACAATTTATA (Gene Tools), was used as a reference.
We exposed 1‐month‐old Tg(ins:H2B‐GFP);Tg(ins:Flag‐NTR) zebrafish, with or without Tg(bactin:igfbp1a), to 1 mM of MTZ and 0.1% DMSO for 24 h to ablate their β cells. We sacrificed the zebrafish 4 days after the MTZ treatment and then analyzed their entire pancreas by confocal microscopy with a 10× objective. With this setup and resolution, a single β cell was typically visualized as 30 adjacent insulin‐positive pixels. Insulin‐positive pixels were quantified with ImageJ software, according to the user guide for statistical measurement of image data. We converted the stack of images from a single pancreas into a flattened projection, in which we then measured the total insulin‐positive area, the number of units comprising adjacent insulin‐positive pixels, and the average size of the units (which ranged from single β cells and β‐cell clusters).
Microarray analysis
Tg(ins:CFP‐NTR);Tg(ins:kaede);Tg(sst:dsRed) or Tg(ins:Flag‐NTR);Tg(sst:dsRed) zebrafish larvae were treated with DMSO or MTZ from 3 to 4 dpf, resulting in ablation of their β cells. The larvae were then washed in PBS+ 10% FBS and dissociated by being passed several times through a 5‐ml syringe with a 21G needle. The larval islets were subsequently picked under a fluorescence microscope, and their RNA was isolated with the RNAqueous‐Micro Total RNA Isolation Kit (Ambion). Triplicates from three independent experiments were analyzed with an Affymetrix Zebrafish Gene 1.0 ST Array.
Cloning
The DNA sequences of the candidate genes were amplified by PCR from zebrafish cDNA and first cloned into the middle‐entry vector pDON221; for example, igfbp1a was cloned using a forward primer containing an attB1 adapter, a Kozak sequence (as denoted by lower case letters in the listed sequences), and the upstream coding sequence of the igfbp1a, 5′‐GGGGACAAGTTTGTACAAAAAAGCAGGCTgccaccATGAACAGACTGCTTCTGAAC‐3′, together with a reverse primer containing an attB2 adapter and the downstream coding sequence of igfbp1a, 5′‐GGGGACCACTTTGTACAAGAAAGCTGGGTTCAGTGGTTGAGTTCCTCGG‐3′. Primers used for amplification of the other candidate genes are listed in Table EV1. Expression constructs were then generated by LR recombination by using p5E‐bactin, p3E‐polyA, the middle‐entry vector containing the candidate genes, and the destination vector pDestTol2CG. All constructs were confirmed by sequencing.
RT–PCR
Total RNA was extracted from individual embryos at 4 or 6 dpf by using Trizol (Invitrogen). cDNA for RT–PCR experiments was synthesized with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), and relative mRNA expression levels were determined by SYBR‐based RT–PCR (Bio‐Rad). mRNA levels were normalized to the eef1a1l1 mRNA levels, as an internal standard. The primers used for RT–PCR were as follows: igfbp1a 5′‐ AAGAGAGCATGAAGGCCAAA‐3′ and 5′‐CGCAGTTTGGCAGGTAGAAT‐3′; eef1a1l1 5′‐GTGCTGTGCTGATTGTTGCT‐3′ and 5′‐TGTATGCGCTGACTTCCTTG‐3′.
Immunofluorescence and EdU staining of zebrafish larvae
Immunohistochemistry was performed according to standard procedures and analyzed with a Leica SP8 confocal microscope. The whole endocrine portion of the pancreas was scanned in all larvae analyzed. Confocal stacks were analyzed with ImageJ software. The contrast was adjusted in most images, and Figs 1G and 7A and B were processed with the “smooth” function. The following antibodies were used: anti‐GFP (1:250, Aves Labs GFP‐1020), anti‐Igfbp1 (1:50, Santa Cruz sc‐13097) anti‐dsRed (1:500, Clontech 632496), anti‐Insulin (1:200, Sigma I8510), anti‐Glucagon (1:200, Sigma G6254), and anti‐Pdx1 (gift from Chris Wright). β‐cell proliferation was assessed by adding 10 mM EdU to eggwater supplemented with 10 mM Hepes (Thermo), and measuring EdU incorporation with the Click‐iT EdU Alexa Fluor 647 imaging kit (Invitrogen), according to the manufacturer's protocol.
Chemical treatment and peptide injection
Zebrafish larvae were either incubated in eggwater containing 10 μM of picropodophyllin (PPP) (Santa Cruz Biotechnology) from 4 to 6 dpf or injected in the pericardial cavity with 8 nl of 6 mM JB1 trifluoroacetate salt (Sigma) at 4 dpf.
Isolating and in vitro culturing mouse and human islets
Male C57Bl/6J mice (11–15 weeks old) fed a standard diet were sacrificed by cervical dislocation. Collagenase P (0.3 mg/ml in Hank's buffered salt solution, Roche) was injected into the common bile duct, such that it was distributed to the whole pancreas. The collagenase‐expanded pancreas was dissected out and digested in a collagenase P‐containing solution for 16 min in a 37 °C water bath. After several washes, the islets were handpicked and incubated in RPMI 1640 media (Gibco) supplemented with 10% FBS (Gibco), 1% penicillin–streptomycin (Gibco), and 1% l‐glutamine (Gibco) for 22 h at 37°C in 5% CO2. The islets were then incubated in RPMI 1640 containing either acetonitrile alone (Sigma‐Aldrich) as vehicle control or different concentrations of recombinant mouse IGFBP1 protein (0.3, 1, 10 or 25 μg/ml) (R&D Systems) for 72 h. For proliferation analyses, 10 μM of EdU (C10640, Life technologies) was added to the media.
All studies involving human islets were performed in accordance with local guidelines and regulations and approved by Regionala etikprövningsnämnden i Stockholm. Human islets from five different donors were obtained from the Uppsala University Hospital (through the JDRF award 31‐2008‐416; ECIT Islet for Basic Research program; the Uppsala University Hospital obtained consent for all participating subjects). The islets were from three female and two male non‐diabetic donors (age 53–77; BMI 27.5–34.7; HbA1c 5.4–5.8; ischemia time 10–23 h before islet isolation). The islets were incubated in CMRL 1066 media (Gibco) supplemented with 10% human serum (H45‐22, Sigma‐Aldrich) at 37°C in 5% CO2 for 22 h once received in the laboratory. The islets were then treated with PBS (vehicle control) or 1 μg/ml of recombinant human IGFBP1 protein (R&D Systems) for 72 h. Thereafter, the islets were snap‐frozen in Tissue‐Tek™ (Sakura) and sectioned. Twenty‐micrometer‐thick sections were placed on glass‐slides (Superfrost®Plus; Thermo Scientific) and used in immunofluorescence studies.
Immunofluorescence staining of mouse and human islets
Mouse islets were fixed in 4% formaldehyde for 20 min, washed twice in PBS supplemented with 2.5% bovine serum albumin, and then incubated in a 5% glycine–PBS solution for 15 min. The islets were next incubated in blocking solution (10% FBS, 0.1% Triton X‐100, 5% DMSO in PBS) for one hour. Primary antibodies, guinea pig anti‐insulin at a concentration of 1:600 (A0564; DAKO), mouse anti‐glucagon at 1:200 (G2654; Sigma‐Aldrich), and rabbit anti‐Ki67 at 1:250 (ab15580; Abcam), were added and the incubation continued overnight at 4°C. Islets were then washed in blocking solution and incubated with Alexa Fluor® secondary antibodies (Life technologies) and DAPI (D1306; Life technologies). The stained islets were mounted in Vectashield® (Vector Laboratories Inc) and imaged with a Leica SP8 confocal microscope.
Non‐consecutive cryosections of human islets were used to ensure analyses at different levels within the islets. The sections were air‐dried, fixed with 4% formaldehyde for 10 min, washed twice with PBS, and permeabilized with PBS containing 10% BSA and 0.3% Triton X‐100 for 30 min. Primary antibodies, guinea pig anti‐insulin at 1:600 and mouse anti‐glucagon at 1:100, were added to the sections and incubated overnight at 4°C. The sections were washed three times in PBS, and then, Alexa Fluor® secondary antibodies and DAPI were added to the sections and incubated for 2 h at room temperature. Sections were mounted with Vectashield® (Vector Laboratories) and imaged with a Leica SP8 confocal microscope.
Quantification of islet cells was performed with ImageJ. All nuclei (DAPI positive), insulin‐positive (ins+) cells, glucagon‐positive (gcg+) cells, and double‐positive cells expressing both insulin and glucagon (ins+gcg+) were counted manually. In mouse islets, the number of ins+gcg+ cells in a whole islet was divided by the number of ins+ cells in the same islet. In each treatment group, islets from 3 to 8 different mice were used. For human islets, we counted the number of ins+gcg+ and ins+ cells in non‐consecutive sections (at least 2,000 ins+ cells per donor) and divided the number of double‐positive ins+gcg+ cells by the number of ins+ cells in the section analyzed.
Human study population, assays, and data analysis
Data on the effectiveness of IGFBP1 in reducing the risk of developing T2D are based on previously published studies (Lewitt et al, 2008, 2010), which are incident case–control studies of Swedish men and women. In these studies, the study sample was part of a population‐based prospective survey, the Stockholm Diabetes Prevention Program, described elsewhere (Eriksson et al, 2008), in which participants (3,128 men and 4,821 women aged 35–56 years) were without known diabetes at baseline. By study design, the baseline sample was enriched for individuals with a family history of diabetes (FHD), defined as known diabetes in at least one first‐degree relative or at least two second‐degree relatives; approximately 50% of the study participants had a FHD. Follow‐up was performed 8–10 years later. Both baseline and follow‐up studies consisted of a questionnaire on lifestyle factors, a health examination, and an oral glucose tolerance test (OGTT).
The case–control studies comprised as cases individuals who had normal glucose tolerance (NGT) at baseline and abnormal glucose tolerance at follow‐up. The controls were pair matched to cases (with regard to FHD and age) by random selection among individuals who had NGT at both baseline and follow‐up. The present analysis includes data from 107 men and 60 women (and their corresponding controls) who developed T2D during the course of the study, diagnosed either between baseline and follow‐up or newly diagnosed by OGTT at follow‐up.
Serum levels of IGFBP1 were measured by an in‐house RIA using a polyclonal antibody and human IGFBP1 as standard, as previously described (Povoa et al, 1984); intra‐ and inter‐assay CVs were 3 and 10%, respectively.
Results are presented as the mean and 95% confidence interval (CI). Serum IGFBP1 levels were log transformed before analysis. Conditional logistic regression was performed to calculate ORs and 95% confidence intervals. Values of IGFBP1 levels are categorized in quartiles according to their distribution within the group of T2D cases and their controls. Tests for linear trends were conducted by assigning median values for IGFBP1 levels in the quartiles as continuous variables. In contrast to the analyses in the published studies (Lewitt et al, 2008, 2010), the lowest quartile of IGFBP1 is used as reference in the analyses presented here. Moreover, in contrast to the analyses in Lewitt et al (2008), in the present study all male subjects with T2D at follow‐up are included in the analyses, as are those with fasting glucose levels ≤ 6.0 mM.
Statistical analysis
P‐values < 0.05 were considered statistically significant. Statistical analyses were carried out by t‐tests or Mann‐Whitney tests when two groups were analyzed and by ANOVA or Kruskal‐Wallis tests when more than two groups were analyzed. For smaller sample sizes, n < 10, nonparametric tests were always used. For sample sizes n < 5, individual values are shown in scatterplots, that is, in Fig EV1.
Author contributions
JL, K‐CL, CK, JC, LR, and OA designed, performed, and analyzed the zebrafish experiments. NS conducted the studies of isolated mouse and human islets. PB provided technical and theoretical input and analyzed data. AH, C‐GÖ, and KB conducted and analyzed the prospective human study. OA conceived the study and wrote the manuscript with help from all coauthors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 7
Acknowledgements
We thank Charlotte Mattsson, Joanna Hård, and Mathilda Lönnäs‐Duncranz for assistance/advice on experiments; Tamsin Lindström for comments and edits on the manuscript; Ryan Anderson and Didier Stainier for transgenic zebrafish. This work was supported by grants from the Ragnar Söderberg's foundation, Swedish Research Council, Swedish Foundation for Strategic Research, Wallenberg Institute for Regenerative Medicine, Strategic Research Programme in Stem Cell Research and Regenerative Medicine at the Karolinska Institutet, and Wenner‐Gren Foundation to O.A. The prospective human study was supported by the Swedish Research Council, Stockholm County Council, the Family Erling‐Persson Foundation, Berth von Kantzows Foundation, the Swedish Council for Working Life and Social Research, the Swedish Diabetes Association, Novo Nordisk Scandinavia and GlaxoSmithKline. J.C. thanks the Carl Trygger′s foundation for a postdoctoral fellowship.
The EMBO Journal (2016) 35: 2026–2044
References
- Ackermann AM, Gannon M (2007) Molecular regulation of pancreatic beta‐cell mass development, maintenance, and expansion. J Mol Endocrinol 38: 193–206 [DOI] [PubMed] [Google Scholar]
- Agudo J, Ayuso E, Jimenez V, Salavert A, Casellas A, Tafuro S, Haurigot V, Ruberte J, Segovia JC, Bueren J, Bosch F (2008) IGF‐I mediates regeneration of endocrine pancreas by increasing beta cell replication through cell cycle protein modulation in mice. Diabetologia 51: 1862–1872 [DOI] [PubMed] [Google Scholar]
- Al‐Hasani K, Pfeifer A, Courtney M, Ben‐Othman N, Gjernes E, Vieira A, Druelle N, Avolio F, Ravassard P, Leuckx G, Lacas‐Gervais S, Ambrosetti D, Benizri E, Hecksher‐Sorensen J, Gounon P, Ferrer J, Gradwohl G, Heimberg H, Mansouri A, Collombat P (2013) Adult duct‐lining cells can reprogram into beta‐like cells able to counter repeated cycles of toxin‐induced diabetes. Dev Cell 26: 86–100 [DOI] [PubMed] [Google Scholar]
- Andersson O, Adams BA, Yoo D, Ellis GC, Gut P, Anderson RM, German MS, Stainier DY (2012) Adenosine signaling promotes regeneration of pancreatic beta cells in vivo . Cell Metab 15: 885–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando R, Hama H, Yamamoto‐Hino M, Mizuno H, Miyawaki A (2002) An optical marker based on the UV‐induced green‐to‐red photoconversion of a fluorescent protein. Proc Natl Acad Sci USA 99: 12651–12656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister‐Lock J, Arvanites AC, Rubin LL, Weir G, Melton DA (2012) Adenosine kinase inhibition selectively promotes rodent and porcine islet beta‐cell replication. Proc Natl Acad Sci USA 109: 3915–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeyens L, Lemper M, Leuckx G, De Groef S, Bonfanti P, Stange G, Shemer R, Nord C, Scheel DW, Pan FC, Ahlgren U, Gu G, Stoffers DA, Dor Y, Ferrer J, Gradwohl G, Wright CV, Van de Casteele M, German MS, Bouwens L et al (2014) Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol 32: 76–83 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Baxter RC (2014) IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer 14: 329–341 [DOI] [PubMed] [Google Scholar]
- Bramswig NC, Everett LJ, Schug J, Dorrell C, Liu C, Luo Y, Streeter PR, Naji A, Grompe M, Kaestner KH (2013) Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J Clin Investig 123: 1275–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brismar K, Grill V, Efendic S, Hall K (1991) The insulin‐like growth factor binding protein‐1 in low and high insulin responders before and during dexamethasone treatment. Metabolism 40: 728–732 [DOI] [PubMed] [Google Scholar]
- Brismar K, Fernqvist‐Forbes E, Wahren J, Hall K (1994) Effect of insulin on the hepatic production of insulin‐like growth factor‐binding protein‐1 (IGFBP‐1), IGFBP‐3, and IGF‐I in insulin‐dependent diabetes. J Clin Endocrinol Metab 79: 872–878 [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner‐Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102–110 [DOI] [PubMed] [Google Scholar]
- Butler AE, Campbell‐Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC (2013) Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon‐producing neuroendocrine tumors. Diabetes 62: 2595–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, Furuyama K, Thorel F, Gribble FM, Reimann F, Herrera PL (2014) Diabetes recovery by age‐dependent conversion of pancreatic delta‐cells into insulin producers. Nature 514: 503–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P, Mansouri A, Hecksher‐Sorensen J, Serup P, Krull J, Gradwohl G, Gruss P (2003) Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev 17: 2591–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P, Xu X, Ravassard P, Sosa‐Pineda B, Dussaud S, Billestrup N, Madsen OD, Serup P, Heimberg H, Mansouri A (2009) The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 138: 449–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben‐Othman N, Pfeifer A, Avolio F, Leuckx G, Lacas‐Gervais S, Burel‐Vandenbos F, Ambrosetti D, Hecksher‐Sorensen J, Ravassard P, Heimberg H, Mansouri A, Collombat P (2013) The inactivation of Arx in pancreatic alpha‐cells triggers their neogenesis and conversion into functional beta‐like cells. PLoS Genet 9: e1003934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossey PA, Jones JS, Miell JP (2000) Dysregulation of the insulin/IGF binding protein‐1 axis in transgenic mice is associated with hyperinsulinemia and glucose intolerance. Diabetes 49: 457–465 [DOI] [PubMed] [Google Scholar]
- Curado S, Anderson RM, Jungblut B, Mumm J, Schroeter E, Stainier DY (2007) Conditional targeted cell ablation in zebrafish: a new tool for regeneration studies. Dev Dyn 236: 1025–1035 [DOI] [PubMed] [Google Scholar]
- Dheen ST, Rajkumar K, Murphy LJ (1996) Effects of insulin‐like growth factors (IGF) on pancreatic islet function in IGF binding protein‐1 transgenic mice. Diabetologia 39: 1249–1254 [DOI] [PubMed] [Google Scholar]
- Dheen ST, Rajkumar K, Murphy LJ (1997) Islet cell proliferation and apoptosis in insulin‐like growth factor binding protein‐1 in transgenic mice. The Journal of endocrinology 155: 551–558 [DOI] [PubMed] [Google Scholar]
- Djiotsa J, Verbruggen V, Giacomotto J, Ishibashi M, Manning E, Rinkwitz S, Manfroid I, Voz ML, Peers B (2012) Pax4 is not essential for beta‐cell differentiation in zebrafish embryos but modulates alpha‐cell generation by repressing arx gene expression. BMC Dev Biol 12: 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson AK, Ekbom A, Granath F, Hilding A, Efendic S, Ostenson CG (2008) Psychological distress and risk of pre‐diabetes and Type 2 diabetes in a prospective study of Swedish middle‐aged men and women. Diabet Med 25: 834–842 [DOI] [PubMed] [Google Scholar]
- Goldman D (2014) Regeneration, morphogenesis and self‐organization. Development 141: 2745–2749 [DOI] [PubMed] [Google Scholar]
- Hanley SC, Austin E, Assouline‐Thomas B, Kapeluto J, Blaichman J, Moosavi M, Petropavlovskaia M, Rosenberg L (2010) {beta}‐Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology 151: 1462–1472 [DOI] [PubMed] [Google Scholar]
- Heald A, Kaushal K, Anderson S, Redpath M, Durrington PN, Selby PL, Gibson MJ (2005) Effects of hormone replacement therapy on insulin‐like growth factor (IGF)‐I, IGF‐II and IGF binding protein (IGFBP)‐1 to IGFBP‐4: implications for cardiovascular risk. Gynecol Endocrinol 20: 176–182 [DOI] [PubMed] [Google Scholar]
- Hesselson D, Anderson RM, Beinat M, Stainier DY (2009) Distinct populations of quiescent and proliferative pancreatic beta‐cells identified by HOTcre mediated labeling. Proc Natl Acad Sci USA 106: 14896–14901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isotton AL, Wender MC, Casagrande A, Rollin G, Czepielewski MA (2012) Effects of oral and transdermal estrogen on IGF1, IGFBP3, IGFBP1, serum lipids, and glucose in patients with hypopituitarism during GH treatment: a randomized study. Eur J Endocrinol 166: 207–213 [DOI] [PubMed] [Google Scholar]
- Jurczyk A, Roy N, Bajwa R, Gut P, Lipson K, Yang C, Covassin L, Racki WJ, Rossini AA, Phillips N, Stainier DY, Greiner DL, Brehm MA, Bortell R, diIorio P (2011) Dynamic glucoregulation and mammalian‐like responses to metabolic and developmental disruption in zebrafish. Gen Comp Endocrinol 170: 334–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley KM, Oh Y, Gargosky SE, Gucev Z, Matsumoto T, Hwa V, Ng L, Simpson DM, Rosenfeld RG (1996) Insulin‐like growth factor‐binding proteins (IGFBPs) and their regulatory dynamics. Int J Biochem Cell Biol 28: 619–637 [DOI] [PubMed] [Google Scholar]
- Kinkel MD, Prince VE (2009) On the diabetic menu: zebrafish as a model for pancreas development and function. BioEssays 31: 139–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotronen A, Lewitt M, Hall K, Brismar K, Yki‐Jarvinen H (2008) Insulin‐like growth factor binding protein 1 as a novel specific marker of hepatic insulin sensitivity. J Clin Endocrinol Metab 93: 4867–4872 [DOI] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB (2007) The Tol2kit: a multisite gateway‐based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 236: 3088–3099 [DOI] [PubMed] [Google Scholar]
- Lee Y, Berglund ED, Yu X, Wang MY, Evans MR, Scherer PE, Holland WL, Charron MJ, Roth MG, Unger RH (2014) Hyperglycemia in rodent models of type 2 diabetes requires insulin‐resistant alpha cells. Proc Natl Acad Sci USA 111: 13217–13222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Crissey MA, Craig LE, Taub R (2003) Impaired hepatocyte DNA synthetic response posthepatectomy in insulin‐like growth factor binding protein 1‐deficient mice with defects in C/EBP beta and mitogen‐activated protein kinase/extracellular signal‐regulated kinase regulation. Mol Cell Biol 23: 1251–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewitt MS, Hilding A, Ostenson CG, Efendic S, Brismar K, Hall K (2008) Insulin‐like growth factor‐binding protein‐1 in the prediction and development of type 2 diabetes in middle‐aged Swedish men. Diabetologia 51: 1135–1145 [DOI] [PubMed] [Google Scholar]
- Lewitt MS, Hilding A, Brismar K, Efendic S, Ostenson CG, Hall K (2010) IGF‐binding protein 1 and abdominal obesity in the development of type 2 diabetes in women. Eur J Endocrinol 163: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wen C, Peng J, Korzh V, Gong Z (2009) Generation of living color transgenic zebrafish to trace somatostatin‐expressing cells and endocrine pancreas organization. Differentiation 77: 128–134 [DOI] [PubMed] [Google Scholar]
- Li J, Chu L, Sun X, Liu Y, Cheng CH (2015) IGFs mediate the action of LH on oocyte maturation in zebrafish. Mol Endocrinol 29: 373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Herrera PL, Guo Y, Sun D, Tang Z, LeRoith D, Liu JL (2004) Pancreatic‐specific inactivation of IGF‐I gene causes enlarged pancreatic islets and significant resistance to diabetes. Diabetes 53: 3131–3141 [DOI] [PubMed] [Google Scholar]
- Mezza T, Muscogiuri G, Sorice GP, Clemente G, Hu J, Pontecorvi A, Holst JJ, Giaccari A, Kulkarni RN (2014) Insulin resistance alters islet morphology in nondiabetic humans. Diabetes 63: 994–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir T, Melton DA, Dor Y (2007) Recovery from diabetes in mice by beta cell regeneration. J Clin Investig 117: 2553–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, Weber GJ, Bowman TV, Jang IH, Grosser T, Fitzgerald GA, Daley GQ, Orkin SH, Zon LI (2007) Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 447: 1007–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisharath H, Rhee JM, Swanson MA, Leach SD, Parsons MJ (2007) Targeted ablation of beta cells in the embryonic zebrafish pancreas using E. coli nitroreductase. Mech Dev 124: 218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat S, Weinberg‐Corem N, Tornovsky‐Babaey S, Schyr‐Ben‐Haroush R, Hija A, Stolovich‐Rain M, Dadon D, Granot Z, Ben‐Hur V, White P, Girard CA, Karni R, Kaestner KH, Ashcroft FM, Magnuson MA, Saada A, Grimsby J, Glaser B, Dor Y (2011) Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab 13: 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povoa G, Roovete A, Hall K (1984) Cross‐reaction of serum somatomedin‐binding protein in a radioimmunoassay developed for somatomedin‐binding protein isolated from human amniotic fluid. Acta Endocrinol (Copenh) 107: 563–570 [DOI] [PubMed] [Google Scholar]
- Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC (2008) Pancreatic beta‐cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 10(Suppl 4): 32–42 [DOI] [PubMed] [Google Scholar]
- Rajwani A, Ezzat V, Smith J, Yuldasheva NY, Duncan ER, Gage M, Cubbon RM, Kahn MB, Imrie H, Abbas A, Viswambharan H, Aziz A, Sukumar P, Vidal‐Puig A, Sethi JK, Xuan S, Shah AM, Grant PJ, Porter KE, Kearney MT et al (2012) Increasing circulating IGFBP1 levels improves insulin sensitivity, promotes nitric oxide production, lowers blood pressure, and protects against atherosclerosis. Diabetes 61: 915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue‐Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H, Miyawaki A (2008) Visualizing spatiotemporal dynamics of multicellular cell‐cycle progression. Cell 132: 487–498 [DOI] [PubMed] [Google Scholar]
- Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andreasson A, Sun X, Picelli S, Sabirsh A, Bjursell M, Smith DM, Kasper M, Ämmälä C, Sandberg R (2016) Single‐cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth A, Stemple DL, Barroso I (2013) The emerging use of zebrafish to model metabolic disease. Dis Model Mech 6: 1080–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL (2010) Conversion of adult pancreatic alpha‐cells to beta‐cells after extreme beta‐cell loss. Nature 464: 1149–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unden AL, Elofsson S, Brismar K (2005) Gender differences in the relation of insulin‐like growth factor binding protein‐1 to cardiovascular risk factors: a population‐based study. Clin Endocrinol 63: 94–102 [DOI] [PubMed] [Google Scholar]
- Wang P, Fiaschi‐Taesch NM, Vasavada RC, Scott DK, Garcia‐Ocana A, Stewart AF (2015) Diabetes mellitus–advances and challenges in human beta‐cell proliferation. Nat Rev Endocrinol 11: 201–212 [DOI] [PubMed] [Google Scholar]
- Weir GC, Bonner‐Weir S (2013) Islet beta cell mass in diabetes and how it relates to function, birth, and death. Ann N Y Acad Sci 1281: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YP, Thorel F, Boyer DF, Herrera PL, Wright CV (2011) Context‐specific alpha‐ to‐beta‐cell reprogramming by forced Pdx1 expression. Genes Dev 25: 1680–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Robertson MA, Hesselson D, Stainier DY, Anderson RM (2015) Glucagon is essential for alpha cell transdifferentiation and beta cell neogenesis. Development 142: 1407–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Robertson MA, Mastracci TL, Anderson RM (2016) An insulin signaling feedback loop regulates pancreas progenitor cell differentiation during islet development and regeneration. Dev Biol 409: 354–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda S, Uno S, Iwahashi H, Fujita Y, Yoshikawa A, Kozawa J, Okita K, Takiuchi D, Eguchi H, Nagano H, Imagawa A, Shimomura I (2013) Predominance of beta‐cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. J Clin Endocrinol Metab 98: 2053–2061 [DOI] [PubMed] [Google Scholar]
- Zhao Z, Low YS, Armstrong NA, Ryu JH, Sun SA, Arvanites AC, Hollister‐Lock J, Shah NH, Weir GC, Annes JP (2014) Repurposing cAMP‐modulating medications to promote beta‐cell replication. Mol Endocrinol 28: 1682–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 7